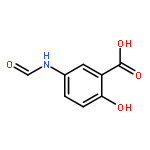
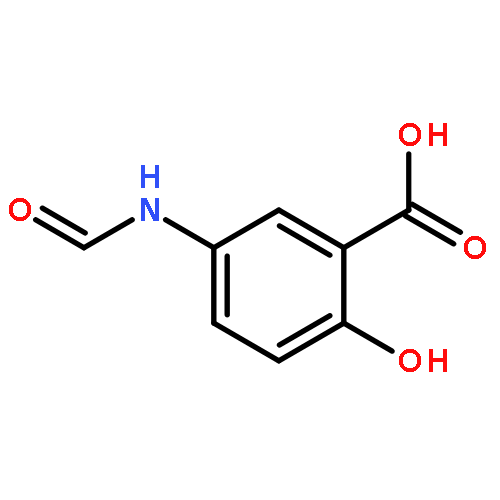
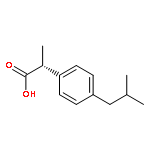
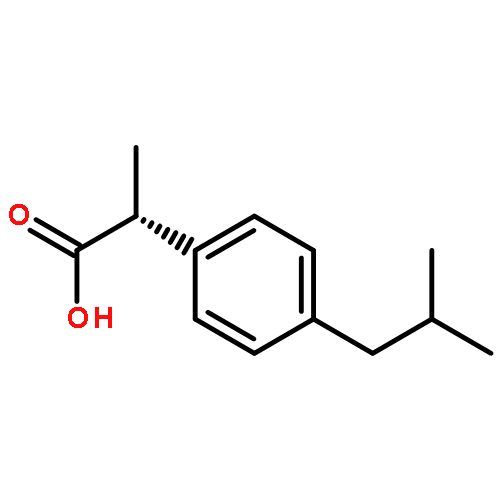
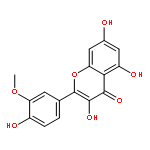
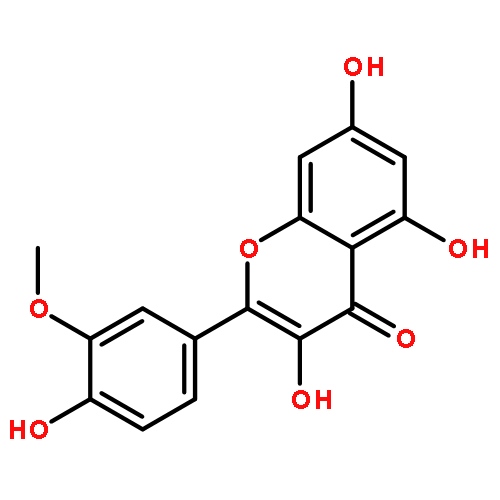
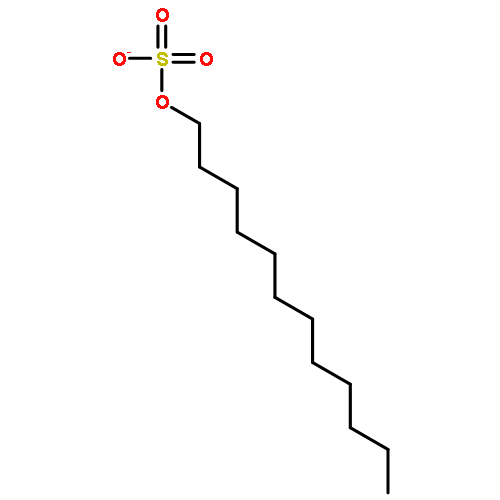
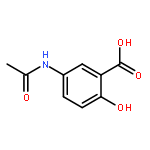
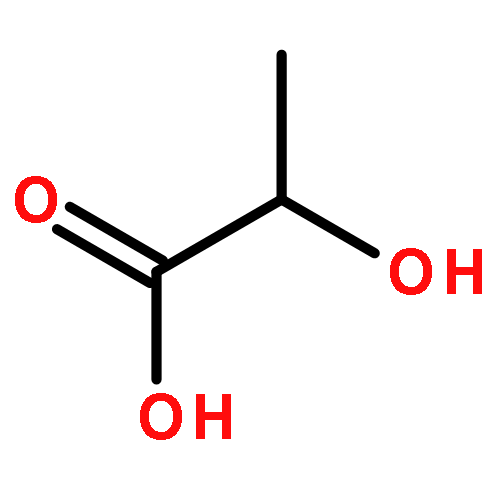
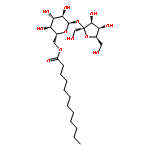
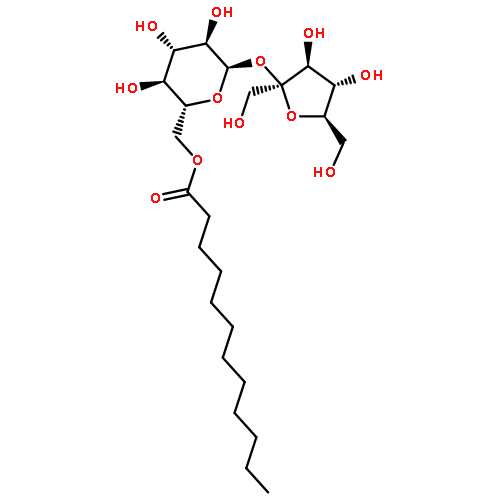
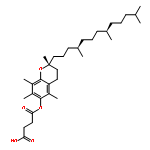
Co-reporter:Dengning Xia;Fude Cui;Yong Gan;Huiling Mu;Mingshi Yang
Journal of Pharmaceutical Sciences 2014 Volume 103( Issue 2) pp:697-705
Publication Date(Web):
DOI:10.1002/jps.23830
The effect of polymorphism of glycerol monostearate (GMS) on drug incorporation and release from lipid matrix particles (LMPs) was investigated using fenofibrate as a model drug. X-ray powder diffraction and differential scanning calorimetry were used to study the polymorphism change of GMS and the drug incorporation in GMS matrix. When medium-chain triglycerides (MCT) was absent, melted GMS was frozen to α-form of GMS with drug molecularly dispersed, whereas β-form of GMS was formed with part of drug crystallized out when the ratio of GMS/MCT in the lipid matrix was 2:1 (w/w). For LMP composed of GMS/MCT (2:1, w/w) prepared, GMS was in α-form when the particles were in nanometer range, whereas GMS was in β-form when lipid particles were in micrometer range. The model drug was molecularly dispread in α-form lipid nanoparticles, whereas part of drug was expulsed out from microparticles because of the denser crystalline packing than α-form of GMS, and caused a faster drug release from lipid microparticles than that from nanoparticles. During the storage, the transformation of GMS from α-form into the more stable β-form promoted drug expulsion and caused drug precipitation. In conclusion, the polymorphism of GMS is an important factor determining particle