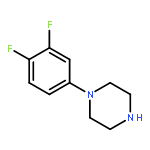
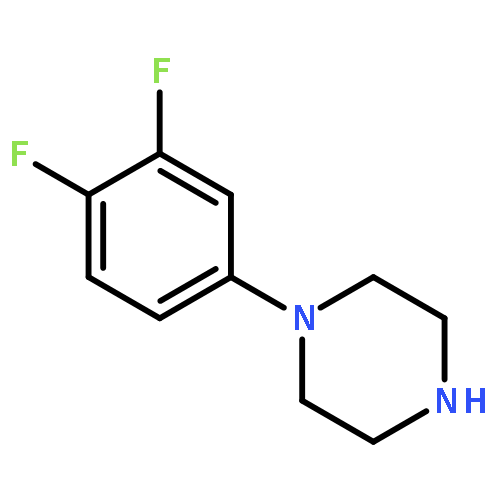
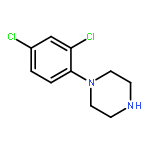
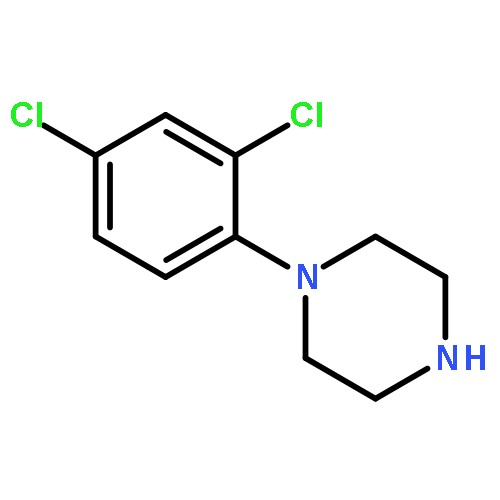
Co-reporter:Jun Yong Choi, Larissa M. Podust, and William R. Roush
Chemical Reviews 2014 Volume 114(Issue 22) pp:11242
Publication Date(Web):October 22, 2014
DOI:10.1021/cr5003134
Co-reporter:Solymar Negretti ; Alison R. H. Narayan ; Karoline C. Chiou ; Petrea M. Kells ; Jessica L. Stachowski ; Douglas A. Hansen ; Larissa M. Podust ; John Montgomery ;David H. Sherman
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:4901-4904
Publication Date(Web):March 14, 2014
DOI:10.1021/ja5016052
Highly regioselective remote hydroxylation of a natural product scaffold is demonstrated by exploiting the anchoring mechanism of the biosynthetic P450 monooxygenase PikCD50N-RhFRED. Previous studies have revealed structural and biochemical evidence for the role of a salt bridge between the desosamine N,N-dimethylamino functionality of the natural substrate YC-17 and carboxylate residues within the active site of the enzyme, and selectivity in subsequent C–H bond functionalization. In the present study, a substrate-engineering approach was conducted that involves replacing desosamine with varied synthetic N,N-dimethylamino anchoring groups. We then determined their ability to mediate enzymatic total turnover numbers approaching or exceeding that of the natural sugar, while enabling ready introduction and removal of these amino anchoring groups from the substrate. The data establish that the size, stereochemistry, and rigidity of the anchoring group influence the regioselectivity of enzymatic hydroxylation. The natural anchoring group desosamine affords a 1:1 mixture of regioisomers, while synthetic anchors shift YC-17 analogue C-10/C-12 hydroxylation from 20:1 to 1:4. The work demonstrates the utility of substrate engineering as an orthogonal approach to protein engineering for modulation of regioselective C–H functionalization in biocatalysis.
