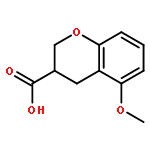
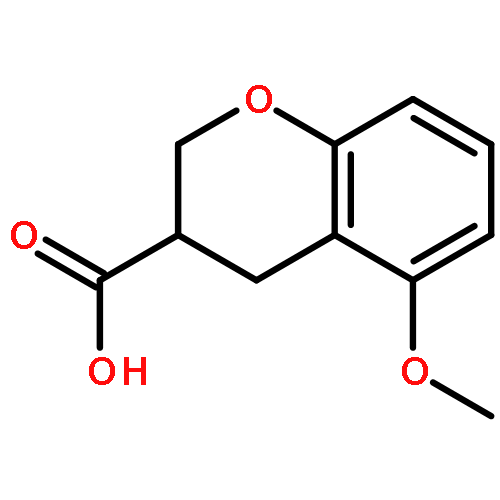
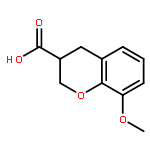
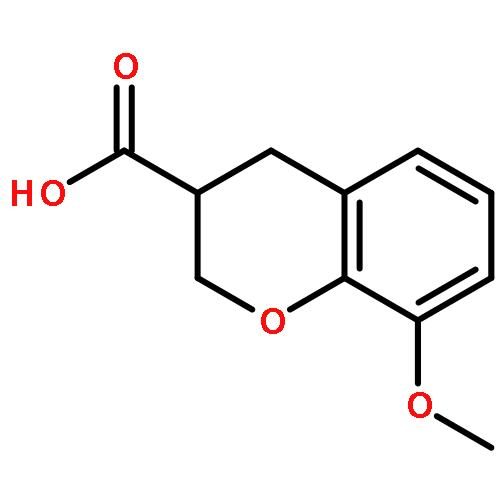
Co-reporter:Ke Zheng, Sarah Iqbal, Pamela Hernandez, HaJeung Park, Philip V. LoGrasso, and Yangbo Feng
Journal of Medicinal Chemistry December 11, 2014 Volume 57(Issue 23) pp:10013-10030
Publication Date(Web):November 13, 2014
DOI:10.1021/jm501256y
The c-jun N-terminal kinase 3 (JNK3) is expressed primarily in the brain. Numerous reports have shown that inhibition of JNK3 is a promising strategy for treatment of neurodegeneration. The optimization of aminopyrazole-based JNK3 inhibitors with improved potency, isoform selectivity, and pharmacological properties by structure–activity relationship (SAR) studies utilizing biochemical and cell-based assays, and structure-based drug design is reported. These inhibitors had high selectivity over JNK1 and p38α, minimal cytotoxicity, potent inhibition of 6-OHDA-induced mitochondrial membrane potential dissipation and ROS generation, and good drug metabolism and pharmacokinetic (DMPK) properties for iv dosing. 26n was profiled against 464 kinases and was found to be highly selective hitting only seven kinases with >80% inhibition at 10 μM. Moreover, 26n showed good solubility, good brain penetration, and good DMPK properties. Finally, the crystal structure of 26k in complex with JNK3 was solved at 1.8 Å to explore the binding mode of aminopyrazole based JNK3 inhibitors.
Co-reporter:Yangbo Feng; Philip V. LoGrasso; Olivier Defert;Rongshi Li
Journal of Medicinal Chemistry 2016 Volume 59(Issue 6) pp:2269-2300
Publication Date(Web):October 20, 2015
DOI:10.1021/acs.jmedchem.5b00683
Rho kinases (ROCKs) belong to the serine–threonine family, the inhibition of which affects the function of many downstream substrates. As such, ROCK inhibitors have potential therapeutic applicability in a wide variety of pathological conditions including asthma, cancer, erectile dysfunction, glaucoma, insulin resistance, kidney failure, neuronal degeneration, and osteoporosis. To date, two ROCK inhibitors have been approved for clinical use in Japan (fasudil and ripasudil) and one in China (fasudil). In 1995 fasudil was approved for the treatment of cerebral vasospasm, and more recently, ripasudil was approved for the treatment of glaucoma in 2014. In this Perspective, we present a comprehensive review of the physiological and biological functions for ROCK, the properties and development of over 170 ROCK inhibitors as well as their therapeutic potential, the current status, and future considerations.
