Co-reporter:Song-Lin Zhang, Hai-Xing Wan, and Wen-Feng Bie
Organic Letters December 1, 2017 Volume 19(Issue 23) pp:6372-6372
Publication Date(Web):November 20, 2017
DOI:10.1021/acs.orglett.7b03229
One-step concurrent fluoro-trifluoromethylation across the triple bond of arylacetylenes in a syn mode is enabled by the collaboration of (phen)CuIII(CF3)3 and CsF that produces chemo-, regio-, and stereoselectively (Z)-α-fluoro-β-CF3 styrenes. This method can be extended to achieve syn-oxy-trifluoromethylation and syn-aryl-trifluoromethylation of alkynes using phenoxides, alkoxides, or phenylboronic acid in place of CsF. It opens up new opportunities for preparing various functionalized trifluoromethylated Z-alkenes and demonstrates the potential of Cu(III)–CF3 complexes in organic synthesis.
Co-reporter:Xian-Jin Wang
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 24) pp:14826-14830
Publication Date(Web):2017/12/04
DOI:10.1039/C7NJ03405A
A friendly protocol is reported to achieve cyanation of aromatic and vinylic boronic acids using nontoxic and readily available α-cyanoacetates as a cyano source under aerobic conditions. Many aryl/vinyl boronic acids (as well as some iodides and bromides) are amenable substrates to give aryl nitriles and acrylonitriles. This cyanation method provides a safe and operationally convenient alternative to traditional ones requiring toxic cyanide salts.
Co-reporter:Song-Lin Zhang;Hai-Xing Wan;Zhu-Qin Deng
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 30) pp:6367-6374
Publication Date(Web):2017/08/02
DOI:10.1039/C7OB01378G
This paper reports a computational study elucidating the reaction mechanism for ynamide-mediated amide bond formation from carboxylic acids and amines. The mechanisms have been studied in detail for ynamide hydrocarboxylation and the subsequent aminolysis of the resulting adduct by an amine. Ynamide hydrocarboxylation is kinetically favorable and thermodynamically irreversible, resulting in the formation of a key low-lying intermediate CP1 featuring geminal vinylic acyloxy and sulfonamide groups. The aminolysis of CP1 by the amine is proposed to be catalyzed by the carboxylic acid itself that imparts favourable bifunctional effects. In the proposed key transition state TSaminolysis-acid-iso2, the amine undergoes direct nucleophilic substitution at the acyl of CP1 to replace the enolate group in a concerted way, which is promoted by secondary hydrogen bonding of carboxylic acid with both the amine and CP1. These secondary interactions are suggested to increase the nucleophilicity of the amine and to activate the Cacyl–O bond to be cleaved, thereby stabilizing the aminolysis transition state. The concerted aminolysis mechanism is competitive with the classic stepwise nucleophilic acyl substitution mechanism that features sequential amine addition to acyl/intramolecular proton transfer/C–O bond cleavage and a key tetrahedral intermediate. Based on the mechanistic model, the carboxylic acid substrate effect and studies of more acidic CF3SO3H as the catalyst are in good agreement with the experimental observations, lending further support for the mechanistic model. The bifunctional catalytic effect of the carboxylic acid substrate may widely play a role in related amide bond-forming reactions and peptide formation chemistry.
Co-reporter:Song-Lin Zhang and Zhu-Qin Deng
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 30) pp:7282-7294
Publication Date(Web):28 Jun 2016
DOI:10.1039/C6OB01198E
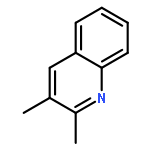
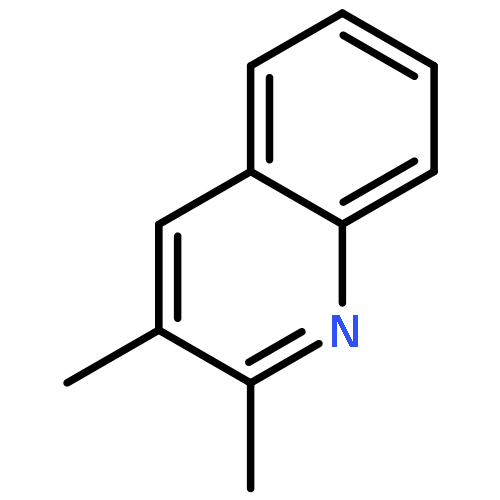
A copper-catalyzed transfer aldol type reaction of β-hydroxy ketones or nitriles with aldehydes is reported, which enables chemo- and stereoselective access to (E)-α,β-unsaturated ketones and (E)-acrylonitriles. A key step of the in situ copper(I)-promoted retro-aldol reaction of β-hydroxy ketones or nitriles is proposed to generate a reactive Cu(I) enolate or cyanomethyl intermediate, which undergoes ensuing aldol condensation with aldehydes to deliver the products. This reaction uses 1.2 mol% Cu(IPr)Cl (IPr denotes 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) as the catalyst in the presence of 6.0 mol% NaOtBu cocatalyst at room temperature or 70 °C. A range of aryl and heteroaryl aldehydes as well as acrylaldehydes are compatible with many useful functional groups being tolerated. Under the mild and weakly basic conditions, competitive Cannizzaro-type reaction of benzaldehydes and side reactions of base-sensitive functional groups can be effectively suppressed, which show synthetic advantages of this reaction compared to classic aldol reactions. The synthetic potential of this reaction is further demonstrated by the one-step synthesis of biologically active quinolines and 1,8-naphthyridine in excellent yields (up to 91%). Finally, a full catalytic cycle for this reaction has been constructed using DFT computational studies in the context of a retro-aldol/aldol two-stage mechanism. A rather flat reaction energy profile is found indicating that both stages are kinetically facile, which is consistent with the mild reaction conditions.