Co-reporter:Debora F. Vieira ; Jun Yong Choi ; Claudia M. Calvet ; Jair Lage Siqueira-Neto ; Jonathan B. Johnston ; Danielle Kellar ; Jiri Gut ; Michael D. Cameron ; James H. McKerrow ; William R. Roush ;Larissa M. Podust
Journal of Medicinal Chemistry 2014 Volume 57(Issue 23) pp:10162-10175
Publication Date(Web):November 13, 2014
DOI:10.1021/jm501568b
Chagas disease is a chronic infection in humans caused by Trypanosoma cruzi and manifested in progressive cardiomyopathy and/or gastrointestinal dysfunction. Limited therapeutic options to prevent and treat Chagas disease put 8 million people infected with T. cruzi worldwide at risk. CYP51, involved in the biosynthesis of the membrane sterol component in eukaryotes, is a promising drug target in T. cruzi. We report the structure–activity relationships (SAR) of an N-arylpiperazine series of N-indolyloxopyridinyl-4-aminopropanyl-based inhibitors designed to probe the impact of substituents in the terminal N-phenyl ring on binding mode, selectivity and potency. Depending on the substituents at C-4, two distinct ring binding modes, buried and solvent-exposed, have been observed by X-ray structure analysis (resolution of 1.95–2.48 Å). The 5-chloro-substituted analogs 9 and 10 with no substituent at C-4 demonstrated improved selectivity and potency, suppressing ≥99.8% parasitemia in mice when administered orally at 25 mg/kg, b.i.d., for 4 days.
Co-reporter:Claudia M. Calvet ; Debora F. Vieira ; Jun Yong Choi ; Danielle Kellar ; Michael D. Cameron ; Jair Lage Siqueira-Neto ; Jiri Gut ; Jonathan B. Johnston ; Li Lin ; Susan Khan ; James H. McKerrow ; William R. Roush ;Larissa M. Podust
Journal of Medicinal Chemistry 2014 Volume 57(Issue 16) pp:6989-7005
Publication Date(Web):August 7, 2014
DOI:10.1021/jm500448u
CYP51 is a P450 enzyme involved in the biosynthesis of the sterol components of eukaryotic cell membranes. CYP51 inhibitors have been developed to treat infections caused by fungi, and more recently the protozoan parasite Trypanosoma cruzi, the causative agent of Chagas disease. To specifically optimize drug candidates for T. cruzi CYP51 (TcCYP51), we explored the structure–activity relationship (SAR) of a N-indolyl-oxopyridinyl-4-aminopropanyl-based scaffold originally identified in a target-based screen. This scaffold evolved via medicinal chemistry to yield orally bioavailable leads with potent anti-T. cruzi activity in vivo. Using an animal model of infection with a transgenic T. cruzi Y luc strain expressing firefly luciferase, we prioritized the biaryl and N-arylpiperazine analogues by oral bioavailability and potency. The drug–target complexes for both scaffold variants were characterized by X-ray structure analysis. Optimization of both binding mode and pharmacokinetic properties of these compounds led to potent inhibitors against experimental T. cruzi infection.
Co-reporter:Jun Yong Choi, Claudia M. Calvet, Debora F. Vieira, Shamila S. Gunatilleke, Michael D. Cameron, James H. McKerrow, Larissa M. Podust, and William R. Roush
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 4) pp:434-439
Publication Date(Web):January 22, 2014
DOI:10.1021/ml500010m
Sterol 14α-demethylase (CYP51) is an important therapeutic target for fungal and parasitic infections due to its key role in the biosynthesis of ergosterol, an essential component of the cell membranes of these pathogenic organisms. We report the development of potent and selective d-tryptophan-derived inhibitors of T. cruzi CYP51. Structural information obtained from the cocrystal structure of CYP51 and (R)-2, which is >1000-fold more potent than its enantiomer (S)-1, was used to guide design of additional analogues. The in vitro efficacy data presented here for (R)-2–(R)-8, together with preliminary in vitro pharmacokinetic data suggest that this new CYP51 inhibitor scaffold series has potential to deliver drug candidates for treatment of T. cruzi infections.Keywords: CYP51; inhibitors; R-configuration; T. cruzi;
Co-reporter:Dr. Debora F. Vieira;Dr. Jun Yong Choi; Dr. William R. Roush; Dr. Larissa M. Podust
ChemBioChem 2014 Volume 15( Issue 8) pp:1111-1120
Publication Date(Web):
DOI:10.1002/cbic.201402027
Abstract
Chagas disease is a chronic infection caused by the protozoan parasite Trypanosoma cruzi, manifested in progressive cardiomyopathy and/or gastrointestinal dysfunction. Therapeutic options to prevent or treat Chagas disease are limited. CYP51, the enzyme key to the biosynthesis of eukaryotic membrane sterols, is a validated drug target in both fungi and T. cruzi. Sulfonamide derivatives of 4-aminopyridyl-based inhibitors of T. cruzi CYP51 (TcCYP51), including the sub-nanomolar compound 3, have molecular structures distinct from other validated CYP51 inhibitors. They augment the biologically relevant chemical space of molecules targeting TcCYP51. In a 2.08 Å X-ray structure, TcCYP51 is in a conformation that has been influenced by compound 3 and is distinct from the previously characterized ground-state conformation of CYP51 drug–target complexes. That the binding site was modulated in response to an incoming inhibitor for the first time characterizes TcCYP51 as a flexible target rather than a rigid template.
