Co-reporter:Yuanjun Li;Yao Tong;Yung H. Wong
Naunyn-Schmiedeberg's Archives of Pharmacology 2015 Volume 388( Issue 2) pp:243-256
Publication Date(Web):2015 February
DOI:10.1007/s00210-014-1066-1
Substantial effort has been directed at elucidating the functions of the products of the Nm23 tumor metastasis suppressor genes over the past two decades, with the ultimate goal of exploring their translational potentials in changing cancer patients’ outcomes. Much attention has been focused on the better-known Nm23-H1, but despite having high sequence similarity, Nm23-H2 functions differently in many aspects. Besides acting as a metastasis suppressor, compelling data suggest that Nm23-H2 may modulate various tumor-associated biological events to enhance tumorigenesis in human solid tumors and hematological malignancies. Linkage to tumorigenesis may occur through the ability of Nm23-H2 to regulate transcription, cell proliferation, apoptosis, differentiation, and telomerase activity. In this review, we examine the linkages of Nm23-H2 to tumorigenesis in terms of its biochemical and structural properties and discuss its potential role in various tumor-associated events.
Co-reporter:Man K. Tse;Christina J. Morris;Mingjie Zhang
Molecular and Cellular Biochemistry 2015 Volume 401( Issue 1-2) pp:27-38
Publication Date(Web):2015 March
DOI:10.1007/s11010-014-2289-7
Activator of G protein signaling 3 (AGS3) is a guanine nucleotide dissociation inhibitor (GDI) which stabilizes the Gαi/o subunits as an AGS3/Gαi/o-GDP complex. It has recently been demonstrated in reconstitution experiments that the AGS3/Gαi/o-GDP complex may act as a substrate of resistance to inhibitors of cholinesterase 8A (Ric-8A), a guanine exchange factor (GEF) for heterotrimeric Gα proteins. Since the ability of Ric-8A to activate Gαi/o subunits that are bound to AGS3 in a cellular environment has not been confirmed, we thus examined the effect of Ric-8A on cAMP accumulation in HEK293 cells expressing different forms of AGS3 and Gαi3. Co-immunoprecipitation assays indicate that full-length AGS3 and its N- and C-terminal truncated mutants can interact with Ric-8A in HEK293 cells. Yeast two-hybrid assay further confirmed that Ric-8A can directly bind to AGS3S, a short form of AGS3 which is endogenously expressed in heart. However, Ric-8A failed to facilitate Gαi-induced suppression of adenylyl cyclase, suggesting that it may not serve as a GEF for AGS3/Gαi/o-GDP complex in a cellular environment.
Co-reporter:Yueqing Hu, Jing Zhu, King H. Chan, Yung H. Wong
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 2) pp:547-552
Publication Date(Web):15 January 2013
DOI:10.1016/j.bmc.2012.10.060
A series of novel and selective N-[3-(6-benzyloxy-3-methoxyphenyl)propyl] amides has recently been shown to possess sub-nanomolar range binding affinity to the type 2 melatonin receptor (MT2). Pharmacokinetics studies suggested that these compounds were subject to vigorous CYP450-mediated metabolism, resulting in a series of metabolites with significantly decreased or diminished binding affinities toward MT2 receptor. The ether bonds were found to be the major positions susceptible to metabolism. In this study, the benzyl ether bond was either removed or replaced with a carbon–carbon bond in an attempt to improve metabolic stability and enhance their resistance towards phase I oxidation. The synthesis, receptor binding affinity, intrinsic potency and metabolic stability of modified structures are reported in this article. By removal or replacement of metabolic labile ether linkerage with carbon linkers, a novel compound was identified with good potency and MT2 selectivity, and with increased metabolic stability.A number of novel compounds were designed, synthesized and evaluated in order to improve metabolic stability of a highly potent and MT2-selective melatonin agonist. Replacement of the ether linkerage with carbon linkers retained the good potency and MT2 selectivity, and removed the metabolic instability at this site.
Co-reporter:Dawna H.T. Kwan;Lisa Y. Yung;Richard D. Ye;Yung H. Wong
Journal of Cellular Biochemistry 2012 Volume 113( Issue 11) pp:3486-3497
Publication Date(Web):
DOI:10.1002/jcb.24225
Abstract
Many Gq-coupled receptors mediate mitogenic signals by stimulating extracellular signal-regulated protein kinases (ERKs) that are typically regulated by the small GTPase Ras. Recent studies have revealed that members of the Gαq family may possess the ability to activate Ras/ERK by interacting with the adaptor protein tetratricopeptide repeat 1 (TPR1). Within the Gαq family, the highly promiscuous Gα14 can relay signals from numerous receptors. Here, we examined if Gα14 interacts with TPR1 to stimulate Ras signaling pathways. Expression of the constitutively active Gα14QL mutant in HEK293 cells led to the formation of GTP-bound Ras as well as increased phosphorylations of downstream signaling molecules including ERK and IκB kinase. Stimulation of endogenous G14-coupled somatostatin type 2 and α2-adrenergic receptors produced similar responses in human hepatocellular HepG2 carcinoma cells. Co-immunoprecipitation assays using HEK293 cells demonstrated a stronger association of TPR1 for Gα14QL than Gα14, suggesting that TPR1 preferentially binds to the GTP-bound form of Gα14. Activated Gα14 also interacted with the Ras guanine nucleotide exchange factors SOS1 and SOS2. Expression of a dominant negative mutant of TPR1 or siRNA-mediated knockdown of TPR1 effectively abolished the ability of Gα14 to induce Ras signaling in native HepG2 or transfected HEK293 cells. Although expression of the dominant negative mutant of TPR1 suppressed Gα14QL-induced phosphorylations of ERK and IκB kinase, it did not affect Gα14QL-induced stimulation of phospholipase Cβ or c-Jun N-terminal kinase. Our results suggest that TPR1 is required for Gα14 to stimulate Ras-dependent signaling pathways, but not for the propagation of signals along Ras-independent pathways. J. Cell. Biochem. 113: 3486–3497, 2012. © 2012 Wiley Periodicals, Inc.