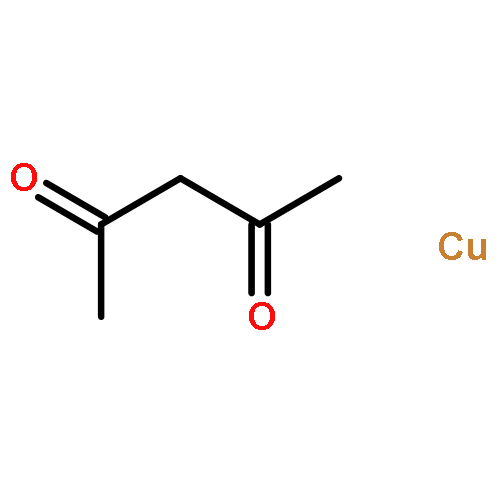
Co-reporter:Song-Lin Zhang and Wen-Feng Bie
RSC Advances 2016 vol. 6(Issue 75) pp:70902-70906
Publication Date(Web):22 Jul 2016
DOI:10.1039/C6RA10302B
The isolation, characterization and reactivity of transition metal trifluoromethyl complexes are fundamental and challenging topics in trifluoromethylation chemistry. We report herein the synthesis and isolation of two new complexes [(phen)CuI(PPh3)2]+[CuIII(CF3)4]− (2) and (phen)CuIII(CF3)3 (3) as well as a known complex (bpy)CuIII(CF3)3 (4) at room temperature. 2 and 3 have been fully characterized using 1H, 19F, 31P NMR, elemental analyses and X-ray crystallography. Reactivity studies indicate that 2 is unreactive toward arylboronic acids. In contrast, 3 and 4 can react with various aryl and heteroaryl boronic acids to deliver trifluoromethylated arenes in good to quantitative yields under mild conditions. The presence of a fluoride additive in DMF under aerobic conditions is crucial to these reactions. This study provides fundamental information about the structure and reactivity of elusive Cu(III) trifluoromethyl complexes that have been proposed as relevant reactive intermediates in many trifluoromethylation reactions.
Co-reporter:Song-Lin Zhang and Ze-Long Yu
The Journal of Organic Chemistry 2016 Volume 81(Issue 1) pp:57-65
Publication Date(Web):December 7, 2015
DOI:10.1021/acs.joc.5b02098
A retro-aldol reaction of two β-hydroxy compounds in synergy with Pd-catalyzed cross-coupling of aryl halides is reported herein, which produces selectively mono-α-arylated ketones and esters in good yields. This reaction is compatible with a broad range of aryl iodides, bromides, chlorides, and triflates and can tolerate an array of functional groups on the aromatic ring. Ready scale-up of this reaction to gram level is applicable without an appreciable decrease in the reaction yield. Furthermore, concise syntheses of biologically active isocoumarin and indole derivatives have been achieved to greatly demonstrate the synthetic value of this retro-aldol reaction. Finally, the reaction mechanism has been discussed on the basis of experimental observations and DFT computational results. A regulated six-membered-ring transition structure has been located for the key retro-aldol C–C cleavage, which constitutes the rate-determining step of a full catalytic cycle. The concept of C–C activation by retro-aldol reaction may also find applications in other fundamental reactions.
Co-reporter:Song-Lin Zhang, Lu Huang and Li-Jun Sun
Dalton Transactions 2015 vol. 44(Issue 10) pp:4613-4622
Publication Date(Web):27 Jan 2015
DOI:10.1039/C4DT03267E
A systematic theoretical study is reported on the mechanisms of reductive elimination from arylPd(II) trifluoromethyl complexes, an important elementary reaction for Pd-catalyzed trifluoromethylation reactions. Various mechanisms leading to the formation of trifluoromethylated products and also competing side pathways have been evaluated. Furthermore, ligand effects are systematically evaluated which provide valuable information about the favourable properties of the ancillary ligands for promoting reductive elimination of trifluoromethylated products from Pd(II) centers.
