Co-reporter:Nathan D. Ricke, Alexander T. Murray, James J. Shepherd, Matthew G. Welborn, Tomohiro Fukushima, Troy Van Voorhis, and Yogesh Surendranath
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7680-7680
Publication Date(Web):September 18, 2017
DOI:10.1021/acscatal.7b03086
Using a combination of experimental and computational investigations, we assemble a consistent mechanistic model for the oxygen reduction reaction (ORR) at molecularly well-defined graphite-conjugated catalyst (GCC) active sites featuring aryl-pyridinium moieties (N+-GCC). ORR catalysis at glassy carbon surfaces modified with N+-GCC fragments displays near-first-order dependence in O2 partial pressure and near-zero-order dependence on electrolyte pH. Tafel analysis suggests an equilibrium one-electron transfer process followed by a rate-limiting chemical step at modest overpotentials that transitions to a rate-limiting electron transfer sequence at higher overpotentials. Finite-cluster computational modeling of the N+-GCC active site reveals preferential O2 adsorption at electrophilic carbons alpha to the pyridinium moiety. Together, the experimental and computational data indicate that ORR proceeds via a proton-decoupled O2 activation sequence involving either concerted or stepwise electron transfer and adsorption of O2, which is then followed by a series of electron/proton transfer steps to generate water and turn over the catalytic cycle. The proposed mechanistic model serves as a roadmap for the bottom-up synthesis of highly active N-doped carbon ORR catalysts.Keywords: density functional theory; electrocatalysis; mechanistic studies; N-doped carbon; oxygen reduction;
Co-reporter:Anna Wuttig, Youngmin Yoon, Jaeyune Ryu, and Yogesh Surendranath
Journal of the American Chemical Society November 29, 2017 Volume 139(Issue 47) pp:17109-17109
Publication Date(Web):October 5, 2017
DOI:10.1021/jacs.7b08345
We show that bicarbonate is neither a general acid nor a reaction partner in the rate-limiting step of electrochemical CO2 reduction catalysis mediated by planar polycrystalline Au surfaces. We formulate microkinetic models and propose diagnostic criteria to distinguish the role of bicarbonate. Comparing these models with the observed zero-order dependence in bicarbonate and simulated interfacial concentration gradients, we conclude that bicarbonate is not a general acid cocatalyst. Instead, it acts as a viable proton donor past the rate-limiting step and a sluggish buffer that maintains the bulk but not local pH in CO2-saturated aqueous electrolytes.
Co-reporter:Matthew E. O’Reilly, R. Soyoung Kim, Seokjoon Oh, and Yogesh Surendranath
ACS Central Science November 22, 2017 Volume 3(Issue 11) pp:1174-1174
Publication Date(Web):October 12, 2017
DOI:10.1021/acscentsci.7b00342
Electrophilic high-valent metal ions are potent intermediates for the catalytic functionalization of methane, but in many cases, their high redox potentials make these intermediates difficult or impossible to access using mild stoichiometric oxidants derived from O2. Herein, we establish electrochemical oxidation as a versatile new strategy for accessing high-valent methane monofunctionalization catalysts. We provide evidence for the electrochemical oxidation of simple PdSO4 in concentrated sulfuric acid electrolytes to generate a putative Pd2III,III species in an all-oxidic ligand field. This electrogenerated high-valent Pd complex rapidly activates methane with a low barrier of 25.9 (±2.6) kcal/mol, generating methanol precursors methyl bisulfate (CH3OSO3H) and methanesulfonic acid (CH3SO3H) via concurrent faradaic and nonfaradaic reaction pathways. This work enables new electrochemical approaches for promoting rapid methane monofunctionalization.
Co-reporter:Sterling B. Chu, Tomohiro Fukushima, and Yogesh Surendranath
Chemistry of Materials 2017 Volume 29(Issue 2) pp:
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.chemmater.6b04930
Co-reporter:Bing Yan;Nolan M. Concannon;Jarrod D. Milshtein; Dr. Fikile R. Brushett; Dr. Yogesh Surendranath
Angewandte Chemie International Edition 2017 Volume 56(Issue 26) pp:7496-7499
Publication Date(Web):2017/06/19
DOI:10.1002/anie.201702578
AbstractPolymer electrolyte membranes employed in contemporary fuel cells severely limit device design and restrict catalyst choice, but are essential for preventing short-circuiting reactions at unselective anode and cathode catalysts. Herein, we report that nickel sulfide Ni3S2 is a highly selective catalyst for the oxygen reduction reaction in the presence of 1.0 m formate. We combine this selective cathode with a carbon-supported palladium (Pd/C) anode to establish a membrane-free, room-temperature formate fuel cell that operates under benign neutral pH conditions. Proof-of-concept cells display open circuit voltages of approximately 0.7 V and peak power values greater than 1 mW cm−2, significantly outperforming the identical device employing an unselective platinum (Pt) cathode. The work establishes the power of selective catalysis to enable versatile membrane-free fuel cells.