Co-reporter:Jun Yong Choi ; Claudia M. Calvet ; Shamila S. Gunatilleke ; Claudia Ruiz ; Michael D. Cameron ; James H. McKerrow ; Larissa M. Podust ;William R. Roush
Journal of Medicinal Chemistry 2013 Volume 56(Issue 19) pp:7651-7668
Publication Date(Web):September 30, 2013
DOI:10.1021/jm401067s
A new series of 4-aminopyridyl-based lead inhibitors targeting Trypanosoma cruzi CYP51 (TcCYP51) has been developed using structure-based drug design as well as structure–property relationship (SPR) analyses. The screening hit starting point, LP10 (KD ≤ 42 nM; EC50 = 0.65 μM), has been optimized to give the potential leads 14t, 27i, 27q, 27r, and 27t, which have low-nanomolar binding affinity to TcCYP51 and significant activity against T. cruzi amastigotes cultured in human myoblasts (EC50 = 14–18 nM for 27i and 27r). Many of the optimized compounds have improved microsome stability, and most are selective against human CYPs 1A2, 2D6, and 3A4 (<50% inhibition at 1 μM). A rationale for the improvement in microsome stability and selectivity of inhibitors against human metabolic CYP enzymes is presented. In addition, the binding mode of 14t with the Trypanosoma brucei CYP51 (TbCYP51) orthologue has been characterized by X-ray structure analysis.
Co-reporter:Larissa M. Podust and David H. Sherman
Natural Product Reports 2012 vol. 29(Issue 10) pp:1251-1266
Publication Date(Web):23 Jul 2012
DOI:10.1039/C2NP20020A
Covering: 1985 to 2012
Diverse oxygenation patterns of natural products generated by secondary metabolic pathways in microorganisms and plants are largely achieved through the tailoring reactions catalysed by cytochrome P450 enzymes (P450s). P450s are a large family of oxidative hemoproteins found in all life forms from prokaryotes to humans. Understanding the reactivity and selectivity of these fascinating C–H bond-activating catalysts will advance their use in generating valuable pharmaceuticals and products for medicine, agriculture and industry. A major strength of this P450 group is its set of established enzyme–substrate relationships, the source of the most detailed knowledge on how P450 enzymes work. Engineering microbial-derived P450 enzymes to accommodate alternative substrates and add new functions continues to be an important near- and long-term practical goal driving the structural characterization of these molecules. Understanding the natural evolution of P450 structure-function should accelerate metabolic engineering and directed evolutionary approaches to enhance diversification of natural product structures and other biosynthetic applications.
Co-reporter:Hugues Ouellet, Petrea M. Kells, Paul R. Ortiz de Montellano, Larissa M. Podust
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 1) pp:332-337
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmcl.2010.11.007
Cytochrome P450 CYP125A1 of Mycobacteriumtuberculosis, a potential therapeutic target for tuberculosis in humans, initiates degradation of the aliphatic chain of host cholesterol and is essential for establishing M. tuberculosis infection in a mouse model of disease. We explored the interactions of CYP125A1 with a reverse type I inhibitor by X-ray structure analysis and UV–vis spectroscopy. Compound LP10 (α-[(4-methylcyclohexyl)carbonyl amino]-N-4-pyridinyl-1H-indole-3-propanamide), previously identified as a potent type II inhibitor of Trypanosomacruzi CYP51, shifts CYP125A1 to a water-coordinated low-spin state upon binding with low micromolar affinity. When LP10 is present in the active site, the crystal structure and spectral characteristics both demonstrate changes in lipophilic and electronic properties favoring coordination of the iron axial water ligand. These results provide an insight into the structural requirements for developing selective CYP125A1 inhibitors.