Co-reporter:Angel K. C. Ip;Prudence H. Tso;Maggie M. K. Lee
Molecular and Cellular Biochemistry 2012 Volume 362( Issue 1-2) pp:159-168
Publication Date(Web):2012 March
DOI:10.1007/s11010-011-1138-1
Regulators of G protein signaling (RGS proteins) serve as GTPase activating proteins for the signal transducing Gα subunits. RGS19, also known as Gα-interacting protein (GAIP), has been shown to subserve other functions such as the regulation of macroautophagy and growth factor signaling. We have recently demonstrated that the expression of RGS19 in human embryonic kidney (HEK) 293 cells resulted in the disruption of serum-induced mitogenic response along the classical Ras/Raf/MEK/ERK pathway. Here, we further examined the effect of RGS19 expression on the stress-activated protein kinases (SAPKs). Both c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) became non-responsive to serum in 293/RGS19 cells, yet the two SAPKs responded to UV irradiation or osmotic stress induced by sorbitol. Kinases upstream of JNK and p38 MAPK, including MKK3/6, MKK4, and MLK3, also failed to respond to serum stimulation in 293/RGS19 cells. Serum-induced activation of the small GTPases Rac1 and Cdc42 was similarly suppressed in these cells. Our results indicate that elevated expression of RGS19 can severely disrupt the regulation of MAPKs by small GTPases.
Co-reporter:Yueqing Hu, Maurice K.C. Ho, King H. Chan, David C. New, Yung H. Wong
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 8) pp:2582-2585
Publication Date(Web):15 April 2010
DOI:10.1016/j.bmcl.2010.02.084
A series of substituted N-[3-(3-methoxyphenyl)propyl] amides were synthesized and their binding affinities towards human melatonin MT1 and MT2 receptors were evaluated. It was discovered that a benzyloxyl substituent incorporated at C6 position of the 3-methoxyphenyl ring dramatically enhanced the MT2 binding affinity and at the same time decreased MT1 binding affinity.A benzyloxyl substituent incorporated at C6 position of the 3-methoxyphenyl ring dramatically enhanced the melatonin MT2-binding affinity.
Co-reporter:Prudence H. Tso;Christina J. Morris;Lisa Y. Yung;Nancy Y. Ip
Neurochemical Research 2009 Volume 34( Issue 6) pp:1101-1112
Publication Date(Web):2009 June
DOI:10.1007/s11064-008-9880-9
Nerve growth factor (NGF)-mediated activation of mitogen-activated protein kinases (MAPK) is critical for differentiation and apoptosis of PC12 cells. Since NGF employs stress-activated c-Jun N-terminal kinase (JNK) to regulate both programmed cell death and neurite outgrowth of PC12 cells, we examined NGF-regulated JNK activity and the role of Gi/o proteins. Induction of JNK phosphorylation by NGF occurred in a time- and dose-dependent manner and was partially inhibited by pertussis toxin (PTX). To discern the participation of various signaling intermediates, PC12 cells were treated with specific inhibitors prior to NGF challenge. NGF-elevated JNK activity was abolished by inhibitors of JNK, p38 MAPK, Src, JAK3 and MEK1/2. NGF-dependent JNK phosphorylation became insensitive to PTX treatment upon transient expressions of Gαz or the PTX-resistant mutants of Gαi1–3 and GαoA. Collectively, these studies indicate that NGF-dependent JNK activity may be mediated via Gi1–3 proteins, JAK3, Src, p38 MAPK and the MEK/ERK cascade.
Co-reporter:May YM Yu;Maurice KC Ho;Andrew MF Liu;Yung H Wong
Journal of Molecular Signaling 2008 Volume 3( Issue 1) pp:
Publication Date(Web):2008 December
DOI:10.1186/1750-2187-3-17
Gα16 can activate phospholipase Cβ (PLCβ) directly like Gαq. It also couples to tetratricopeptide repeat 1 (TPR1) which is linked to Ras activation. It is unknown whether PLCβ and TPR1 interact with the same regions on Gα16. Previous studies on Gαq have defined two minimal clusters of amino acids that are essential for the coupling to PLCβ. Cognate residues in Gα16 might also be essential for interacting with PLCβ, and possibly contribute to TPR1 interaction and other signaling events.Alanine mutations were introduced to the two amino acid clusters (246–248 and 259–260) in the switch III region and α3 helix of Gα16. Regulations of PLCβ and STAT3 were partially weakened by each cluster mutant. A mutant harboring mutations at both clusters generally produced stronger suppressions. Activation of Jun N-terminal kinase (JNK) by Gα16 was completely abolished by mutating either clusters. Contrastingly, phosphorylations of extracellular signal-regulated kinase (ERK) and nuclear factor κB (NF-κB) were not significantly affected by these mutations. The interactions between the mutants and PLCβ2 and TPR1 were also reduced in co-immunoprecipitation assays. Coupling between G16 and different categories of receptors was impaired by the mutations, with the effect of switch III mutations being more pronounced than those in the α3 helix. Mutations of both clusters almost completely abolished the receptor coupling and prevent receptor-induced Gβγ release.The integrity of the switch III region and α3 helix of Gα16 is critical for the activation of PLCβ, STAT3, and JNK but not ERK or NF-κB. Binding of Gα16 to PLCβ2 or TPR1 was reduced by the mutations of either cluster. The same region could also differentially affect the effectiveness of receptor coupling to G16. The studied region was shown to bear multiple functionally important roles of G16.