Co-reporter:Song-Lin Zhang and Lu Huang
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 39) pp:9963-9968
Publication Date(Web):13 Aug 2015
DOI:10.1039/C5OB01675D
A Cu(I)-catalyzed cyanation reaction of aryl iodides with α-cyanoacetates is reported herein, which uses α-cyanoacetates as the nontoxic and easy-handling CN source through copper-mediated C–CN bond cleavage. This reaction enables access to aryl nitriles with an array of functional groups on the aromatic ring in good to excellent yields.
Co-reporter:Song-Lin Zhang, Lu Huang, and Wen-Feng Bie
Organometallics 2014 Volume 33(Issue 12) pp:3030-3039
Publication Date(Web):June 10, 2014
DOI:10.1021/om500294b
A theoretical study of reaction mechanisms for C–CN bond activation of nitriles by a RhIII–silyl complex is reported. Various mechanisms including direct oxidative addition, insertion of the cyanide C≡N triple bond into the RhIII–silyl bond followed by β-carbon elimination, insertion of the cyanide C≡N triple bond into the RhIII–silyl bond followed by α-carbon elimination (deisocyanide), radical mechanisms, and other possible alternatives have been evaluated. Our results provide strong evidence for the sequential mechanism of cyano insertion/α-C elimination (deisocyanide). The cyanide insertion step should be the rate-limiting step, while the deisocyanide step is facile. The intermediate from cyanide insertion, i.e., the Rh(III) η2-iminoacyl complex, has been identified and is in good agreement with the experimentally characterized X-ray crystal structure. The oxidative addition and cyanide insertion/β-C elimination mechanisms are kinetically inhibited due to extremely high activation barriers. Radical mechanisms are also kinetically unfavorable due to the electrophilic nature of the cationic Rh(III) complex. These findings distinguish the cationic RhIII–silyl complex from the electron-rich Ni(0) systems frequently exploited for the activation of cyanide C–CN bonds, where an oxidative addition mechanism should be operative. Furthermore, the rate-limiting step of cyano insertion into the Rh–silyl bond has been examined for various nitriles. The reactivity trend for these nitriles is also in good agreement with experimental observations, which show significant steric effects but small electronic effects for the RCN R group. The origin of the favorable insertion to give a Rh(III) η2-iminoacyl complex versus the formation of a Rh(III) η1-imino complex has been elucidated by using natural charge population analyses. It is attributed to the presence of two pairs of favorable stabilizing Rh···C and Si···N interactions in the transition state TS1 for cyano insertion in the insertion/deisocyanide mechanism. However, this effect is replaced by detrimental repulsive Rh···N and Si···C interactions in the isomeric transition state TS1′ with the reverse orientation of Rh–Si versus C≡N bonds.
Co-reporter:Song-Lin Zhang, Wen-Feng Bie, and Lu Huang
Organometallics 2014 Volume 33(Issue 19) pp:5263-5271
Publication Date(Web):September 2, 2014
DOI:10.1021/om500588s
A theoretical study on reaction mechanisms for copper-catalyzed Ullmann-type C–P coupling of diphenylphosphine with aryl halides is reported herein. The equilibria and consequent relative concentrations of possible copper species in the reaction solution were evaluated to determine probable active catalytic species in the presence of several typical ancillary ligands in toluene and DMSO solvents. Subsequently, reactivity of these key copper species with aryl halides were studied in the context of commonly proposed mechanisms for copper(I)-catalyzed Ullmann reactions, including oxidative addition/reductive elimination, σ-bond metathesis, single electron transfer (SET), and halogen atom transfer (HAT) mechanisms. On the basis of these studies, we propose that for phenanthroline and 1,2-ethylenediamine types of bidentate ligands, the active catalyst should be the neutral form LCu(I)-PPh2 in nonpolar toluene, while the Cu(PPh2)2– anion should be significant in highly polar DMSO. In contrast, for phosphine type ligands, the active catalytic species should be the neutral LCu(I)-PPh2 complexes in both toluene and DMSO. More interestingly, for both neutral LCu(I)-PPh2 and anionic Cu(PPh2)2– complexes, HAT mechanism is proposed to be kinetically the most favorable in toluene. However, in DMSO, the operative reaction mechanism should be influenced by the ancillary ligand used. For phosphine ligand, HAT mechanism is still the most favored with the LCu(I)-PPh2 as the active catalyst. For phen and diamine ligands, SET mechanism has been shown to be the most favored for anionic Cu(PPh2)2– complex, and HAT mechanism is proposed for neutral LCu(I)-PPh2 complexes; both contribute to the activation of aryl halides in reaction solution. Therefore, a combined effect of solvent polarity and ancillary ligand has been recognized on determining the identity of the active catalytic species and the operative reaction mechanism for Ullmann-type P-arylation reactions. These results and findings distinguish the reaction mechanism for Ullmann P-arylation reactions from the other type of Ullmann reactions, such as N- and O-arylation reactions.