Co-reporter:Yan Yin; Ke Zheng; Nibal Eid; Shannon Howard; Ji-Hak Jeong; Fei Yi∞; Jia Guo∞; Chul Min Park; Mathieu Bibian; Weilin Wu; Pamela Hernandez; HaJeung Park; Yuntao Wu∞; Jun-Li Luo; Philip V. LoGrasso
Journal of Medicinal Chemistry 2015 Volume 58(Issue 4) pp:1846-1861
Publication Date(Web):January 26, 2015
DOI:10.1021/jm501680m
The discovery/optimization of bis-aryl ureas as Limk inhibitors to obtain high potency and selectivity and appropriate pharmacokinetic properties through systematic SAR studies is reported. Docking studies supported the observed SAR. Optimized Limk inhibitors had high biochemical potency (IC50 < 25 nM), excellent selectivity against ROCK and JNK kinases (>400-fold), potent inhibition of cofilin phosphorylation in A7r5, PC-3, and CEM-SS T cells (IC50 < 1 μM), and good in vitro and in vivo pharmacokinetic properties. In the profiling against a panel of 61 kinases, compound 18b at 1 μM inhibited only Limk1 and STK16 with ≥80% inhibition. Compounds 18b and 18f were highly efficient in inhibiting cell-invasion/migration in PC-3 cells. In addition, compound 18w was demonstrated to be effective on reducing intraocular pressure (IOP) on rat eyes. Taken together, these data demonstrated that we had developed a novel class of bis-aryl urea derived potent and selective Limk inhibitors.
Co-reporter:Ding Mei, Yan Yin, Fanhong Wu, Jiaxing Cui, Hong Zhou, Guofeng Sun, Yu Jiang, Yangbo Feng
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 10) pp:2505-2517
Publication Date(Web):15 May 2015
DOI:10.1016/j.bmc.2015.03.047
An activity model and a selectivity model from 3D-QSAR studies were established by CoMFA and CoMSIA to explore the SAR. Then docking was used to study the binding modes between ligand and kinases (ROCK2 and PKA), and the molecular docking results were further validated by MD simulations. Computational results suggested that substitution containing positive charge attached to the middle phenyl ring, or electropositive group in urea linker was favored for both activity and ROCK2/PKA selectivity. Finally, three compounds were designed, and biological evaluation demonstrated that these molecular models were effective for guiding the design of potent and selective ROCK inhibitors.
Co-reporter:Ke Zheng, Chul Min Park, Sarah Iqbal, Pamela Hernandez, HaJeung Park, Philip V. LoGrasso, and Yangbo Feng
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 4) pp:413
Publication Date(Web):March 2, 2015
DOI:10.1021/ml500474d
A novel series of 2-aminopyridopyrimidinone based JNK (c-jun N-terminal kinase) inhibitors were discovered and developed. Structure–activity relationships (SARs) were systematically developed utilizing biochemical and cell based assays and in vitro and in vivo drug metabolism and pharmacokinetic (DMPK) studies. Through the optimization of lead compound 1, several potent and selective JNK inhibitors with high oral bioavailability were developed. Inhibitor 13 was a potent JNK3 inhibitor (IC50 = 15 nM), had high selectivity against p38 (IC50 > 10 μM), had high potency in functional cell based assays, and had high stability in human liver microsome (t1/2 = 76 min), a clean CYP-450 inhibition profile, and excellent oral bioavailability (%F = 87). Moreover, cocrystal structures of compounds 13 and 22 in JNK3 were solved at 2.0 Å. These structures elucidated the binding mode (Type-I binding) and can pave the way for further inhibitor design of this pyridopyrimidinone scaffold for JNK inhibition.Keywords: JNK3; kinase inhibitor; mitochondrial dysfunction; Parkinson’s disease (PD); pyridopyrimidinone
Co-reporter:Yan Yin ; Li Lin ; Claudia Ruiz ; Susan Khan ; Michael D. Cameron ¥; Wayne Grant ; Jennifer Pocas ; Nibal Eid ; HaJeung Park ∞; Thomas Schröter ; Philip V. LoGrasso ¥
Journal of Medicinal Chemistry 2013 Volume 56(Issue 9) pp:3568-3581
Publication Date(Web):April 9, 2013
DOI:10.1021/jm400062r
RhoA and its downstream effector ROCK mediate stress fiber formation and cell contraction through their effects on the phosphorylation of myosin light chain (MLC). Inhibition of the RhoA/ROCK pathway has proven to be a promising strategy for several indications such as cardiovascular disease, glaucoma, and inflammatory disease. In 2010, our group reported urea-based ROCK inhibitors as potential antiglaucoma agents. These compounds showed potent IC50 values in enzymatic and cell-based assays and significant intraocular pressure (IOP)-lowering effects in rats (∼7 mmHg).(22) To develop more advanced ROCK inhibitors targeting various potential applications (such as myocardial infarction, erectile dysfunction, multiple sclerosis, etc.) in addition to glaucoma, a thorough SAR for this urea-based scaffold was studied. The detailed optimization process, counter-screening, and in vitro and in vivo DMPK studies are discussed. Potent and selective ROCK inhibitors with various in vivo pharmacokinetic properties were discovered.