Co-reporter:Dawna H. T. Kwan;Angel Y. F. Kam;Yung H. Wong
Neurochemical Research 2008 Volume 33( Issue 1) pp:125-133
Publication Date(Web):2008 January
DOI:10.1007/s11064-007-9425-7
The human formyl peptide receptor like 1 (FPRL-1) is a variant of the Gi-coupled formyl-peptide receptor. Functional FPRL-1 is endogenously expressed in the U87 astrocytoma cell line and there is accumulating evidence to suggest that FPRL-1 may be involved in neuroinflammation associated with the pathogenesis of Alzheimer’s disease. In this study, we examined the ability of FPRL-1 to mobilize intracellular Ca2+ in U87 astrocytoma cells, as well as in Chinese hamster ovary (CHO) cells stably expressing FPRL-1. We showed that Trp–Lys–Tyr–Met–Val–Met–NH2 (WKYMVM), a specific agonist for FPRL-1, stimulated Ca2+ influx in both U87 and FPRL-1/CHO cells. These effects can be inhibited by the FPRL-1 selective antagonist, WRW4. Involvement of Gi proteins was demonstrated with the use of pertussis toxin, while inhibitors of store-operated channels (SOC) including 1-[2-(4-methoxyphenyl)]-2-[3-(4-methpxyphenyl)propoxy]ethyl-1H-imidazole hydrochloride (SKF96365) and 2-aminoethoxydiphenyl borate (2-APB) were found to abolish the WKYMVM-induced Ca2+ increase. However, intracellular Ca2+ mobilization in both cell lines were unaffected by the phospholipase Cβ inhibitor U73122 or selective ryanodine receptor inhibitors. Our data demonstrated that activation of Gi-coupled FPRL-1 can lead to Ca2+ influx possibly via SOCs in U87 and FPRL-1/CHO cells.
Co-reporter:Andrew M. F. Liu;Yung H. Wong;Angel Y. F. Kam
Journal of Neurochemistry 2007 Volume 103(Issue 4) pp:1553-1566
Publication Date(Web):30 JUL 2007
DOI:10.1111/j.1471-4159.2007.04876.x
Formyl peptide-receptor like-1 (FPRL-1) may possess critical roles in Alzheimer’s diseases, chemotaxis and release of neurotoxins, possibly through its regulation of nuclear factor-κB (NFκB). Here we illustrate that activation of FPRL-1 in human U87 astrocytoma or Chinese hamster ovary cells stably expressing the receptor resulted in the phosphorylations of inhibitor-κB kinase (IKK), an onset kinase for NFκB signaling cascade. FPRL-1 selective hexapeptide Trp-Lys-Tyr-Met-Val-Met (WKYMVM) promoted IKK phosphorylations in time- and dose-dependent manners while pre-treatment of pertussis toxin abrogated the Gαi/o-dependent stimulations. The FPRL-1-mediated IKK phosphorylation required extracellular signal-regulated protein kinase (ERK), phosphatidylinositol 3-kinase and cellular Src (c-Src), but not c-Jun N-terminal kinase and p38 mitogen-activated protein kinase. Despite its ability to mobilize Ca2+, WKYMVM did not require Ca2+ for the modulation of IKK phosphorylation. Activation of FPRL-1 also induced NFκB-driven luciferase expression. Interestingly, cholesterol depletion from plasma membrane by methyl-β-cyclodextrin abolished the FPRL-1-stimulated IKK phosphorylation, denoting the important role of lipid raft integrity in the FPRL-1 to IKK signaling. Furthermore, we demonstrated that in U87 cells, several signaling intermediates in the FPRL-1-IKK pathway including Gαi2, c-Src and ERK were constitutively localized at the raft microdomains. WKYMVM administration not only resulted in higher amount of ERK recruitment to the raft region, but also specifically stimulated raft-associated c-Src and ERK phosphorylations. Taken together, these results demonstrate that FPRL-1 is capable of activating NFκB signaling through IKK phosphorylation and this may serve as a useful therapeutical target for FPRL-1-related diseases.