Co-reporter:Bing Yan;Nolan M. Concannon;Jarrod D. Milshtein; Dr. Fikile R. Brushett; Dr. Yogesh Surendranath
Angewandte Chemie International Edition 2017 Volume 56(Issue 26) pp:7322-7322
Publication Date(Web):2017/06/19
DOI:10.1002/anie.201704346
A membrane-free fuel cell relies on the selectivity of catalysts at both the anode and the cathode. In their Communication on page 7496 ff., Y. Surendranath et al. show that the sulfide N3S2 selectively catalyzes the oxygen reduction reaction (ORR) in the presence of a high concentration of formate. Paired with the known formate oxidation catalyst Pd/C, the selective ORR catalyst Ni3S2 enables the construction of a membrane-free formate fuel cell that operates at neutral pH and outperforms the Pt–Pd device.
Co-reporter:Seokjoon Oh; James R. Gallagher; Jeffrey T. Miller
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:1820-1823
Publication Date(Web):January 23, 2016
DOI:10.1021/jacs.5b13080
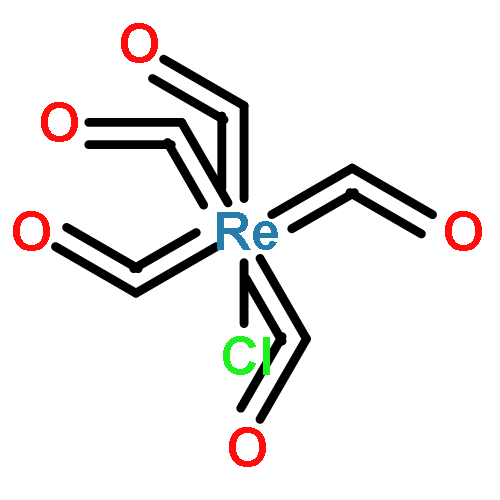
Condensation of fac-Re(5,6-diamino-1,10-phenanthroline)(CO)3Cl to o-quinone edge defects on graphitic carbon surfaces generates graphite-conjugated rhenium (GCC-Re) catalysts that are highly active for CO2 reduction to CO in acetonitrile electrolyte. X-ray photoelectron and X-ray absorption spectroscopies establish the formation of surface-bound Re centers with well-defined coordination environments. GCC-Re species on glassy carbon surfaces display catalytic currents greater than 50 mA cm−2 with 96 ± 3% Faradaic efficiency for CO production. Normalized for the number of Re active sites, GCC-Re catalysts exhibit higher turnover frequencies than that of a soluble molecular analogue, fac-Re(1,10-phenanthroline)(CO)3Cl, and turnover numbers greater than 12,000. In contrast to the molecular analogue, GCC-Re surfaces display a Tafel slope of 150 mV/decade, indicative of a catalytic mechanism involving rate-limiting one-electron transfer. This work establishes graphite-conjugation as a powerful strategy for generating well-defined, tunable, heterogeneous electrocatalysts on ubiquitous graphitic carbon surfaces.
Co-reporter:Megan N. Jackson
Journal of the American Chemical Society 2016 Volume 138(Issue 9) pp:3228-3234
Publication Date(Web):February 10, 2016
DOI:10.1021/jacs.6b00167
The effect of the proton donor on the kinetics of interfacial concerted proton–electron transfer (CPET) to polycrystalline Au was probed indirectly by studying the rate of hydrogen evolution from trialkylammonium donors with different steric profiles, but the same pKa. Detailed kinetic studies point to a mechanism for HER catalysis that involves rate-limiting CPET from the proton donor to the electrode surface, allowing this catalytic reaction to serve as a proxy for the rate of interfacial CPET. In acetonitrile electrolyte, triethylammonium (TEAH+) displays up to 20-fold faster CPET kinetics than diisopropylethylammonium (DIPEAH+) at all measured potentials. In aqueous electrolyte, this steric constraint is largely lifted, suggesting a key role for water in mediating interfacial CPET. In acetonitrile, TEAH+ also displays a much larger transfer coefficient (β = 0.7) than DIPEAH+ (β = 0.4), and TEAH+ displays a potential-dependent H/D kinetic isotope effect that is not observed for DIPEAH+. These results demonstrate that proton donor structure strongly impacts the free energy landscape for CPET to extended solid surfaces and highlight the crucial role of the proton donor in the kinetics of electrocatalytic energy conversion reactions.