stability, drug incorporation, and the release of the drug from LMP. Critical attention should be paid on the investigation as well as control of the lipid polymorphism when formulating lipid-based matrix particles. © 2013 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 103:697–705, 2014
Co-reporter:Su-Ting Huang, Yong-Zhong Du, Hong Yuan, Xing-Guo Zhang, Jing Miao, Fu-De Cui, Fu-Qiang Hu
Carbohydrate Polymers 2011 Volume 83(Issue 4) pp:1715-1722
Publication Date(Web):1 February 2011
DOI:10.1016/j.carbpol.2010.10.032
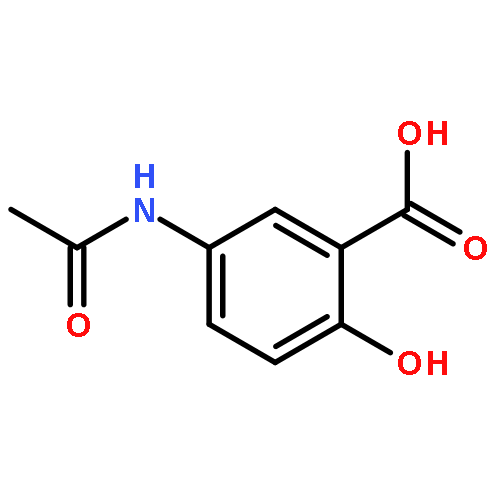
The controlled release of chemotherapeutical reagent with high water solubility was a challenge for targeting drug delivery. In this article, an antiviral agent, acyclovir was conjugated to chitosan-g-stearate via a succinate linker. Chitosan-g-stearate was synthesized by the reaction between the amino group of chitosan oligosaccharide and the carboxyl group of stearic acid. Both chitosan-g-stearate and acyclovir-chitosan-g-stearate could self aggregate to form micelles in aqueous solution. Acyclovir-chitosan-g-stearate micelle had smaller size (24.9 ± 1.1 nm), lower positive zeta potential (24.4 mV) and higher critical micelle concentration (123.23 mg mL−1) in distilled water, compared with those of chitosan-g-stearate (34.2 ± 3.8 nm, 46.9 ± 6.2 mV and 90.07 mg mL−1, respectively). Acyclovir release from acyclovir-chitosan-g-stearate micelles could prolong to 24 h in vitro. For the free acyclovir and acyclovir-chitosan-g-stearate micelle with acyclovir concentration of 0.044 μM mL−1, the inhibition of acyclovir on hepatitis B surface antigen was increased from 12.7% to 22.3% from 5 d to 9 d, while the inhibition of acyclovir-chitosan-g-stearate was increased from 58.2% to 80.3% from 5 d to 9 d. The cellular uptake and antiviral activity of acyclovir was successfully increased and improved through chemical conjugation of acyclovir to chitosan-g-stearate.