Co-reporter:Anthony S. L. Chan;Linda W. C. Ng;Lydia S. W. Poon;Winnie W. Y. Chan
Apoptosis 2007 Volume 12( Issue 1) pp:167-179
Publication Date(Web):2007 January
DOI:10.1007/s10495-006-0524-8
Dopamine and norepinephrine are neurotransmitters which participate in various regulatory functions of the human brain. These functions are lost in neurodegenerative diseases including Parkinson’s disease and Alzheimer’s disease. In this study, we used SK-N-MC neuroblastoma cells to investigate the cytotoxicities of high concentrations of dopamine and norepinephrine on neuronal cells. Dopamine, norepinephrine, as well as their corresponding synthetic agonists (SKF38393 and isoproterenol, respectively) triggered SK-N-MC cell death when applied at 50–100 μM persistently for 2 days. This catecholamine-induced cell death appears to be neuronal specific, as demonstrated by their inabilities of triggering apoptosis of A549 lung carcinoma cells and Cos-7 kidney fibroblasts. By pretreating SK-N-MC cells with target-specific inhibitors before administration of catecholamine, components of G protein signaling (i.e. Gs/cAMP/PKA), monoamine oxidases, nitric oxide synthase, c-Jun N-terminal kinase and oxidative stress were found to be involved in this dopamine/norepinephrine-induced cytotoxicity, which subsequently led to caspase-dependent and -independent apoptotic responses as well as DNA degradation. In contrast, agonists of Gi-coupled dopamine receptors and adrenergic receptors (quinpirole and UK14,304, respectively) were incapable of triggering apoptosis of SK-N-MC cells. Our results suggest that both G protein (Gs)-mediated signaling cascade and oxidative stress participate in the dopamine/norepinephrine-induced neuronal apoptosis.
Co-reporter:David C New;Yung H Wong
Journal of Molecular Signaling 2007 Volume 2( Issue 1) pp:
Publication Date(Web):2007 December
DOI:10.1186/1750-2187-2-2
G protein-coupled receptors are key regulators of cellular communication, mediating the efficient coordination of a cell's responses to extracellular stimuli. When stimulated these receptors modulate the activity of a wide range of intracellular signalling pathways that facilitate the ordered development, growth and reproduction of the organism. There is now a growing body of evidence examining the mechanisms by which G protein-coupled receptors are able to regulate the expression, activity, localization and stability of cell cycle regulatory proteins that either promote or inhibit the initiation of DNA synthesis. In this review, we will detail the intracellular pathways that mediate the G protein-coupled receptor regulation of cellular proliferation, specifically the progression from the G1 phase to the S phase of the cell cycle.
Co-reporter:E. C. H. Yip;A. S. L. Chan;H. Pang;Y. K. Tam;Y. H. Wong
Cell Biology and Toxicology 2007 Volume 23( Issue 2) pp:139
Publication Date(Web):2007 March
DOI:10.1007/s10565-006-9000-z
Co-reporter:Anthony S L Chan;Yung H Wong
British Journal of Pharmacology 2004 Volume 142(Issue 4) pp:
Publication Date(Web):29 JAN 2009
DOI:10.1038/sj.bjp.0705851
Signaling networks involving different receptor systems allow extracellular signals to be integrated and transformed into various biological activities. In this report, we studied the activity of the c-Jun N-terminal kinase (JNK) subgroup of mitogen-activated protein kinases (MAPKs), in response to stimulation by G protein-coupled receptors (GPCRs) and co-activation with epithermal growth factor receptor (EGFR).
Stimulation of exogenous GPCRs in Cos-7 cells induced JNK activation of different magnitudes depending on their G-protein coupling specificities (Gq>Gi>Gs), and a moderate JNK activation was linked to stimulation of endogenous EGFR by EGF.
Co-stimulation with GPCR agonists and EGF resulted in differential augmentation of JNK activities, with Gi-coupled receptors associated with a synergistic JNK activation upon co-stimulation with EGF, while Gq- and Gs-coupled receptors were incapable of triggering this effect.
This Gi/EGF-induced synergistic JNK activation was inhibited by pertussis toxin and AG1478, and may involve Src family tyrosine kinases, PI3 K, Ca2+/calmodulin and small GTPases as important intermediates, while Ca2+ mobilization was triggered by the stimulation of Gq-coupled receptor or EGF treatment, but not by the Gi- or Gs-coupled receptors.
Transient expression of Gβγ subunits with EGF treatment, or co-activation of exogenous Gi-coupled receptor with thapsigargin also resulted in a synergistic JNK activation. Activation of Gi-coupled receptor accompanied with EGF treatment enhanced the expression level and activity of MAPK phosphatase type I, which occurred after the maximal synergistic JNK activation.
Our results support a mechanistic model where EGF signaling may differentially regulate the JNK activities triggered by GPCRs of different coupling specificities.
Signaling networks involving different receptor systems allow extracellular signals to be integrated and transformed into various biological activities. In this report, we studied the activity of the c-Jun N-terminal kinase (JNK) subgroup of mitogen-activated protein kinases (MAPKs), in response to stimulation by G protein-coupled receptors (GPCRs) and co-activation with epithermal growth factor receptor (EGFR).
Stimulation of exogenous GPCRs in Cos-7 cells induced JNK activation of different magnitudes depending on their G-protein coupling specificities (Gq>Gi>Gs), and a moderate JNK activation was linked to stimulation of endogenous EGFR by EGF.
Co-stimulation with GPCR agonists and EGF resulted in differential augmentation of JNK activities, with Gi-coupled receptors associated with a synergistic JNK activation upon co-stimulation with EGF, while Gq- and Gs-coupled receptors were incapable of triggering this effect.
This Gi/EGF-induced synergistic JNK activation was inhibited by pertussis toxin and AG1478, and may involve Src family tyrosine kinases, PI3 K, Ca2+/calmodulin and small GTPases as important intermediates, while Ca2+ mobilization was triggered by the stimulation of Gq-coupled receptor or EGF treatment, but not by the Gi- or Gs-coupled receptors.