Co-reporter:Anna Wuttig, Can Liu, Qiling Peng, Momo Yaguchi, Christopher H. Hendon, Kenta Motobayashi, Shen Ye, Masatoshi Osawa, and Yogesh Surendranath
ACS Central Science 2016 Volume 2(Issue 8) pp:522
Publication Date(Web):August 8, 2016
DOI:10.1021/acscentsci.6b00155
Rational design of selective CO2-to-fuels electrocatalysts requires direct knowledge of the electrode surface structure during turnover. Metallic Cu is the most versatile CO2-to-fuels catalyst, capable of generating a wide array of value-added products, including methane, ethylene, and ethanol. All of these products are postulated to form via a common surface-bound CO intermediate. Therefore, the kinetics and thermodynamics of CO adsorption to Cu play a central role in determining fuel-formation selectivity and efficiency, highlighting the need for direct observation of CO surface binding equilibria under catalytic conditions. Here, we synthesize nanostructured Cu films adhered to IR-transparent Si prisms, and we find that these Cu surfaces enhance IR absorption of bound molecules. Using these films as electrodes, we examine Cu-catalyzed CO2 reduction in situ via IR spectroelectrochemistry. We observe that Cu surfaces bind electrogenerated CO, derived from CO2, beginning at −0.60 V vs RHE with increasing surface population at more negative potentials. Adsorbed CO is in dynamic equilibrium with dissolved 13CO and exchanges rapidly under catalytic conditions. The CO adsorption profiles are pH independent, but adsorbed CO species undergo a reversible transformation on the surface in modestly alkaline electrolytes. These studies establish the potential, concentration, and pH dependencies of the CO surface population on Cu, which serve to maintain a pool of this vital intermediate primed for further reduction to higher order fuel products.
Co-reporter:Anna Wuttig;Momo Yaguchi;Masatoshi Osawa;Kenta Motobayashi
PNAS 2016 Volume 113 (Issue 32 ) pp:E4585-E4593
Publication Date(Web):2016-08-09
DOI:10.1073/pnas.1602984113
CO2 reduction in aqueous electrolytes suffers efficiency losses because of the simultaneous reduction of water to H2. We combine in situ surface-enhanced IR absorption spectroscopy (SEIRAS) and electrochemical kinetic studies to probe the
mechanistic basis for kinetic bifurcation between H2 and CO production on polycrystalline Au electrodes. Under the conditions of CO2 reduction catalysis, electrogenerated CO species are irreversibly bound to Au in a bridging mode at a surface coverage of
∼0.2 and act as kinetically inert spectators. Electrokinetic data are consistent with a mechanism of CO production involving
rate-limiting, single-electron transfer to CO2 with concomitant adsorption to surface active sites followed by rapid one-electron, two-proton transfer and CO liberation
from the surface. In contrast, the data suggest an H2 evolution mechanism involving rate-limiting, single-electron transfer coupled with proton transfer from bicarbonate, hydronium,
and/or carbonic acid to form adsorbed H species followed by rapid one-electron, one-proton, or H recombination reactions.
The disparate proton coupling requirements for CO and H2 production establish a mechanistic basis for reaction selectivity in electrocatalytic fuel formation, and the high population
of spectator CO species highlights the complex heterogeneity of electrode surfaces under conditions of fuel-forming electrocatalysis.
Co-reporter:Joseph M. Falkowski; Nolan M. Concannon; Bing Yan
Journal of the American Chemical Society 2015 Volume 137(Issue 25) pp:7978-7981
Publication Date(Web):June 23, 2015
DOI:10.1021/jacs.5b03426
Electrodeposited thin films and nanoparticles of Ni3S2 are highly active, poison- and corrosion-resistant catalysts for oxygen reduction to water at neutral pH. In pH 7 phosphate buffer, Ni3S2 displays catalytic onset at 0.8 V versus the reversible hydrogen electrode, a Tafel slope of 109 mV decade–1, and high faradaic efficiency for four-electron reduction of O2 to water. Under these conditions, the activity and stability of Ni3S2 exceeds that of polycrystalline platinum and manganese, nickel, and cobalt oxides, illustrating the catalytic potential of pairing labile first-row transition metal active sites with a more covalent sulfide host lattice.