Co-reporter:Yangbo Feng, Jeremy W. Chambers, Sarah Iqbal, Marcel Koenig, HaJeung Park, Lisa Cherry, Pamela Hernandez, Mariana Figuera-Losada, and Philip V. LoGrasso
ACS Chemical Biology 2013 Volume 8(Issue 8) pp:1747
Publication Date(Web):June 10, 2013
DOI:10.1021/cb3006165
Both JNK and LRRK2 are associated with Parkinson’s disease (PD). Here we report a reasonably selective and potent kinase inhibitor (compound 6) that bound to both JNK and LRRK2 (a dual inhibitor). A bidentate-binding strategy that simultaneously utilized the ATP hinge binding and a unique protein surface site outside of the ATP pocket was applied to the design and identification of this kind of inhibitor. Compound 6 was a potent JNK3 and modest LRRK2 dual inhibitor with an enzyme IC50 value of 12 nM and 99 nM (LRRK2-G2019S), respectively. Compound 6 also exhibited good cell potency, inhibited LRRK2:G2019S-induced mitochondrial dysfunction in SHSY5Y cells, and was demonstrated to be reasonably selective against a panel of 116 kinases from representative kinase families. Design of such a probe molecule may help enable testing if dual JNK and LRRK2 inhibitions have added or synergistic efficacy in protecting against neurodegeneration in PD.
Co-reporter:Sarwat Chowdhury, Yen Ting Chen, Xingang Fang, Wayne Grant, Jennifer Pocas, Michael D. Cameron, Claudia Ruiz, Li Lin, HaJeung Park, Thomas Schröter, Thomas D. Bannister, Philip V. LoGrasso, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 6) pp:1592-1599
Publication Date(Web):15 March 2013
DOI:10.1016/j.bmcl.2013.01.109
SAR and lead optimization studies for Rock inhibitors based on amino acid-derived quinazolines are described. Studies demonstrated that these amino acid derived quinazolinones were mainly pan-Rock (I & II) inhibitors. While selectivity against other kinases could be achieved, selectivity for most of these compounds against PKA was not achieved. This is distinct from Rock inhibitors based on non-amino acid derived quinazolinones, where high selectivity against PKA could be obtained.22 The inhibitors presented here in some cases possessed sub-nanomolar inhibition of Rock, nanomolar potency in ppMLC cell based assays, low to fair cytochrome P-450 inhibition, and good human microsomal stability.
Co-reporter:Xingang Fang, Yen Ting Chen, E. Hampton Sessions, Sarwat Chowdhury, Tomas Vojkovsky, Yan Yin, Jennifer R. Pocas, Wayne Grant, Thomas Schröter, Li Lin, Claudia Ruiz, Michael D. Cameron, Philip LoGrasso, Thomas D. Bannister, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 6) pp:1844-1848
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmcl.2011.01.039
Rho kinase (ROCK) is an attractive therapeutic target for various diseases including glaucoma, hypertension, and spinal cord injury. Herein, we report the development of a series of ROCK-II inhibitors based on 4-quinazolinone and quinazoline scaffolds. SAR studies at three positions of the quinazoline core led to the identification of analogs with high potency against ROCK-II and good selectivity over protein kinase A (PKA).
Co-reporter:Sarwat Chowdhury, E. Hampton Sessions, Jennifer R. Pocas, Wayne Grant, Thomas Schröter, Li Lin, Claudia Ruiz, Michael D. Cameron, Stephan Schürer, Philip LoGrasso, Thomas D. Bannister, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7107-7112
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.083
Rho kinase (ROCK) inhibitors are potential therapeutic agents to treat disorders such as hypertension, multiple sclerosis, cancers, and glaucoma. Here, we disclose the synthesis, optimization, biological evaluation of potent indole and 7-azaindole based ROCK inhibitors that have high potency on ROCK (IC50 = 1 nM) with 740-fold selectivity over PKA (47). Moreover, 47 showed very good DMPK properties making it a good candidate for further development. Finally, docking studies with a homology model of ROCK-II were performed to rationalize the binding mode of these compounds and showed the compounds bound in both orientations to take advantage to H-bonds with Lys-121 of ROCK-II.