Transient expression of Gβγ subunits with EGF treatment, or co-activation of exogenous Gi-coupled receptor with thapsigargin also resulted in a synergistic JNK activation. Activation of Gi-coupled receptor accompanied with EGF treatment enhanced the expression level and activity of MAPK phosphatase type I, which occurred after the maximal synergistic JNK activation.
Our results support a mechanistic model where EGF signaling may differentially regulate the JNK activities triggered by GPCRs of different coupling specificities.
British Journal of Pharmacology (2004) 142, 635–646. doi:10.1038/sj.bjp.0705851
Co-reporter:Maurice K C Ho;Lisa Y Yung;Joy S C Chan;Jasmine H P Chan;Cecilia S S Wong;Yung H Wong
British Journal of Pharmacology 2001 Volume 132(Issue 7) pp:
Publication Date(Web):29 JAN 2009
DOI:10.1038/sj.bjp.0703933
The bovine Gα14 is a member of the Gq subfamily of G proteins that can regulate phospholipase Cβ isoforms but the extent to which Gα14 recognizes different receptor classes is not known.
Gα14 was cotransfected with a variety of receptors in COS-7 cells, and agonist-induced stimulation of phospholipase C was then measured.
Activation of the type 2 but not type 1 somatostatin receptor in cells coexpressing Gα14 stimulated the accumulation of inositol phosphates; functional expression of both subtypes of somatostatin receptors was determined by the ability of somatostatin to inhibit cyclic AMP accumulation.
Among the three opioid receptors (μ, δ, and κ), only the δ receptor was capable of stimulating IP formation when coexpressed with Gα14 in COS-7 cells.
A panel of Gi- and Gs-linked receptors was screened for their ability to stimulate IP accumulation via Gα14. The adenosine A1, complement C5a, dopamine D1, D2 and D5, formyl peptide, luteinizing hormone, secretin, and the three subtypes of melatonin (mt1, MT2, and Xenopus) receptors were all incapable of activating Gα14, while the α2- and β2-adrenoceptors were able to do so.
Gα14-mediated stimulation of phospholipase Cβ was agonist dose-dependent. These data demonstrate that although Gα14 can interact with different classes of receptors, it is much less promiscuous than Gα15 or Gα16.
The bovine Gα14 is a member of the Gq subfamily of G proteins that can regulate phospholipase Cβ isoforms but the extent to which Gα14 recognizes different receptor classes is not known.
Gα14 was cotransfected with a variety of receptors in COS-7 cells, and agonist-induced stimulation of phospholipase C was then measured.
Activation of the type 2 but not type 1 somatostatin receptor in cells coexpressing Gα14 stimulated the accumulation of inositol phosphates; functional expression of both subtypes of somatostatin receptors was determined by the ability of somatostatin to inhibit cyclic AMP accumulation.
Among the three opioid receptors (μ, δ, and κ), only the δ receptor was capable of stimulating IP formation when coexpressed with Gα14 in COS-7 cells.
A panel of Gi- and Gs-linked receptors was screened for their ability to stimulate IP accumulation via Gα14. The adenosine A1, complement C5a, dopamine D1, D2 and D5, formyl peptide, luteinizing hormone, secretin, and the three subtypes of melatonin (mt1, MT2, and Xenopus) receptors were all incapable of activating Gα14, while the α2- and β2-adrenoceptors were able to do so.
Gα14-mediated stimulation of phospholipase Cβ was agonist dose-dependent. These data demonstrate that although Gα14 can interact with different classes of receptors, it is much less promiscuous than Gα15 or Gα16.
British Journal of Pharmacology (2001) 132, 1431–1440; doi:10.1038/sj.bjp.0703933
Co-reporter:Wendy W.S. Yeung, Yung H. Wong
Cellular Signalling (September 2010) Volume 22(Issue 9) pp:1379-1387
Publication Date(Web):1 September 2010
DOI:10.1016/j.cellsig.2010.05.008
Phosphatidylinositol 3-kinase (PI3K) mediates receptor tyrosine kinase and G protein coupled receptor (GPCR) signaling by phosphorylating phosphoinositides to elicit various biological responses. Gαq has previously been shown to inhibit class IA PI3K by interacting with the p110α subunit. However, it is not known if PI3Ks can associate with other Gαq family members such as Gα16. Here, we demonstrated that class IA PI3Ks, p85/p110α and p85/p110β, could form stable complexes with wild type Gα16 and its constitutively active mutant (Gα16QL) in HEK293 cells. In contrast, no interaction between Gα16 and class IB PI3K was observed. The Gα16/p110α signaling complex could be detected in hematopoietic cells that endogenously express Gα16. Overexpression of class I PI3Ks did not inhibit Gα16QL-induced IP3 production and, unlike p63RhoGEF, class IA PI3Ks did not attenuate the binding of PLCβ2 to Gα16QL. On the contrary, the function of class IA PI3Ks was suppressed by Gα16QL as revealed by diminished production of PIP3 as well as inhibition of EGF-induced Akt phosphorylation. Taken together, these results suggest that Gα16 can bind to class IA PI3Ks and inhibit the PI3K signaling pathway.