Co-reporter:Fu-de Cui;An-jin Tao;Dong-mei Cun;Li-qiang Zhang;Kai Shi
Journal of Pharmaceutical Sciences 2007 Volume 96(Issue 2) pp:421-427
Publication Date(Web):18 OCT 2006
DOI:10.1002/jps.20750
The aim of the present work was to investigate the preparation of PLGA nanoparticles (PNP) and PLGA-Hp55 nanoparticles (PHNP) as potential drug carriers for oral insulin delivery. The nanoparticles were prepared by a modified emulsion solvent diffusion method in water, and their physicochemical characteristics, drug release in vitro and hypoglycemic effects in diabetic rats were evaluated. The particle sizes of the PNP and PHNP were 150 ± 17 and 169 ± 16 nm, respectively, and the drug recoveries of the nanoparticles were 50.30 ± 3.1 and 65.41 ± 2.3%, respectively. The initial release of insulin from the nanoparticles in simulated gastric fluid over 1 h was 50.46 ± 6.31 and 19.77 ± 3.15%, respectively. The relative bioavailability of PNP and PHNP compared with subcutaneous (s.c.) injection (1 IU/kg) in diabetic rats was 3.68 ± 0.29 and 6.27 ± 0.42%, respectively. The results show that the use of insulin-loaded PHNP is an effective method of reducing serum glucose levels. ©2006 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 96:421–427, 2007
Co-reporter:Dengning Xia, Fude Cui, Yong Gan, Huiling Mu, Mingshi Yang
Journal of Pharmaceutical Sciences (February 2014) Volume 103(Issue 2) pp:697-705
Publication Date(Web):1 February 2014
DOI:10.1002/jps.23830
The effect of polymorphism of glycerol monostearate (GMS) on drug incorporation and release from lipid matrix particles (LMPs) was investigated using fenofibrate as a model drug. X-ray powder diffraction and differential scanning calorimetry were used to study the polymorphism change of GMS and the drug incorporation in GMS matrix. When medium-chain triglycerides (MCT) was absent, melted GMS was frozen to α-form of GMS with drug molecularly dispersed, whereas β-form of GMS was formed with part of drug crystallized out when the ratio of GMS/MCT in the lipid matrix was 2:1 (w/w). For LMP composed of GMS/MCT (2:1, w/w) prepared, GMS was in α-form when the particles were in nanometer range, whereas GMS was in β-form when lipid particles were in micrometer range. The model drug was molecularly dispread in α-form lipid nanoparticles, whereas part of drug was expulsed out from microparticles because of the denser crystalline packing than α-form of GMS, and caused a faster drug release from lipid microparticles than that from nanoparticles. During the storage, the transformation of GMS from α-form into the more stable β-form promoted drug expulsion and caused drug precipitation. In conclusion, the polymorphism of GMS is an important factor determining particle stability, drug incorporation, and the release of the drug from LMP. Critical attention should be paid on the investigation as well as control of the lipid polymorphism when formulating lipid-based matrix particles.
Co-reporter:M. Niu, K. Shi, Y. Sun, J. Wang, F. Cui
Journal of Drug Delivery Science and Technology (2008) Volume 18(Issue 4) pp:293-297
Publication Date(Web):1 January 2008
DOI:10.1016/S1773-2247(08)50055-4
Cyclosporine A as a model drug, drug loaded solid lipid nanoparticles (SLNs) were prepared with glyceryl monostearate and Compritol ATO 888 using the ultrasonication method. The characteristics of SLN were assessed by particle size analysis, zeta potential measurement, entrapment efficiency, physical stability andpre-ocular retention study. It demonstrated that the particle size and stability of the SLNs were greatly influenced by the formulation including the composition of lipid matrix, surfactant, charge modifier and the drug amount. The CyA-loaded SLNs produced by optimized formulation showed a good particle size distribution, the average particle size, zeta potential and entrapment efficiency was 122 nm, + 23 mv and 93%, respectively, as well as good physical stability. The ocular retention study indicated that the SLNs coated with plus charge modifier enhanced ocular retention time on the cornea surface compared with SLNs without surface modifier.