Co-reporter:E. Hampton Sessions, Sarwat Chowdhury, Yan Yin, Jennifer R. Pocas, Wayne Grant, Thomas Schröter, Li Lin, Claudia Ruiz, Michael D. Cameron, Philip LoGrasso, Thomas D. Bannister, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7113-7118
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.084
Therapeutic interventions with Rho kinase (ROCK) inhibitors may effectively treat several disorders such as hypertension, stroke, cancer, and glaucoma. Herein we disclose the optimization and biological evaluation of potent novel ROCK inhibitors based on substituted indole and 7-azaindole core scaffolds. Substitutions on the indole C3 position and on the indole NH and/or amide NH positions all yielded potent and selective ROCK inhibitors (25, 42, and 50). Improvement of aqueous solubility and tailoring of in vitro and in vivo DMPK properties could be achieved through these substitutions.
Co-reporter:Yen Ting Chen, Tomas Vojkovsky, Xingang Fang, Jennifer R. Pocas, Wayne Grant, Amiee M. W. Handy, Thomas Schröter, Philip LoGrasso, Thomas D. Bannister and Yangbo Feng
MedChemComm 2011 vol. 2(Issue 1) pp:73-75
Publication Date(Web):06 Dec 2010
DOI:10.1039/C0MD00194E
Rho kinase (ROCK) is currently investigated as a target for various diseases such as glaucoma and spinal cord injury. Herein, we report the asymmetric synthesis of chroman 1, a highly potent ROCK inhibitor, and its analogs. The inhibitory properties of these compounds for ROCK-II and a selected set of highly homologous kinases are also discussed.
Co-reporter:Xingang Fang ; Yan Yin ; Yen Ting Chen ; Lei Yao ; Bo Wang ; Michael D. Cameron ; Li Lin ; Susan Khan ; Claudia Ruiz ; Thomas Schröter ; Wayne Grant ; Amiee Weiser ; Jennifer Pocas ; Alok Pachori ; Stephan Schürer ; Philip LoGrasso
Journal of Medicinal Chemistry 2010 Volume 53(Issue 15) pp:5727-5737
Publication Date(Web):July 20, 2010
DOI:10.1021/jm100579r
Rho kinase (ROCK) is a promising drug target for the treatment of many diseases including hypertension, multiple sclerosis, cancer, and glaucoma. The structure−activity relationships (SAR) around a series of tetrahydroisoquinolines were evaluated utilizing biochemical and cell-based assays to measure ROCK inhibition. These novel ROCK inhibitors possess high potency, high selectivity, and appropriate pharmacokinetic properties for glaucoma applications. The lead compound, 35, had subnanomolar potency in enzyme ROCK-II assays as well as excellent cell-based potency (IC50 = 51 nM). In a kinase panel profiling, 35 had an off-target hit rate of only 1.6% against 442 kinases. Pharmacology studies showed that compound 35 was efficacious in reducing intraocular pressure (IOP) in rats with reasonably long duration of action. These results suggest that compound 35 may serve as a promising agent for further development in the treatment of glaucoma.
Co-reporter:Yan Yin, Michael D. Cameron, Li Lin, Susan Khan, Thomas Schröter, Wayne Grant, Jennifer Pocas, Yen Ting Chen, Stephan Schürer, Alok Pachori, Philip LoGrasso and Yangbo Feng
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 4) pp:175
Publication Date(Web):April 5, 2010
DOI:10.1021/ml1000382
A series of urea-based Rho kinase (ROCK) inhibitors were designed and evaluated. The discovered compounds had excellent enzyme and cellular potency, high kinase selectivity, high aqueous solubility, good porcine corneal penetration, and appropriate DMPK profiles for topical applications as antiglaucoma therapeutics.Keywords (keywords): Glaucoma; IOP; kinase inhibitor; pyrazole; ROCK; urea
Co-reporter:Yan Yin, Li Lin, Claudia Ruiz, Michael D. Cameron, Jennifer Pocas, Wayne Grant, Thomas Schröter, Weimin Chen, Derek Duckett, Stephan Schürer, Philip LoGrasso, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6686-6690
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.09.115
A series of benzothiazole derivatives as ROCK inhibitors have been discovered. Compounds with good biochemical and cellular potency, and sufficient kinase selectivity have been identified.ROCK inhibitors have been developed from 2-chromanyl- and 2-carboxamido-benzothiazoles. SAR studies and lead optimizations are described, which result in novel ROCK-II inhibitors with sub-nanomolar IC50s and good kinase selectivity.