Co-reporter:Lei Yang, Maggie M.K. Lee, Manton M.H. Leung, Yung H. Wong
Cellular Signalling (November 2016) Volume 28(Issue 11) pp:1663-1672
Publication Date(Web):1 November 2016
DOI:10.1016/j.cellsig.2016.07.017
•Expression of RGS20 enhanced tumor cell aggregation, migration, and invasion.•shRNA-mediated knockdown of RGS20 in tumor cell lines impaired these responses.•RGS20 elevated the expression of vimentin but reduced the expression of E-cadherin.Several RGS (regulator of G protein signaling) proteins are known to be upregulated in a variety of tumors but their roles in modulating tumorigenesis remain undefined. Since the expression of RGS20 is elevated in metastatic melanoma and breast tumors, we examined the effects of RGS20 overexpression and knockdown on the cell mobility and adhesive properties of different human cancer cell lines, including cervical cancer HeLa, breast adenocarcinoma MDA-MB-231, and non-small cell lung carcinoma H1299 and A549 cells. Expression of RGS20 enhanced cell aggregation, migration, invasion and adhesion as determined by hanging drop aggregation, wound healing, transwell chamber migration and invasion assays. Conversely, shRNA-mediated knockdown of endogenous RGS20 impaired these responses. In addition, RGS20 elevated the expression of vimentin (a mesenchymal cell marker) but down-regulated the expression of E-cadherin, two indicators commonly associated with metastasis. These results suggest that the expression of RGS20 may promote metastasis of tumor cells.
Co-reporter:Prudence H. Tso, Yingchun Wang, Sivia Y.S. Wong, Lydia S.W. Poon, Anthony S.L. Chan, Yung H. Wong
Cellular Signalling (November 2010) Volume 22(Issue 11) pp:1700-1707
Publication Date(Web):1 November 2010
DOI:10.1016/j.cellsig.2010.06.010
Regulator of G protein signaling 19 (RGS19), also known as Gα-interacting protein (GAIP), is a GTPase activating protein (GAP) for Gαi subunits. Apart from its GAP function, RGS19 has been implicated in growth factor signaling through binding to GAIP-interacting protein C-terminus (GIPC) via its C-terminal PDZ-binding motif. To gain additional insight on its function, we have stably expressed RGS19 in a number of mammalian cell lines and examined its effect on cell proliferation. Interestingly, overexpression of RGS19 stimulated the growth of HEK293, PC12, Caco2, and NIH3T3 cells. This growth promoting effect was not shared by other RGS proteins including RGS4, RGS10 and RGS20. Despite its ability to stimulate cell proliferation, RGS19 failed to induce neoplastic transformation in NIH3T3 cells as determined by focus formation and soft-agar assays, and it did not induce tumor growth in athymic nude mice. Deletion mutants of RGS19 lacking the PDZ-binding motif failed to complex with GIPC and did not exhibit any growth promoting effect. Overexpression of GIPC alone in HEK293 cells stimulated cell proliferation whereas its knockdown in H1299 non-small cell lung carcinomas suppressed cell proliferation. This study demonstrates that RGS19, in addition to acting as a GAP, is able to stimulate cell proliferation in a GIPC-dependent manner.
Co-reporter:Prudence H. Tso, Yingchun Wang, Lisa Y. Yung, Yao Tong, Maggie M.K. Lee, Yung H. Wong
Cellular Signalling (May 2013) Volume 25(Issue 5) pp:1064-1074
Publication Date(Web):1 May 2013
DOI:10.1016/j.cellsig.2013.02.010
Besides serving as signal terminators for G protein pathways, several regulators of G protein signaling (RGS) can also modulate cell proliferation. RGS19 has previously been shown to enhance Akt signaling despite impaired Ras signaling. The present study examines the mechanism by which RGS19 inhibits Ras signaling. In HEK293 cells stably expressing RGS19, serum-induced Ras activation and phosphorylations of Raf/MEK/ERK were significantly inhibited, while cells expressing RGS2, 4, 7, 8, 10, or 20 did not exhibit this inhibitory phenotype. Conversely, siRNA-mediated knockdown of RGS19 enabled partial recovery of serum-induced ERK phosphorylation. Interestingly, two isoforms of the tumor metastasis suppressor Nm23 (H1 and H2) were upregulated in 293/RGS19 cells. As a nucleoside diphosphate kinase, Nm23H1 can phosphorylate the kinase suppressor of Ras (KSR). Elevated levels of phosphorylated KSR were indeed detected in the nuclear fractions of 293/RGS19 cells. Co-immunoprecipitation assays revealed that Nm23H1/2 can form complexes with RGS19, Ras, or KSR. siRNA-mediated knockdown of Nm23H1/2 allowed 293/RGS19 cells to partially recover their ERK responses to serum treatment, while overexpression of Nm23H1/2 in HEK293 cells suppressed the serum-induced ERK response. This study demonstrates that expression of RGS19 can suppress Ras-mediated signaling via upregulation of Nm23.Highlights►Raf/MEK/ERK phosphorylations were inhibited in cells expressing RGS19. ► Nm23 (H1 and H2) were upregulated in the same cells. ► Elevated levels of phosphorylated KSR were detected in the nuclear fractions. ► Nm23 H1/2 formed complexes with RGS19, Ras and KSR. ► RGS19 expression could suppress Ras-mediated signaling via upregulation of Nm23.