Co-reporter:Liang Li, Feng Wu, Songlin Zhang, Dawei Wang, Yuqiang Ding and Zhenzhong Zhu
Dalton Transactions 2013 vol. 42(Issue 13) pp:4539-4543
Publication Date(Web):17 Dec 2012
DOI:10.1039/C2DT32800C
In this paper, a C–F bond activation reaction of a chloro-bridged iridium(III) dimer (dfppy)2Ir(μ-Cl)2Ir(dfppy)2 (1) (dfppy denotes 2-(4,6-difluorophenyl)pyridyl) in the presence of sodium methoxide has been reported, leading to the formation of a heteroleptic cyclometalated iridium(III) fluorophenylpyridine complex 2. HPLC-mass analysis confirmed the release of formaldehyde in the product mixture. When sodium benzyloxide was used as the base, complex 2 was also generated with the release of a benzaldehyde derivative. Complex 2 has been fully characterized by 1H-NMR, 19F-NMR and X-ray crystallographic methods, confirming the partial loss of one of the fluorine atoms on one of the cyclometalated phenylpyridyl ligands. Photophysical studies of complex 2 show that it has a similar absorption spectrum to that of Ir(III)(dfppy)3. However, the emission spectrum shows a red shift maximum emission band at 478 nm due to the loss of a single fluorine atom, highlighting the critical effect of fluorine on the photoluminescence of these Ir(III) complexes. Finally, intensive mechanistic studies including HPLC-mass analysis, 1H-NMR, and 19F-NMR studies demonstrate that the formation of complex 2 should involve a critical β-hydride elimination of Ir(III)-alkoxide intermediate and the participation of Ir-hydride and/or Ir-fluoride intermediates.
Co-reporter:Song-Lin Zhang and Hui-Jun Fan
Organometallics 2013 Volume 32(Issue 17) pp:4944-4951
Publication Date(Web):August 23, 2013
DOI:10.1021/om4006615
A systematic theoretical study on reaction mechanisms for copper-catalyzed Ullmann S-arylation reactions of thiophenols with aryl halides is reported herein. The equilibriums and consequent relative concentrations of possible copper species in the reaction solution were carefully evaluated to determine the most probable active catalytic forms. Subsequently, reactivity studies of these copper species with aryl halides were performed in the context of several commonly proposed mechanisms for copper(I)-catalyzed Ullmann reactions, such as oxidative addition/reductive elimination, σ-bond metathesis, single electron transfer (SET), and halogen atom transfer (HAT) mechanisms. On the basis of these intensive studies, we propose for the first time that the active copper catalyst should be neutral (L)Cu(I)-SAr species (L denotes a neutral ancillary ligand; SAr denotes a thiophenolato type ligand) in nonpolar or less polar solvent and anionic Cu(SAr)2— species in highly polar solvent. These two kinds of species are in equilibrium with each other. For both copper species, the HAT mechanism is the most favored among all the possible mechanisms examined. Under the HAT mechanism, a critical halogen atom transfer from aryl halide to Cu(I) center occurs and accordingly involves the formation of intermediate Cu(II)(SAr)(X) (X denotes a halide ligand) species as well as an aryl radical. Subsequent direct and rapid attack of the aryl radical to the thiophenolato ligand in Cu(II)(SAr)(X) delivers the coupling product. Aryl halide substrate effect studies reveal that various kinds of aryl halides follow the reactivity trend of ArI > ArBr > ArCl under this HAT mechanism. This trend prediction is in good agreement with experimental observations that aryl iodides are generally more reactive than aryl bromides and chlorides for such Ullmann S-arylation reactions and thus lends further support for this HAT mechanism. Given the mechanistic proposals for Ullmann N- and O-arylation reactions, Ullmann S-arylation reactions should probably follow an analogous mechanism to that of O-arylation reactions, which is in distinct contrast with the oxidative addition mechanism proposed for N-arylation reactions. This highlights once again that the reaction mechanism of such copper(I)-catalyzed Ullmann reactions is dependent on the nature of the nucleophiles employed. Nucleophiles with reactive centers from different groups in the periodic table may possibly be involved in different mechanisms, and vice versa. These insights should therefore be valuable for the understanding of the mechanism of Ullmann S-arylation reactions and further development of orthogonal or selective Ullmann reactions involving multifunctional nucleophiles.