Co-reporter:Ke Zheng ; Sarah Iqbal ; Pamela Hernandez ; HaJeung Park ; Philip V. LoGrasso
Journal of Medicinal Chemistry () pp:
Publication Date(Web):November 13, 2014
DOI:10.1021/jm501256y
The c-jun N-terminal kinase 3 (JNK3) is expressed primarily in the brain. Numerous reports have shown that inhibition of JNK3 is a promising strategy for treatment of neurodegeneration. The optimization of aminopyrazole-based JNK3 inhibitors with improved potency, isoform selectivity, and pharmacological properties by structure–activity relationship (SAR) studies utilizing biochemical and cell-based assays, and structure-based drug design is reported. These inhibitors had high selectivity over JNK1 and p38α, minimal cytotoxicity, potent inhibition of 6-OHDA-induced mitochondrial membrane potential dissipation and ROS generation, and good drug metabolism and pharmacokinetic (DMPK) properties for iv dosing. 26n was profiled against 464 kinases and was found to be highly selective hitting only seven kinases with >80% inhibition at 10 μM. Moreover, 26n showed good solubility, good brain penetration, and good DMPK properties. Finally, the crystal structure of 26k in complex with JNK3 was solved at 1.8 Å to explore the binding mode of aminopyrazole based JNK3 inhibitors.
.jpg)
![Pyrido[2,3-d]pyrimidine, 7-chloro-2-(methylthio)-](http://img.cochemist.com/ccimg/352400/352328-41-7.png)
![Pyrido[2,3-d]pyrimidine, 7-chloro-2-(methylthio)-](http://img.cochemist.com/ccimg/352400/352328-41-7_b.png)
![4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline](http://img.cochemist.com/ccimg/223700/223645-67-8.png)
![4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline](http://img.cochemist.com/ccimg/223700/223645-67-8_b.png)




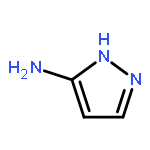
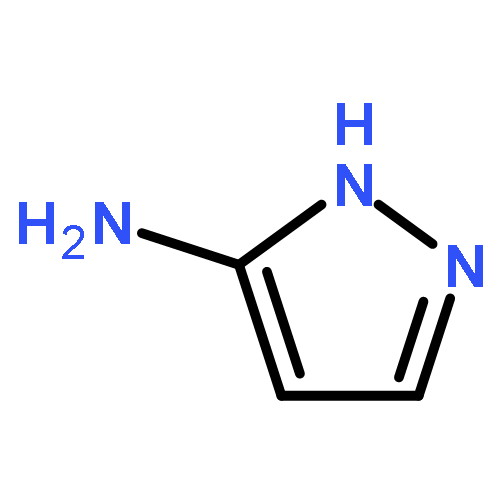
