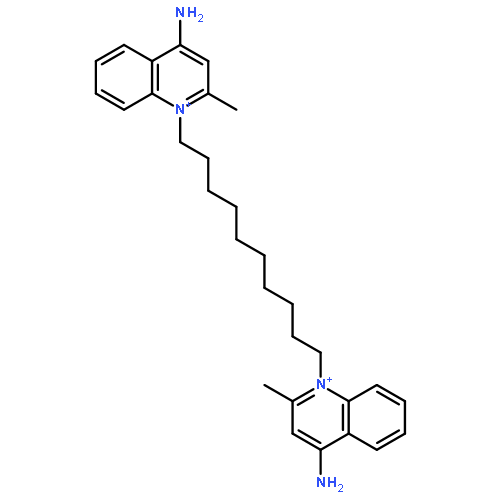
Co-reporter:Jia Ju, Meng-Lei Huan, Ning Wan, Yi-Lin Hou, Xi-Xi Ma, Yi-Yang Jia, Chen Li, Si-Yuan Zhou, Bang-Le Zhang
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 10) pp:2401-2407
Publication Date(Web):15 May 2016
DOI:10.1016/j.bmcl.2016.04.007
Cholesterol derivatives M1–M6 as synthetic cationic lipids were designed and the biological evaluation of the cationic liposomes based on them as non-viral gene delivery vectors were described. Plasmid pEGFP-N1, used as model gene, was transferred into 293T cells by cationic liposomes formed with M1–M6 and transfection efficiency and GFP expression were tested. Cationic liposomes prepared with cationic lipids M1–M6 exhibited good transfection activity, and the transfection activity was parallel (M2 and M4) or superior (M1 and M6) to that of DC-Chol derived from the same backbone. Among them, the transfection efficiency of cationic lipid M6 was parallel to that of the commercially available Lipofectamine2000. The optimal formulation of M1 and M6 were found to be at a mol ratio of 1:0.5 for cationic lipid/DOPE, and at a N/P charge mol ratio of 3:1 for liposome/DNA. Under optimized conditions, the efficiency of M1 and M6 is greater than that of all the tested commercial liposomes DC-Chol and Lipofectamine2000, even in the presence of serum. The results indicated that M1 and M6 exhibited low cytotoxicity, good serum compatibility and efficient transfection performance, having the potential of being excellent non-viral vectors for gene delivery.
Co-reporter:Zi-Wei Jing, Yi-Yang Jia, Ning Wan, Min Luo, Meng-Lei Huan, Tai-Bin Kang, Si-Yuan Zhou, Bang-Le Zhang
Biomaterials 2016 84() pp: 276-285
Publication Date(Web):April 2016
DOI:10.1016/j.biomaterials.2016.01.045
The covalently modified ureido-conjugated chitosan/TPP multifunctional nanoparticles have been developed as targeted nanomedicine delivery system for eradication of Helicobacter pylori. H. pylori can specifically express the urea transport protein on its membrane to transport urea into cytoplasm for urease to produce ammonia, which protects the bacterium in the acid milieu of stomach. The clinical applicability of topical antimicrobial agent is needed to eradicate H. pylori in the infected fundal area. In this study, we designed and synthesized two ureido-conjugated chitosan derivatives UCCs-1 and UCCs-2 for preparation of multifunctional nanoparticles. The process was optimized in order to prepare UCCs/TPP nanoparticles for encapsulation of amoxicillin. The results showed that the amoxicillin-UCCs/TPP nanoparticles exhibited favorable pH-sensitive characteristics, which could procrastinate the release of amoxicillin at gastric acids and enable the drug to deliver and target to H. pylori at its survival region effectively. Compared with unmodified amoxicillin-chitosan/TPP nanoparticles, a more specific and effective H. pylori growth inhibition was observed for amoxicillin-UCCs/TPP nanoparticles. Drug uptake analysis tested by flow cytometry and confocal laser scanning microscopy verified that the uptake of FITC-UCCs-2/TPP nanoparticles was associated with urea transport protein on the membrane of H. pylori and reduced with the addition of urea as competitive transport substrate. These findings suggest that the multifunctional amoxicillin-loaded nanoparticles have great potential for effective therapy of H. pylori infection. They may also serve as pharmacologically effective nanocarriers for oral targeted delivery of other therapeutic drugs to treat H. pylori.
Co-reporter:Hong-Fei Liu, Ning Wan, Meng-Lei Huan, Yi-Yang Jia, Xiao-Feng Yuan, Si-Yuan Zhou, Bang-Le Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 20) pp:4794-4797