Co-reporter:D. Cun, F. Cui, L. Yang, M. Yang, ... R. Yang
Journal of Drug Delivery Science and Technology (2008) Volume 18(Issue 4) pp:267-272
Publication Date(Web):1 January 2008
DOI:10.1016/S1773-2247(08)50051-7
The purpose of this work was to study the physicochemical characteristics of a series of melittin-loaded poly (d,l-lactide-co-glycolide) (PLGA) microspheres (MS) intended for intra-articular injection and the mechanism of melittin release. Melittin-loaded MS were prepared by a double emulsion solvent evaporation method and characterized in terms of drug loading, size, surface morphology, in vitro and in vivo in rat air-pouch (pseudo synovial fluid) drug release profiles. The effects of molecular weight of PLGA and osmotic pressure gradient between the inner and the outer water phase were investigated. The melittin MS were round in shape and with the size in the range of 4-15 μm which is suitable for intraarticular injection. The encapsulation efficiency varied between 65 and 96%. In vitro melittin release occurred in typical tri-phase pattern. The in vitro release data were fitted to certain theoretical models and it was concluded that the erosion of polymer is the main mechanism that controls the release of melittin after initial burst release. Melittin could be released continuously and completely from the lower molecular weight PLGA for one month both in vitro and in vivo. There is a satisfactory correlation between in vitro and in vivo release behavior. The present work demonstrated that the lower molecular weight PLGA is the best candidate for a one month long-term sustained release dosage form of melittin.
Co-reporter:S. Sun, F. Cui, Y. Kawashima, N. Liang, ... Y. Yu
Journal of Drug Delivery Science and Technology (2008) Volume 18(Issue 4) pp:239-243
Publication Date(Web):1 January 2008
DOI:10.1016/S1773-2247(08)50047-5
The aim of this study was to prepare a novel insulin-sodium oleate (Ins/SO) complex and to evaluate the characteristics of the resultant complex. The Ins/SO complex was prepared using the hydrophobic ion pairing (HIP) method and the structure characters were assessed by the FTIR and DSC analysis method. It was found that the Ins/SO complex was synthesized by the HIP method. The insulin complexation efficiency was reached up to 96.6 ± 0.41% and mean diameter of the Ins/SO complex was sized about 80 nm under optimal conditions. The insulin bioactivity of the complex was evaluated by in vivo test. When 1 IU/kg insulin or insulin equivalent Ins/SO complex was given to normal rats by subcutaneous injection, the plasma glucose level was reduced by almost the same percentage (41.0 ± 8.19% for insulin and 44.0 ± 6.29% for Ins/SO complex, respectively) over 1 h. It was showed that insulin was not out of the bioactivity during the complexation process. In order to evaluate the bioactivity of the complex for oral administration, 20 IU/kg insulin or insulin equivalent Ins/SO complex was given to rats by intragastric administration, respectively. In the case of the Ins/SO complex, the plasma glucose level reduced to 59.5 ± 6.29% of the initial one within the first 4 h and gradually recovered to 88.9 ± 8.73% within 16 h, while there was almost no hypoglycemic effect for native insulin. The results suggested that the complex has potential for oral candidates.
Co-reporter:Qian Li, Yong-Zhong Du, Hong Yuan, Xing-Guo Zhang, Jing Miao, Fu-De Cui, Fu-Qiang Hu
European Journal of Pharmaceutical Sciences (20 November 2010) Volume 41(Issues 3–4) pp:498-507
Publication Date(Web):20 November 2010
DOI:10.1016/j.ejps.2010.08.004
To increase lipophilicity of water-soluble antiviral drug, the prodrug of Lamivudine (LA), Lamivudine stearate (LAS) was synthesized via ester linkage between LA and stearic acid. After the esterification, the octanol–water partition coefficient (log P) of LA increased from −0.95 to 1.82. Stearic acid-g-chitosan oligosaccharide (CSO-SA) micelles have demonstrated fast internalization and accumulation ability to tumor cells. Herein, the CSO-SA with 3.79% amino substitution degree (SD) was prepared for loading LAS. The critical micelle concentration (CMC) of CSO-SA was about 0.032 mg/ml. The micelles with 1 mg/ml CSO-SA concentration had 460.8 nm average diameters with a narrow size distribution and 29.7 mV surface zeta potential. After LAS was incorporated, the micellar size decreased and the zeta potential increased. The LAS loaded CSO-SA micelles (CSO-SA/LAS) possessed high entrapment efficiency and drug loading. LA release from CSO-SA/LAS showed a pH-dependent behavior. The release rate of LA from CSO-SA/LAS increased significantly as the pH of release medium reduced from 7.4 to 6.2. CSO-SA/LAS presented a low cytotoxicity and high cellular uptake percentage of LAS against HBV transfected tumor cells (HepG2.2.15). In vitro anti-HBV activities of CSO-SA/LAS presented more conspicuous inhibitory effects on antigen expression and DNA replication compared with LA and LAS.