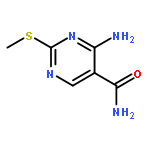
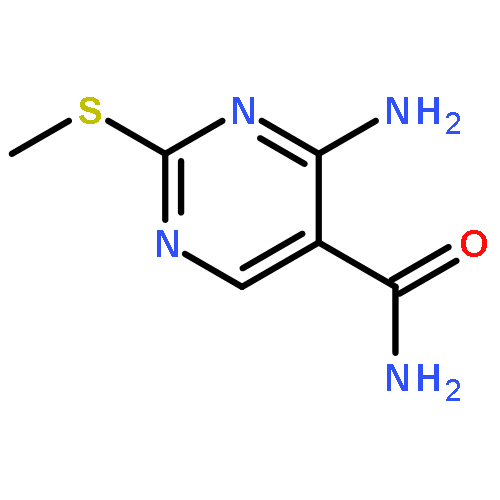
![4-chloro-5,6-dimethyl-7H-Pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/82800/82703-38-6.png)
![4-chloro-5,6-dimethyl-7H-Pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/82800/82703-38-6_b.png)
![METHYL 2-OXO-1,1',2,2',5',6',7',7A'-OCTAHYDROSPIRO[INDOLE-3,3'-PY<WBR />RROLIZINE]-2'-CARBOXYLATE](http://img.cochemist.com/ccimg/74500/74487-97-1.png)
![METHYL 2-OXO-1,1',2,2',5',6',7',7A'-OCTAHYDROSPIRO[INDOLE-3,3'-PY<WBR />RROLIZINE]-2'-CARBOXYLATE](http://img.cochemist.com/ccimg/74500/74487-97-1_b.png)
![ETHANOL, 2-[(2-FLUOROPHENYL)AMINO]-](http://img.cochemist.com/ccimg/73400/73339-01-2.png)
![ETHANOL, 2-[(2-FLUOROPHENYL)AMINO]-](http://img.cochemist.com/ccimg/73400/73339-01-2_b.png)
![4-Chloro-6-methyl-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/35900/35808-68-5.png)
![4-Chloro-6-methyl-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/35900/35808-68-5_b.png)
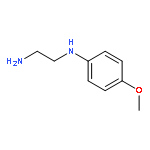
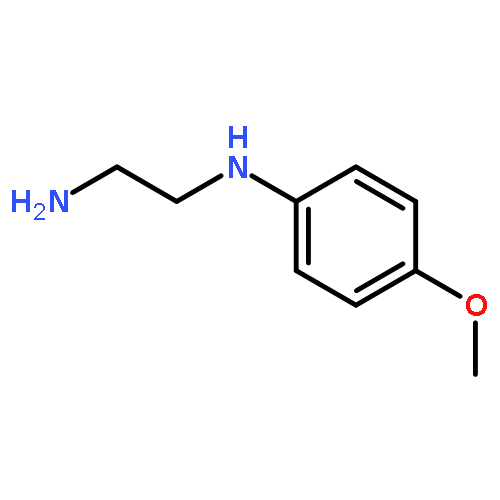


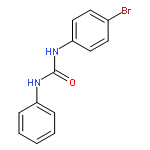
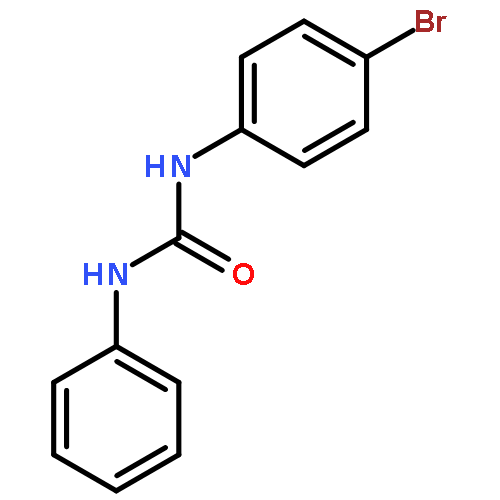


![Ethanol, 2-[(3-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/6400/6393-19-7.png)
![Ethanol, 2-[(3-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/6400/6393-19-7_b.png)





![Ethanol, 2-[(4-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-81-5.png)
![Ethanol, 2-[(4-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-81-5_b.png)
![Ethanol, 2-[(4-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-77-9.png)
![Ethanol, 2-[(4-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-77-9_b.png)
![Ethanol,2-[(3-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-76-8.png)
![Ethanol,2-[(3-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-76-8_b.png)