Co-reporter:Hua Mei, Maurice K.C. Ho, Lisa Y. Yung, Zhenguo Wu, Nancy Y. Ip, Yung H. Wong
Cellular Signalling (February 2011) Volume 23(Issue 2) pp:389-397
Publication Date(Web):1 February 2011
DOI:10.1016/j.cellsig.2010.10.009
The recent identification of Gαz expression in C2C12 myoblasts and its demonstrated interaction with the transcription factor Eya2 inferred an unanticipated role of Gαz in muscle development. In the present study, endogenous Gαz mRNA and protein expressions in C2C12 cells increased upon commencement of myogenesis and peaked at around 4–6 days after induction but were undetectable in adult skeletal muscle. Surprisingly, stable expression of recombinant Gαz in C2C12 myoblasts strongly suppressed myotube formation upon serum deprivation, and the constitutively active mutant GαzQL exerted more pronounced effects. Transcriptional activities of reporter genes responsive to early (MyoD, MEF2 and myogenin) and late (muscle creatine kinase and myosin heavy chain) myogenic markers were reduced by transiently expressed GαzQL. Membrane attachment of Gαz was apparently required for the suppressive effects because a fatty acylation-deficient Gαz mutant could not inhibit myogenin expression. Introduction of siRNA against Gαz enhanced myogenin-driven luciferase activity and increased myosin heavy chain expression. Immunostaining of C2C12 cells over-expressing Gαz showed delayed nuclear expression of myogenin and severe myotube deformation. Gαz expression was accompanied by reduced levels of Rock2, RhoA and RhoGAP, enhanced expression of Rnd3, and a reduction of serum-responsive factor-driven reporter activity. These results support a novel role of Gαz in restraining myogenic differentiation through the disruption of Rho signaling.
Co-reporter:Hao Zuo, Anthony S.L. Chan, Hermann Ammer, Yung H. Wong
Cellular Signalling (December 2013) Volume 25(Issue 12) pp:2440-2452
Publication Date(Web):1 December 2013
DOI:10.1016/j.cellsig.2013.08.019
•Fhit expression is up-regulated specifically by Gq-coupled receptors.•Gq up-regulates Fhit expression by increasing Fhit translation through PKC/MEK.•Activated Gaq increases cell-cell adhesion through Fhit.The tumor suppressor Fhit protein is defective or absent in many tumor cells due to methylation, mutation or deletion of the FHIT gene. Despite numerous attempts to unravel the functions of Fhit, the mechanisms by which the function and expression of Fhit are regulated remain poorly understood. We have recently shown that activated Gαq subunits interact directly with Fhit and enhance its inhibitory effect on cell growth. Here we investigated the regulation of Fhit expression by Gq. Our results showed that Fhit was up-regulated specifically by activating Gα subunits of the Gq subfamily but not by those of the other G protein subfamilies. This up-regulation effect was mediated by a PKC/MEK pathway independent of Src-mediated Fhit Tyr114 phosphorylation. We further demonstrated that elevated Fhit expression was due to the specific regulation of Fhit protein synthesis in the ribosome by activated Gαq, where the regulations of cap-dependent protein synthesis were apparently not required. Moreover, we showed that activated Gαq could increase cell–cell adhesion through Fhit. These findings provide a possible handle to modulate the level of the Fhit tumor suppressor by manipulating the activity of Gq-coupled receptors.
Co-reporter:Wendy W.S. Yeung, Yung H. Wong
Cellular Signalling (August 2009) Volume 21(Issue 8) pp:1317-1325
Publication Date(Web):1 August 2009
DOI:10.1016/j.cellsig.2009.03.017
Heterotrimeric G proteins regulate diverse physiological processes by modulating the activities of intracellular effectors. Members of the Gαq family link G protein-coupled receptor activation to phospholipase Cβ (PLCβ) activity and intracellular calcium signaling cascades. However, they differ markedly in biochemical properties as well as tissue distribution. Recent findings have shown that some of the cellular activities of Gαq family members are independent of PLCβ activation. A guanine nucleotide exchange factor, p63RhoGEF, has been shown to interact with Gαq proteins and thus provides linkage to RhoA activation. However, it is not known if p63RhoGEF can associate with other Gαq family members such as Gα16. In the present study, we employed co-immunoprecipitation studies in HEK293 cells to demonstrate that p63RhoGEF can form a stable complex with the constitutively active mutant of Gα16 (Gα16QL). Interestingly, overexpression of p63RhoGEF inhibited Gα16QL-induced IP3 production in a concentration-dependent manner. The binding of PLCβ2 to Gα16QL could be displaced by p63RhoGEF. Similarly, p63RhoGEF inhibited the binding of tetratricopeptide repeat 1 to Gα16QL, leading to a suppression of Gα16QL-induced Ras activation. In the presence of p63RhoGEF, Gα16QL-induced STAT3 phosphorylation was significantly reduced and Gα16QL-mediated SRE transcriptional activation was attenuated. Taken together, these results suggest that p63RhoGEF binds to activated Gα16 and inhibits its signaling pathways.