Co-reporter:Songlin Zhang, Zhenzhong Zhu and Yuqiang Ding
Dalton Transactions 2012 vol. 41(Issue 45) pp:13832-13840
Publication Date(Web):13 Aug 2012
DOI:10.1039/C2DT31500A
A systematic theoretical study on reaction mechanisms for copper(I)-catalyzed C–O coupling of phenols with aryl bromides is reported herein. Through evaluation of the relative concentrations of possible copper species in reaction solution and reactivity study of these copper species with aryl halides in the context of several commonly proposed mechanisms for copper(I)-catalyzed Ullmann reactions, we propose that the active copper catalyst should be a neutral (L)Cu(I)–OAr (L denotes an ancillary ligand; OAr denotes an aryloxide ligand) species in nonpolar solvent and Cu(OAr)2− anion in highly polar solvent. In the reaction solution, these two kinds of copper species should be in equilibrium, the direction of which is highly dependent on the polarity of the solvent. For both kinds of copper species, a halogen atom transfer mechanism is favored where an initial halogen atom transfer from phenyl bromide to the Cu(I) center occurs, resulting in the formation of Cu(II)(OAr)(Br) and a phenyl radical. Subsequent rapid attack of this phenyl radical to the aryloxide ligand bound to copper(II) would yield the coupling product and Cu(I)(Br) species, which can be readily converted to the active Cu(I)–OAr species in the presence of phenols and base. Other mechanisms such as oxidative addition, single electron transfer and σ-bond metathesis mechanisms all possess activation barriers which are too high, rendering them kinetically unfavorable. Electronic effects on phenol rings reveal that electron-donating substituents accelerate the coupling of (phen)Cu(I)(OAr) with aryl halides whereas electron-withdrawing substituents lead to much higher activation barriers, which is consistent with experimental findings and thus lends further support for such a halogen atom transfer mechanism. In view of the widely accepted oxidative addition/reductive elimination mechanism for analogous copper(I)-catalyzed coupling of N-nucleophiles with aryl halides, our results here highlight that the reaction mechanism of copper(I)-catalyzed Ullmann reactions is highly dependent on the nature of the nucleophile and different kinds of nucleophiles can be involved in different mechanisms.
Co-reporter:Songlin Zhang ; Li Shi ;Yuqiang Ding
Journal of the American Chemical Society 2011 Volume 133(Issue 50) pp:20218-20229
Publication Date(Web):November 23, 2011
DOI:10.1021/ja205294y
A systematic theoretical study is carried out on the mechanism for Pd(II)-catalyzed oxidative cross-coupling between electron-deficient arenes and alkenes. Two types of reaction pathways involving either a sequence of initial arene C–H activation followed by alkene activation, or the reverse sequence of initial alkene C–H activation followed by arene activation are evaluated. Several types of C–H activation mechanisms are discussed including oxidative addition, σ-bond metathesis, concerted metalation/deprotonation, and Heck-type alkene insertion. It is proposed that the most favored reaction pathway should involve an initial concerted metalation/deprotonation step for arene C–H activation by (L)Pd(OAc)2 (L denotes pyridine type ancillary ligand) to generate a (L)(HOAc)Pd(II)–aryl intermediate, followed by substitution of the ancillary pyridine ligand by alkene substrate and direct insertion of alkene double bond into Pd(II)–aryl bond. The rate- and regio-determining step of the catalytic cycle is concerted metalation/deprotonation of arene C–H bond featuring a six-membered ring transition state. Other mechanism alternatives possess much higher activation barriers, and thus are kinetically less competitive. Possible competing homocoupling pathways have also been shown to be kinetically unfavorable. On the basis of the proposed reaction pathway, the regioselectivity predicted for a number of monosubstituted benzenes is in excellent agreement with experimental observations, thus, lending further support for our proposed mechanism. Additionally, the origins of the regioselectivity of C–H bond activation is elucidated to be caused by a major steric repulsion effect of the ancillary pyridine type ligand with ligands on palladium center and a minor electronic effect of the preinstalled substituent on the benzene ring on the cleaving C–H bond. This would finally lead to the formation of a mixture of meta and para C–H activation products with meta products dominating while no ortho products were detected. Finally, the multiple roles of the ancillary pyridine type ligand have been discussed. These insights are valuable for our understanding and further development of more efficient and selective transition metal-catalyzed oxidative C–H/C–H coupling reactions.
Co-reporter:Song-Lin Zhang and Zhu-Qin Deng
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 38) pp:NaN8970-8970
Publication Date(Web):2016/08/25
DOI:10.1039/C6OB01452F
A Cu(I)-catalyzed retro-aldol reaction of β-hydroxyketones with ortho-aminobenzaldehydes and nicotinaldehydes is reported that produces a range of quinolines and naphthyridines with high efficiency and selectivity. This reaction uses β-hydroxyketones as a regiospecific ketone-protected enolate source via copper-catalyzed retro-aldol Cα–Cβ bond cleavage. The in situ generated copper enolate undergoes kinetically favorable cyclization with ortho-amino aryl aldehydes to produce quinolines and naphthyridines in a chemo- and regioselective manner. The mild and weakly basic reaction conditions also suppress possible side reactions of benzaldehydes under strongly basic conditions, resulting in improved reaction yields.