Publication Date(Web):15 October 2014
DOI:10.1016/j.bmcl.2014.09.007
PC407 is an effective COX-2 inhibitor in non-steroidal anti-inflammatory drug development but the poor solubility limits their usefulness. The aim of the study was to prepare and evaluate 4-oxo-4-[4-(5-(naphthalen-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzenesulfonamido]butyrate disodium, a derivative of PC407 with enhanced water solubility for injectable formulation. The prepared derivative displayed interesting high aqueous solubility (20.3 mg/mL, much superior to the parent compound PC407, 1.6 μg/mL) with confirmed in vivo analgesic activity. This derivative represents the profiles of prodrug and potential candidate of PC407 for the development of injectable COX-2 inhibitor due to extraordinary water solubility, low toxicity, and impressive analgesic activity.Synthesis and biological evaluation of a potent prodrug 3b for injectable formulation are described. The disodium salt 3b displayed interesting high aqueous solubility with confirmed prodrug profiles and in vivo analgesic activity.
Co-reporter:Bang-le Zhang, Wei He, Xin Shi, Meng-lei Huan, Qiu-ju Huang, Si-yuan Zhou
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:805-808
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.104
An efficient total synthesis of (R) and (S)-3-methyl 5-pentyl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate in high optical purities is reported. The useful step is the resolution of racemic 2, 6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylic acid by using commercially available Cinchona alkaloids cinchonidine and quinidine as the resolving agents. Under the optimum conditions, the optical purities for R- and S-enantiomers are extremely high (ee >99.5%). The further dihydropyridine receptor binding activity assay shows that the S-enantiomer is more potent than R-enantiomer both in rat cardiac (approximately 19 times) and cerebral cortex membrane (12 times).The synthesis of the calcium modulator (R) and (S)-1 in extremely high optical purities (ee >99.5%) is reported. (S)-1 was defined as active isomer which is more potent than (R)-1 both in rat cardiac and cerebral cortex membrane.
Co-reporter:Zi-Wei Jing, Zhi-Wei Ma, Chen Li, Yi-Yang Jia, Min Luo, Xi-Xi Ma, Si-Yuan Zhou, Bang-Le Zhang
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2016.12.072
The covalently cross-linked chitosan-poly(ethylene glycol)1540 derivatives have been developed as a controlled release system with potential for the delivery of protein drug. The swelling characteristics of the hydrogels based on these derivatives as the function of different PEG content and the release profiles of a model protein (bovine serum albumin, BSA) from the hydrogels were evaluated in simulated gastric fluid with or without enzyme in order to simulate the gastrointestinal tract conditions. The derivatives cross-linked with difunctional PEG1540-dialdehyde via reductive amination can swell in alkaline pH and remain insoluble in acidic medium. The cumulative release amount of BSA was relatively low in the initial 2 h and increased significantly at pH 7.4 with intestinal lysozyme for additional 12 h. The results proved that the release-and-hold behavior of the cross-linked CS–PEG1540H-CS hydrogel provided a swell and intestinal enzyme controlled release carrier system, which is suitable for oral protein drug delivery.The covalently cross-linked chitosan-poly(ethylene glycol)1540 hydrogels have been developed as the swell and intestinal enzyme controlled release system with potential for the oral delivery of protein drug.