![Ethanol,2-[(2-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-75-7.png)
![Ethanol,2-[(2-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/3000/2933-75-7_b.png)
![2-[(4-methylphenyl)amino]ethanol](http://img.cochemist.com/ccimg/3000/2933-74-6.png)
![2-[(4-methylphenyl)amino]ethanol](http://img.cochemist.com/ccimg/3000/2933-74-6_b.png)
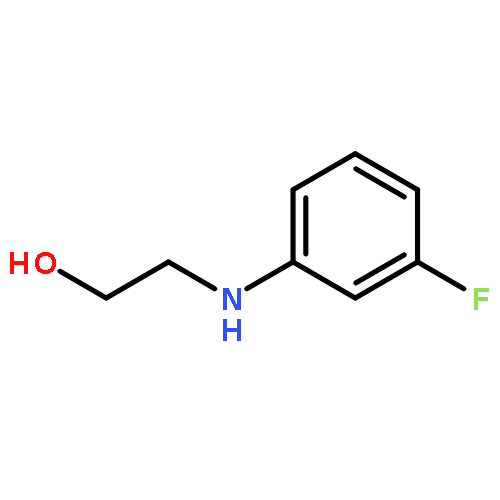
![Ethanol,2-[(3-methylphenyl)amino]-](http://img.cochemist.com/ccimg/200/102-41-0.png)
![Ethanol,2-[(3-methylphenyl)amino]-](http://img.cochemist.com/ccimg/200/102-41-0_b.png)
![Ethanol,2-[(2-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/100/94-87-1.png)
![Ethanol,2-[(2-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/100/94-87-1_b.png)












![N-(4'-Cyano[1,1'-biphenyl]-4-yl)-N'-[4-(3H-imidazo[4,5-b]pyridin-7-yl)phenyl] Urea](http://img.cochemist.com/ccimg/1361500/1361414-26-7.png)
![N-(4'-Cyano[1,1'-biphenyl]-4-yl)-N'-[4-(3H-imidazo[4,5-b]pyridin-7-yl)phenyl] Urea](http://img.cochemist.com/ccimg/1361500/1361414-26-7_b.png)


![6-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/915200/915140-96-4.png)
![6-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/915200/915140-96-4_b.png)


![Carbamic acid, [(5-cyano-1H-benzimidazol-2-yl)methyl]-, 1,1-dimethylethyl ester (9CI)](/data/chemimg/127100/852180-82-6.png)
![Carbamic acid, [(5-cyano-1H-benzimidazol-2-yl)methyl]-, 1,1-dimethylethyl ester (9CI)](/data/chemimg/127100/852180-82-6_b.png)

![6-Chloro-1H-pyrrolo[3,2-c]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800401-54-1.png)
![6-Chloro-1H-pyrrolo[3,2-c]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800401-54-1_b.png)
![(5-Chloro-1H-benzo[d]imidazol-2-yl)methanamine hydrochloride](http://img.cochemist.com/ccimg/712300/712275-25-7.png)