Co-reporter:Song-Lin Zhang, Hai-Xing Wan and Zhu-Qin Deng
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 30) pp:NaN6374-6374
Publication Date(Web):2017/07/07
DOI:10.1039/C7OB01378G
This paper reports a computational study elucidating the reaction mechanism for ynamide-mediated amide bond formation from carboxylic acids and amines. The mechanisms have been studied in detail for ynamide hydrocarboxylation and the subsequent aminolysis of the resulting adduct by an amine. Ynamide hydrocarboxylation is kinetically favorable and thermodynamically irreversible, resulting in the formation of a key low-lying intermediate CP1 featuring geminal vinylic acyloxy and sulfonamide groups. The aminolysis of CP1 by the amine is proposed to be catalyzed by the carboxylic acid itself that imparts favourable bifunctional effects. In the proposed key transition state TSaminolysis-acid-iso2, the amine undergoes direct nucleophilic substitution at the acyl of CP1 to replace the enolate group in a concerted way, which is promoted by secondary hydrogen bonding of carboxylic acid with both the amine and CP1. These secondary interactions are suggested to increase the nucleophilicity of the amine and to activate the Cacyl–O bond to be cleaved, thereby stabilizing the aminolysis transition state. The concerted aminolysis mechanism is competitive with the classic stepwise nucleophilic acyl substitution mechanism that features sequential amine addition to acyl/intramolecular proton transfer/C–O bond cleavage and a key tetrahedral intermediate. Based on the mechanistic model, the carboxylic acid substrate effect and studies of more acidic CF3SO3H as the catalyst are in good agreement with the experimental observations, lending further support for the mechanistic model. The bifunctional catalytic effect of the carboxylic acid substrate may widely play a role in related amide bond-forming reactions and peptide formation chemistry.
Co-reporter:Song-Lin Zhang and Zhu-Qin Deng
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 48) pp:NaN32667-32667
Publication Date(Web):2016/11/18
DOI:10.1039/C6CP07093K
Reductive elimination from Pd(II) aryl trifluoromethyl complexes is a challenging and elusive step which is accompanied by a number of kinetically more favorable side reactions giving rising to a complex mixture. We report herein the synthesis and isolation of several arylPd(II) trifluoromethyl complexes (2a–c) and study their electronic structures, photophysical properties and reductive elimination reactivities. A remarkable concentration effect on chemoselectivity is observed for thermal decomposition of (Xantphos)Pd(II)(Ar)(CF3) (2c) that favors the formation of Ar–CF3 at lower concentrations, but gives increasingly more Ar–Ar homocoupling product to a dominant extent as the concentration of 2c increases. This is solid evidence for the involvement of an intermolecular Ar/CF3 ligand exchange/Ar–Ar reductive elimination mechanism that has been proposed based on DFT computational studies. The interplay between theory and experiment provides valuable insights into the mechanism and kinetics of the key elementary reaction of reductive elimination at Pd(II), and may thus prompt the design of more efficient Pd-mediated nucleophilic trifluoromethylation reactions.
Co-reporter:Liang Li, Feng Wu, Songlin Zhang, Dawei Wang, Yuqiang Ding and Zhenzhong Zhu
Dalton Transactions 2013 - vol. 42(Issue 13) pp:NaN4543-4543
Publication Date(Web):2012/12/17
DOI:10.1039/C2DT32800C
In this paper, a C–F bond activation reaction of a chloro-bridged iridium(III) dimer (dfppy)2Ir(μ-Cl)2Ir(dfppy)2 (1) (dfppy denotes 2-(4,6-difluorophenyl)pyridyl) in the presence of sodium methoxide has been reported, leading to the formation of a heteroleptic cyclometalated iridium(III) fluorophenylpyridine complex 2. HPLC-mass analysis confirmed the release of formaldehyde in the product mixture. When sodium benzyloxide was used as the base, complex 2 was also generated with the release of a benzaldehyde derivative. Complex 2 has been fully characterized by 1H-NMR, 19F-NMR and X-ray crystallographic methods, confirming the partial loss of one of the fluorine atoms on one of the cyclometalated phenylpyridyl ligands. Photophysical studies of complex 2 show that it has a similar absorption spectrum to that of Ir(III)(dfppy)3. However, the emission spectrum shows a red shift maximum emission band at 478 nm due to the loss of a single fluorine atom, highlighting the critical effect of fluorine on the photoluminescence of these Ir(III) complexes. Finally, intensive mechanistic studies including HPLC-mass analysis, 1H-NMR, and 19F-NMR studies demonstrate that the formation of complex 2 should involve a critical β-hydride elimination of Ir(III)-alkoxide intermediate and the participation of Ir-hydride and/or Ir-fluoride intermediates.
Co-reporter:Song-Lin Zhang and Lu Huang
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 39) pp:NaN9968-9968
Publication Date(Web):2015/08/13
DOI:10.1039/C5OB01675D
A Cu(I)-catalyzed cyanation reaction of aryl iodides with α-cyanoacetates is reported herein, which uses α-cyanoacetates as the nontoxic and easy-handling CN source through copper-mediated C–CN bond cleavage. This reaction enables access to aryl nitriles with an array of functional groups on the aromatic ring in good to excellent yields.