Co-reporter:Anthony S.L. Chan, Winnie W.I. Lau, Aydan C.H. Szeto, Jiuling Wang, Yung H. Wong
Journal of Molecular Biology (25 September 2016) Volume 428(Issue 19) pp:3869-3884
Publication Date(Web):25 September 2016
DOI:10.1016/j.jmb.2016.03.026
•Activations of specific Gα subunits can elevate the productions of CXCL1, CXCL5 and CXCL8, but not CXCL6 and CXCL7.•Gαq triggers CXCL8 production effectively, while Gαs and Gα12 elicit weaker responses, and Gαi being totally ineffective.•However, in the presence of Gβγ-responsive PLCβ2/3, activations of Gi-coupled receptors can trigger CXCL8 production.•Co-activation of Gi and Gq results in a synergistic CXCL8 production that requires functional PLCβ2/3, Src, ERK and STAT3.•The induced CXCL8 can influence CXCR2-expressing host cells and neighbouring cells in an autocrine or paracrine fashion.CXCL8 (also known as interleukin-8 or IL-8) is a proinflammatory chemokine that not only modulates the inflammatory and immune responses, but whose upregulation is often associated with diseases including various types of cancer. Although numerous ligands for G protein-coupled receptors (GPCRs) have been shown to stimulate the production of CXCL8, the specificity of the G protein signal remains undefined. By expressing the constitutively active Gα subunits in HEK293 cells, CXCL8 production was herein demonstrated to be most effectively stimulated by Gαq family members, while those of Gαs and Gα12 elicited much weaker activities, and Gαi being totally ineffective. However, in cell lines such as HepG2, HeLa, and MCF-7 that endogenously express Gβγ-responsive phospholipase Cβ isoforms (PLCβ2/3), activation of the Gi-coupled α2-adrenoceptor significantly stimulated CXCL8 production. This Gi-induced CXCL8 production was apparently mediated via specific Gβγ dimers and required the presence of PLCβ2/3. Co-activation of Gi-coupled α2-adrenoceptor and Gq-coupled bradykinin receptor resulted in a synergistic CXCL8 production, with Gβγ-responsive PLCβ2/3, Src, ERK, and STAT3 serving as critical signaling intermediates. The treatment of HepG2 and B-10 endothelial cells with bradykinin stimulated CXCL8 production and cell proliferation. Interestingly, the latter response was driven by CXCL8 autocrine signaling because it was abolished by SB225002, an antagonist that prevents CXCL8 from binding to CXCR2. Collectively, our results provide a mechanistic basis for various G protein subfamilies to regulate the production of CXCL8, which may then lead to paracrine and/or autocrine signaling with major implications in both normal physiology and pathophysiological conditions.Download high-res image (181KB)Download full-size image
Co-reporter:Jing Zhu, Stephanie Lee, Maurice K.C. Ho, Yueqing Hu, ... Yung H. Wong
Drug Metabolism and Pharmacokinetics (2010) Volume 25(Issue 5) pp:477-486
Publication Date(Web):1 January 2010
DOI:10.2133/dmpk.DMPK-10-RG-037
Cycloastragenol (CAG) is the aglycone derivative of astragaloside IV which has recently been demonstrated to activate telomerase and represents a potential drug candidate for the treatment of degenerative diseases. In the present study, intestinal absorption and metabolism of CAG were examined using the Caco-2 model and liver microsomes, respectively. The results showed that CAG rapidly passes through the Caco-2 cell monolayer by passive diffusion. Four different glucuronide conjugates and two oxidized CAG metabolites were found in the apical and basolateral sides of Caco-2 monolayer, suggesting that first-pass intestinal metabolism of CAG might occur upon passage through the intestinal epithelium. CAG underwent extensive metabolism in rat and human liver microsomes with only 17.4% and 8.2%, respectively, of the starting amount of CAG remaining after 30 min of incubation. Monohydroxylation of the parent and oxidization of the hydroxylated CAG were found in the liver samples. The present study indicates that CAG is efficiently absorbed through intestinal epithelium. However, extensive first-pass hepatic metabolism would limit the oral bioavailability of this compound.

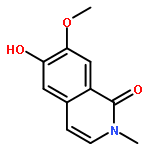
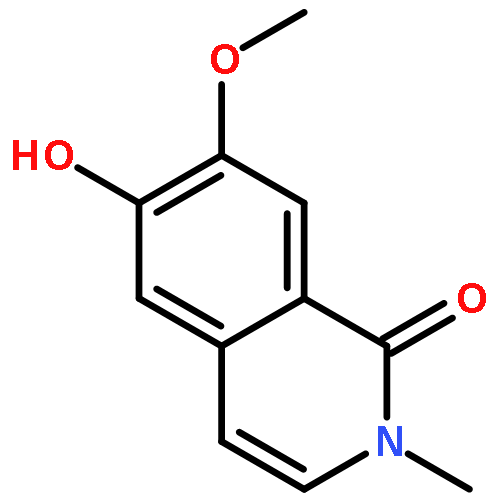




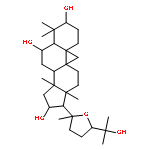


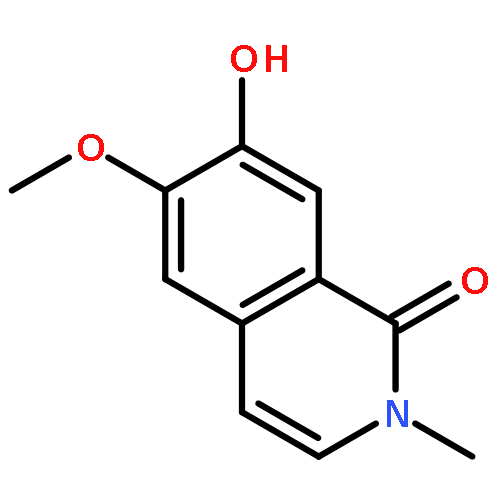












![5,7-dihydroxy-2-(4-hydroxyphenyl)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/500/480-10-4.png)
![5,7-dihydroxy-2-(4-hydroxyphenyl)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/500/480-10-4_b.png)