![(5-Chloro-1H-benzo[d]imidazol-2-yl)methanamine hydrochloride](http://img.cochemist.com/ccimg/712300/712275-25-7_b.png)
![tert-Butyl ((6-chloro-1H-benzo[d]imidazol-2-yl)methyl)carbamate](http://img.cochemist.com/ccimg/712300/712275-17-7.png)
![tert-Butyl ((6-chloro-1H-benzo[d]imidazol-2-yl)methyl)carbamate](http://img.cochemist.com/ccimg/712300/712275-17-7_b.png)














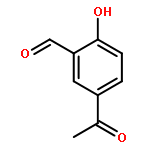
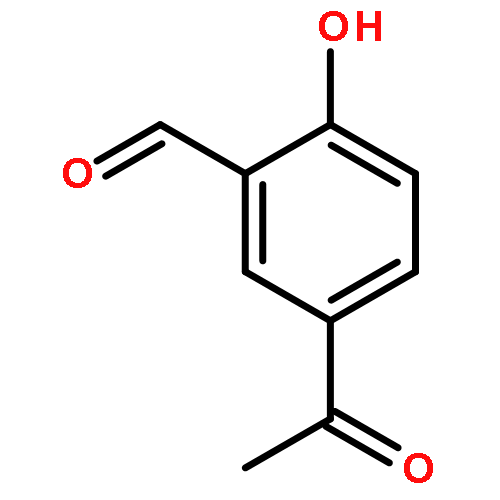
![2-[(3-methoxybenzyl)amino]ethanol](http://img.cochemist.com/ccimg/64900/64834-62-4.png)
![2-[(3-methoxybenzyl)amino]ethanol](http://img.cochemist.com/ccimg/64900/64834-62-4_b.png)
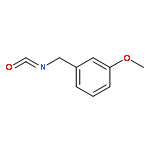
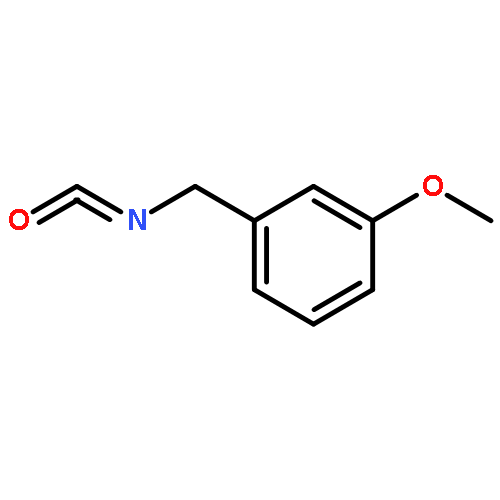
![2-(4-methoxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic Acid](http://img.cochemist.com/ccimg/55400/55362-76-0.png)
![2-(4-methoxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic Acid](http://img.cochemist.com/ccimg/55400/55362-76-0_b.png)
![ETHANOL, 2-[(4-BROMOPHENYL)AMINO]-](http://img.cochemist.com/ccimg/55200/55110-99-1.png)
![ETHANOL, 2-[(4-BROMOPHENYL)AMINO]-](http://img.cochemist.com/ccimg/55200/55110-99-1_b.png)


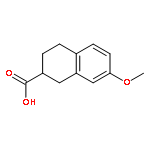
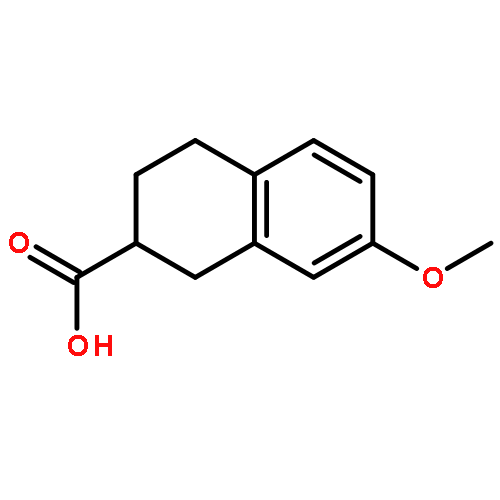
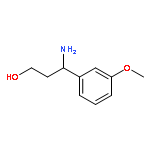
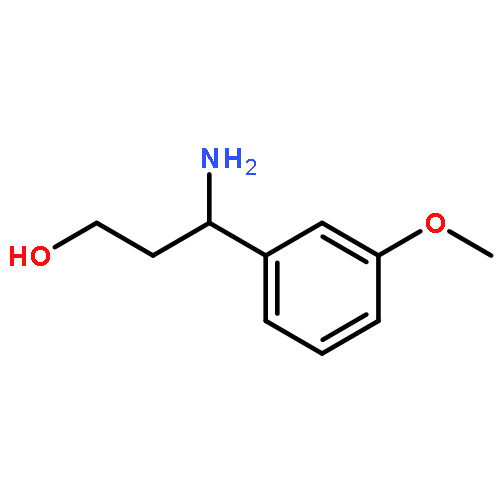


![7-Chloro-1H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/7000/6980-11-6.png)
![7-Chloro-1H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/7000/6980-11-6_b.png)







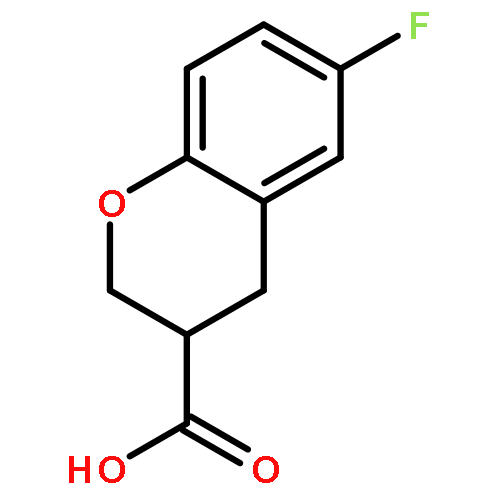
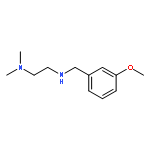
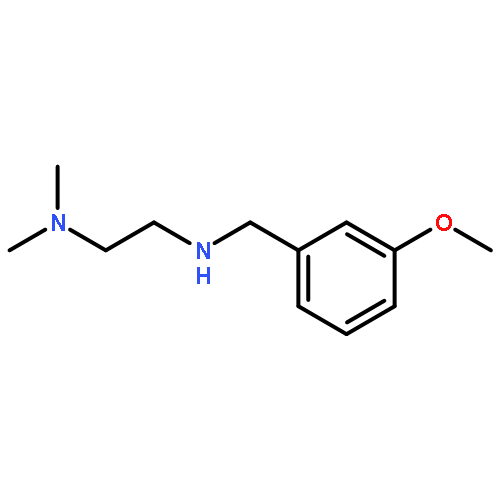
![6-Chloro-1H-pyrrolo[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800402-07-7.png)
![6-Chloro-1H-pyrrolo[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800402-07-7_b.png)