Co-reporter:Song-Lin Zhang and Zhu-Qin Deng
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 30) pp:NaN7294-7294
Publication Date(Web):2016/06/28
DOI:10.1039/C6OB01198E
A copper-catalyzed transfer aldol type reaction of β-hydroxy ketones or nitriles with aldehydes is reported, which enables chemo- and stereoselective access to (E)-α,β-unsaturated ketones and (E)-acrylonitriles. A key step of the in situ copper(I)-promoted retro-aldol reaction of β-hydroxy ketones or nitriles is proposed to generate a reactive Cu(I) enolate or cyanomethyl intermediate, which undergoes ensuing aldol condensation with aldehydes to deliver the products. This reaction uses 1.2 mol% Cu(IPr)Cl (IPr denotes 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) as the catalyst in the presence of 6.0 mol% NaOtBu cocatalyst at room temperature or 70 °C. A range of aryl and heteroaryl aldehydes as well as acrylaldehydes are compatible with many useful functional groups being tolerated. Under the mild and weakly basic conditions, competitive Cannizzaro-type reaction of benzaldehydes and side reactions of base-sensitive functional groups can be effectively suppressed, which show synthetic advantages of this reaction compared to classic aldol reactions. The synthetic potential of this reaction is further demonstrated by the one-step synthesis of biologically active quinolines and 1,8-naphthyridine in excellent yields (up to 91%). Finally, a full catalytic cycle for this reaction has been constructed using DFT computational studies in the context of a retro-aldol/aldol two-stage mechanism. A rather flat reaction energy profile is found indicating that both stages are kinetically facile, which is consistent with the mild reaction conditions.
Co-reporter:Song-Lin Zhang and Ze-Long Yu
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 44) pp:NaN10515-10515
Publication Date(Web):2016/10/15
DOI:10.1039/C6OB01979J
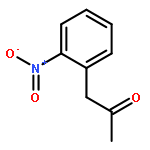
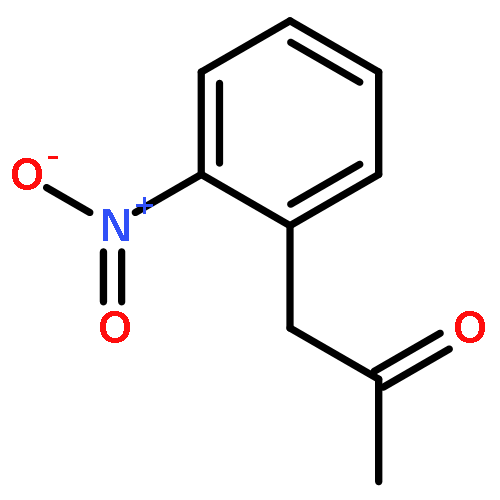
Divergent synthesis of indoles, oxindoles, isocoumarins and isoquinolinones is described in this report by using a general Pd-catalyzed tandem reaction of β-hydroxy carbonyl compounds with aryl halides bearing an ortho-nitro, -ester or -cyano substituent. A key retro-aldol/α-arylation reaction is involved that merges classic Pd cross-coupling chemistry with novel Pd-promoted retro-aldol C–C activation to produce α-arylated ketones or esters. Subsequent intramolecular condensation of the carbonyl with the ortho-synthon gives target heterocycles. The use of common, commercially available and cheap substrates and catalyst system adds additional synthetic advantages to the conceptual significance.
Co-reporter:Song-Lin Zhang and Wen-Feng Bie
Dalton Transactions 2016 - vol. 45(Issue 44) pp:NaN17592-17592
Publication Date(Web):2016/08/26
DOI:10.1039/C6DT03016E
We report novel ion-pair bisphosphine CuI/CuIII trifluoromethyl complexes [(P2)2CuI]+[CuIII(CF3)4]− (P2 = DPPE, BINAP or Xantphos) featuring tetrahedral CuI and square planar CuIII units that are prepared via oxidative trifluoromethylation of CuI with CF3SiMe3 in the presence of AgF. The bisphosphine CuI/CuIII CF3 complexes are highly reactive for aerobic trifluoromethylation of arylboronic acids to produce trifluoromethylated arenes in good to quantitative yields, which is in sharp contrast to the CuI/CuIII complex with phen/PPh3 ligands (1′). These results not only provide strong evidence that both the neutral and ion-pair CuIII CF3 complexes are competent catalytic species for Cu-mediated oxidative trifluoromethylation reactions, but also have important mechanistic implications that the active catalyst and reaction mechanism should be distinct and ligand-dependent for trifluoromethylation reactions with different types of ancillary ligands.