![N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-Glutamic acid 5-(2,5-dioxo-1-pyrrolidinyl) ester](http://img.cochemist.com/ccimg/153500/153445-05-7.png)
![N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-Glutamic acid 5-(2,5-dioxo-1-pyrrolidinyl) ester](http://img.cochemist.com/ccimg/153500/153445-05-7_b.png)


















![2-Propenoyl chloride, 3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/82600/82592-68-5.png)
![2-Propenoyl chloride, 3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/82600/82592-68-5_b.png)
![[3-[HYDROXY(2-HYDROXYETHOXY)PHOSPHORYL]OXY-2-[(E)-OCTADEC-9-ENOYL]OXYPROPYL] (E)-OCTADEC-9-ENOATE](http://img.cochemist.com/ccimg/76400/76391-83-8.png)
![[3-[HYDROXY(2-HYDROXYETHOXY)PHOSPHORYL]OXY-2-[(E)-OCTADEC-9-ENOYL]OXYPROPYL] (E)-OCTADEC-9-ENOATE](http://img.cochemist.com/ccimg/76400/76391-83-8_b.png)


![1-Hexanol, 6-[[(3b)-cholest-5-en-3-yl]oxy]-](http://img.cochemist.com/ccimg/68400/68354-84-7.png)
![1-Hexanol, 6-[[(3b)-cholest-5-en-3-yl]oxy]-](http://img.cochemist.com/ccimg/68400/68354-84-7_b.png)

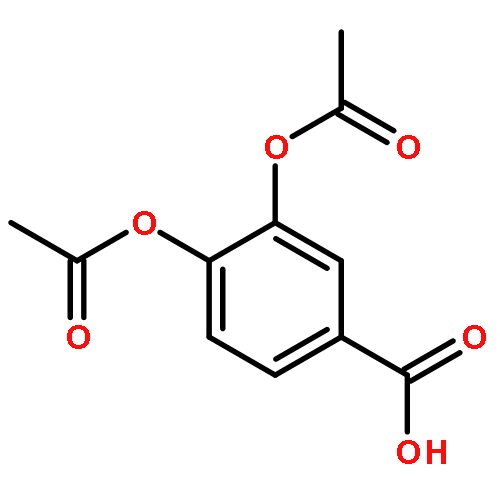


![2-PROPENOYL CHLORIDE, 3-[4-(ACETYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/54000/53901-95-4.png)
![2-PROPENOYL CHLORIDE, 3-[4-(ACETYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/54000/53901-95-4_b.png)
![2-Propenoyl chloride, 3-[4-(acetyloxy)-3-methoxyphenyl]-](http://img.cochemist.com/ccimg/51000/50906-12-2.png)
![2-Propenoyl chloride, 3-[4-(acetyloxy)-3-methoxyphenyl]-](http://img.cochemist.com/ccimg/51000/50906-12-2_b.png)
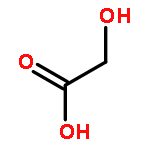
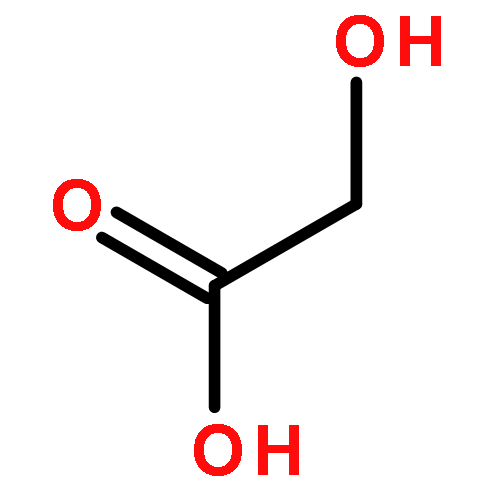
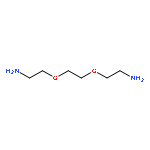
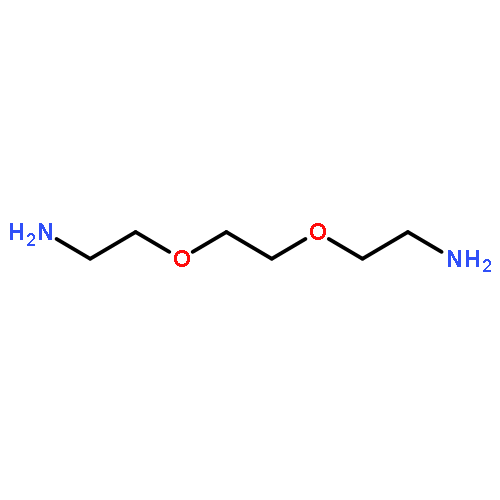
![Hexanoic acid, 6-[(4-methoxybenzoyl)amino]-](http://img.cochemist.com/ccimg/22900/22834-47-5.png)
![Hexanoic acid, 6-[(4-methoxybenzoyl)amino]-](http://img.cochemist.com/ccimg/22900/22834-47-5_b.png)
![2-Propenoic acid,3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/13800/13788-48-2.png)
![2-Propenoic acid,3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/13800/13788-48-2_b.png)