Co-reporter:Petrea M. Kells, Hugues Ouellet, Javier Santos-Aberturas, Jesus F. Aparicio, Larissa M. Podust
Chemistry & Biology 2010 Volume 17(Issue 8) pp:841-851
Publication Date(Web):27 August 2010
DOI:10.1016/j.chembiol.2010.05.026
We present the X-ray structure of PimD, both substrate-free and in complex with 4,5-desepoxypimaricin. PimD is a cytochrome P450 monooxygenase with native epoxidase activity that is critical in the biosynthesis of the polyene macrolide antibiotic pimaricin. Intervention in this secondary metabolic pathway could advance the development of drugs with improved pharmacologic properties. Epoxidation by P450 typically includes formation of a charge-transfer complex between an oxoferryl π-cation radical species (Compound I) and the olefin π-bond as the initial intermediate. Catalytic and structural evidence presented here suggest that epoxidation of 4,5-desepoxypimaricin proceeds via a hydroperoxoferric intermediate (Compound 0). The oxygen atom of Compound 0 distal to the heme iron may insert into the double bond of the substrate to make an epoxide ring. Stereoelectronic features of the putative transition state suggest substrate-assisted proton delivery.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (253 K)Download as PowerPoint slideHighlights► Binding mode of polyene macrolide to P450 PimD is revealed by X-ray crystallography ► Epoxidation of 4,5-deepoxipimaricin via Compound 0 is suggested ► Steric and stereoelectric factors drive epoxidation down the less favorable pathway
Co-reporter:Alexis S. Ivanov, Oksana V. Gnedenko, Andrey A. Molnar, Alexander I. Archakov and Larissa M. Podust
ACS Chemical Biology 2010 Volume 5(Issue 8) pp:767
Publication Date(Web):June 17, 2010
DOI:10.1021/cb100055v
NADPH-cytochrome P450 reductase (CPR) transfers two reducing equivalents derived from NADPH via FAD and FMN to microsomal P450 monooxygenases in one-electron transfer steps. The crystal structure of yeast CPR (yCPR) contains a surface-exposed FMN binding site (FMN2 site) at the interface of the FMN binding and connecting domains, in addition to the single buried site that has been observed in rat CPR. This finding provides a testable hypothesis of how intramolecular (between FAD and FMN) and intermolecular (between FMN and P450) electron transfer may occur in CPR. To verify that occupancy of the FMN2 site is not an artifact of crystallization, a surface plasmon resonance (SPR) biosensor technique has been applied to probe the selectivity of this site under functional conditions. A series of kinetic and equilibrium binding experiments involving yCPR immobilized on different sensor chip surfaces was performed using FMN and FAD, as well as FMN-derived compounds, including riboflavin, dimethylalloxazine, and alloxazine, and other molecules that resemble the planar isoalloxazine ring structure. Only FMN and FAD showed stoichiometric binding responses. Binding affinity for FMN was in the submicromolar range, 30 times higher than that for FAD. Association kinetic rates for the yCPR/FMN complex were up to 60-fold higher than for the yCPR/FAD complex. Taken together, these data indicate that (i) the surface-exposed site in yCPR is highly selective toward binding flavins, (ii) binding of FMN in this site is notably favored, and finally, (iii) both the phosphate group and the isoalloxazine ring of FMN are essential for binding.

![2-fluoro-4-[4-fluoro-3-(trifluoromethyl)phenyl]benzoic Acid](http://img.cochemist.com/ccimg/1262000/1261916-27-1.png)
![2-fluoro-4-[4-fluoro-3-(trifluoromethyl)phenyl]benzoic Acid](http://img.cochemist.com/ccimg/1262000/1261916-27-1_b.png)
![2',3,5'-Trifluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/1184600/1184513-36-7.png)
![2',3,5'-Trifluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/1184600/1184513-36-7_b.png)
![3,3',4'-Trifluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/1179200/1179174-66-3.png)
![3,3',4'-Trifluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/1179200/1179174-66-3_b.png)
![3-Fluoro-3'-(trifluoromethyl)-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/926300/926222-59-5.png)
![3-Fluoro-3'-(trifluoromethyl)-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/926300/926222-59-5_b.png)