Co-reporter:Tomohiro Fukushima; Walter Drisdell; Junko Yano
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:10926-10929
Publication Date(Web):August 21, 2015
DOI:10.1021/jacs.5b06737
Condensation of ortho-phenylenediamine derivatives with ortho-quinone moieties at edge planes of graphitic carbon generates graphite-conjugated pyrazines (GCPs) that are active for oxygen reduction electrocatalysis in alkaline aqueous electrolyte. Catalytic rates of oxygen reduction are positively correlated with the electrophilicity of the active site pyrazine unit and can be tuned by over 70-fold by appending electron-withdrawing substituents to the phenylenediamine precursors. Discrete molecular analogs containing pyrazine moieties display no activity above background under identical conditions. This simple bottom up method for constructing molecularly well-defined active sites on ubiquitous graphitic solids enables the rational design of tunable heterogeneous catalysts.
Co-reporter:Anthony Shoji Hall; Youngmin Yoon; Anna Wuttig
Journal of the American Chemical Society 2015 Volume 137(Issue 47) pp:14834-14837
Publication Date(Web):November 4, 2015
DOI:10.1021/jacs.5b08259
Gold inverse opal (Au-IO) thin films are active for CO2 reduction to CO with high efficiency at modest overpotentials and high selectivity relative to hydrogen evolution. The specific activity for hydrogen evolution diminishes by 10-fold with increasing porous film thickness, while CO evolution activity is largely unchanged. We demonstrate that the origin of hydrogen suppression in Au-IO films stems from the generation of diffusional gradients within the pores of the mesostructured electrode rather than changes in surface faceting or Au grain size. For electrodes with optimal mesoporosity, 99% selectivity for CO evolution can be obtained at overpotentials as low as 0.4 V. These results establish electrode mesostructuring as a complementary method for tuning selectivity in CO2-to-fuels catalysis.
Co-reporter:Anna Wuttig and Yogesh Surendranath
ACS Catalysis 2015 Volume 5(Issue 7) pp:4479
Publication Date(Web):June 24, 2015
DOI:10.1021/acscatal.5b00808
Herein, we show that group 11 CO2 reduction catalysts are rapidly poisoned by progressive deposition of trace metal ion impurities present in high purity electrolytes. Metal impurity deposition was characterized by XPS and in situ stripping voltammetry and is coincident with loss of catalytic activity and selectivity for CO2 reduction, favoring hydrogen evolution on poisoned surfaces. Metal deposition can be suppressed by complexing trace metal ion impurities with ethylenediaminetetraacetic acid or solid-supported iminodiacetate resins. Metal ion complexation allows for reproducible, sustained catalytic activity and selectivity for CO2 reduction on Au, Ag, and Cu electrodes. Together, this study establishes the principal mode by which group 11 CO2 reduction catalysts are poisoned and lays out a general approach for extending the lifetime of electrocatalysts subject to impurity metal deposition.Keywords: CO2 reduction; electrocatalysis; energy storage; metal impurities; surface poisoning
Co-reporter:Joseph M. Falkowski and Yogesh Surendranath
ACS Catalysis 2015 Volume 5(Issue 6) pp:3411
Publication Date(Web):April 21, 2015
DOI:10.1021/acscatal.5b00449
The systematic development of improved electrocatalysts requires strategies for preparing candidate materials as well-defined thin-film electrodes that are amenable to straightforward characterization of reaction mechanism and catalyst specific activity. While numerous thin film preparation methods are established for transition metals and metal alloys, few strategies exist for transition metal chalcogenides, despite growing recognition of their role as potent electrocatalysts. Herein we show that electrochemical atomic layer deposition (E-ALD) is a powerful tool for accessing well-defined metal chalcogenide electrocatalysts, by synthesizing, for the first time, crystalline conformal films of Co9S8, a promising earth-abundant oxygen reduction catalyst, with tunable nanoscale thickness. The as-prepared nanofilms display relatively high activity for the oxygen reduction reaction and provide a robust platform for detailed mechanistic investigations. Initial mechanistic studies reveal that oxygen reduction on Co9S8 nanofilms proceeds via rate-limiting one-electron transfer to O2 with a specific activity of 20.6 μA cm–2 at 600 mV vs RHE. This study opens the door to the systematic application of E-ALD to investigate chalcogenide electrocatalysts across the transition series.Keywords: E-ALD; electrocatalysis; mechanistic studies; metal chalcogenide; oxygen reduction; thin films
.jpg)




![Dibenzo[a,c]phenazine-11-carboxylic acid](/data/chemimg/3753600/134859-17-9.png)
![Dibenzo[a,c]phenazine-11-carboxylic acid](/data/chemimg/3753600/134859-17-9_b.png)