Co-reporter:Songlin Zhang, Zhenzhong Zhu and Yuqiang Ding
Dalton Transactions 2012 - vol. 41(Issue 45) pp:NaN13840-13840
Publication Date(Web):2012/08/13
DOI:10.1039/C2DT31500A
A systematic theoretical study on reaction mechanisms for copper(I)-catalyzed C–O coupling of phenols with aryl bromides is reported herein. Through evaluation of the relative concentrations of possible copper species in reaction solution and reactivity study of these copper species with aryl halides in the context of several commonly proposed mechanisms for copper(I)-catalyzed Ullmann reactions, we propose that the active copper catalyst should be a neutral (L)Cu(I)–OAr (L denotes an ancillary ligand; OAr denotes an aryloxide ligand) species in nonpolar solvent and Cu(OAr)2− anion in highly polar solvent. In the reaction solution, these two kinds of copper species should be in equilibrium, the direction of which is highly dependent on the polarity of the solvent. For both kinds of copper species, a halogen atom transfer mechanism is favored where an initial halogen atom transfer from phenyl bromide to the Cu(I) center occurs, resulting in the formation of Cu(II)(OAr)(Br) and a phenyl radical. Subsequent rapid attack of this phenyl radical to the aryloxide ligand bound to copper(II) would yield the coupling product and Cu(I)(Br) species, which can be readily converted to the active Cu(I)–OAr species in the presence of phenols and base. Other mechanisms such as oxidative addition, single electron transfer and σ-bond metathesis mechanisms all possess activation barriers which are too high, rendering them kinetically unfavorable. Electronic effects on phenol rings reveal that electron-donating substituents accelerate the coupling of (phen)Cu(I)(OAr) with aryl halides whereas electron-withdrawing substituents lead to much higher activation barriers, which is consistent with experimental findings and thus lends further support for such a halogen atom transfer mechanism. In view of the widely accepted oxidative addition/reductive elimination mechanism for analogous copper(I)-catalyzed coupling of N-nucleophiles with aryl halides, our results here highlight that the reaction mechanism of copper(I)-catalyzed Ullmann reactions is highly dependent on the nature of the nucleophile and different kinds of nucleophiles can be involved in different mechanisms.
Co-reporter:Song-Lin Zhang, Lu Huang and Li-Jun Sun
Dalton Transactions 2015 - vol. 44(Issue 10) pp:NaN4622-4622
Publication Date(Web):2015/01/27
DOI:10.1039/C4DT03267E
A systematic theoretical study is reported on the mechanisms of reductive elimination from arylPd(II) trifluoromethyl complexes, an important elementary reaction for Pd-catalyzed trifluoromethylation reactions. Various mechanisms leading to the formation of trifluoromethylated products and also competing side pathways have been evaluated. Furthermore, ligand effects are systematically evaluated which provide valuable information about the favourable properties of the ancillary ligands for promoting reductive elimination of trifluoromethylated products from Pd(II) centers.





![Dichlorotetrakis[2-(2-Pyridyl)Phenyl]Diiridium(Iii)](http://img.cochemist.com/ccimg/92300/92220-65-0.png)
![Dichlorotetrakis[2-(2-Pyridyl)Phenyl]Diiridium(Iii)](http://img.cochemist.com/ccimg/92300/92220-65-0_b.png)








![3-Buten-2-one, 4-[2-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/852600/852527-28-7.png)
![3-Buten-2-one, 4-[2-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/852600/852527-28-7_b.png)
![Benzaldehyde, 4-[(1E)-3-oxo-1-butenyl]-](http://img.cochemist.com/ccimg/803000/802954-26-3.png)
![Benzaldehyde, 4-[(1E)-3-oxo-1-butenyl]-](http://img.cochemist.com/ccimg/803000/802954-26-3_b.png)









![3-Buten-2-one, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/115700/115665-92-4.png)
![3-Buten-2-one, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/115700/115665-92-4_b.png)

















![2-Propenenitrile, 3-[4-(trifluoromethyl)phenyl]-, (2Z)-](http://img.cochemist.com/ccimg/57200/57103-25-0.png)
![2-Propenenitrile, 3-[4-(trifluoromethyl)phenyl]-, (2Z)-](http://img.cochemist.com/ccimg/57200/57103-25-0_b.png)


![2-Propenenitrile, 3-[4-(trifluoromethyl)phenyl]-, (E)-](http://img.cochemist.com/ccimg/51800/51791-29-8.png)
![2-Propenenitrile, 3-[4-(trifluoromethyl)phenyl]-, (E)-](http://img.cochemist.com/ccimg/51800/51791-29-8_b.png)












![[1,1'-Biphenyl]-4-carbonitrile,4'-nitro-](http://img.cochemist.com/ccimg/31900/31835-63-9.png)
![[1,1'-Biphenyl]-4-carbonitrile,4'-nitro-](http://img.cochemist.com/ccimg/31900/31835-63-9_b.png)

![4-[4-(DIMETHYLAMINO)PHENYL]BUT-3-EN-2-ONE](http://img.cochemist.com/ccimg/30700/30625-58-2.png)
![4-[4-(DIMETHYLAMINO)PHENYL]BUT-3-EN-2-ONE](http://img.cochemist.com/ccimg/30700/30625-58-2_b.png)



















![2-Propenenitrile, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/4900/4854-85-7.png)
![2-Propenenitrile, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/4900/4854-85-7_b.png)