Co-reporter:Yuting Cui, Jianhui Liu
Tetrahedron Letters 2017 Volume 58, Issue 16(Issue 16) pp:
Publication Date(Web):19 April 2017
DOI:10.1016/j.tetlet.2017.03.018
•A dimethylacridines and dicyanobenzene based molecule has been designed and synthesized as a novel TADF emitter.•The optical, electrochemical and thermal properties were investigated.•The molecule shows a singlet-triplet energy gap as small as 0.04 eV.•The doped OLEDs exhibited a high EQE of 16.5%.A novel compound was designed and synthesized by connecting a dicyanobenzene acceptor and two 9,9-dimethyl-9,10-dihydroacridine donors to the 1,3,5-position of a phenyl ring by meta-position connection. This compound, which is a novel emitter for OLED devices, exhibits preferable heat stability. Moreover, the energy gap between its singlet and triplet states is as small as 0.04 eV, resulting in this molecule possesses thermally activated delayed fluorescence. Therefore, the corresponding device showed efficient electroluminescent performances. The maximum external quantum efficiency, maximum current efficiency, maximum power efficiency and maximum luminance were 16.5%, 40.8 cd A−1, 45.8 lm W−1 and 5120 cd m−2, respectively. In addition, the CIEx,y only changed from (0.22, 0.38) to (0.22, 0.39) over the entire operating voltage range, which confirms that the device possesses highly stable chromaticity with respect to the current density. Based on these experimental results, meta-connected type structures may provide a new approach for developing high-performance TADF emitters for OLED applications.Download high-res image (54KB)Download full-size image
Co-reporter:Xiangbin Cao, Jianhui Liu, Pan Hong, Guanglan Li, Ce Hao
Journal of Photochemistry and Photobiology A: Chemistry 2017 Volume 346(Volume 346) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jphotochem.2017.05.035
•Two novel styrylcyanine dyes were developed based on ICT.•The probes exhibited red-emission and large Stokes shift.•The probes displayed high sensitivity to viscosity.•The fluorescence intensity was affected by intramolecular rotation.Based on the mechanism of intramolecular charge transfer (ICT), two new styrylcyanine dyes, DPA-1 and DPA-2, composed of an electron-rich N-phenylaniline and a cationic benzothiazene connected with ethylene(s) bridge, were designed and synthesized. These two dyes exhibited red-emission (653/607 nm) and simultaneously impressive large Stokes shift (111/92 nm) due to intramolecular charge transfer effect, twisted geometry and extended conjugation system. When their solution viscosity increased from 1.01 cP to 234 cP in the water-glycerol system, the fluorescence intensity of the synthetic dyes was enhanced by 81-fold and 64-fold, respectively. Additionally, a favorable linear relationship between the fluorescence intensity and the environmental viscosity was observed within the above viscosity range for the obtained samples (R2 > 0.99), which led to the establishment of a method for the quantitative determination of the solution viscosity. Such dyes with improved photophysical properties, including emission wavelength and Stokes shift, could be used as promising candidates for intracellular viscosity detection. Moreover, the mechanism of fluorescence emission of the resultant products toward viscosity was further investigated. Of the two probes studies, DPA-1 is better than DPA-2 in terms of emission wavelength, Stokes shift and the sensitivity of the fluorescence intensity to viscosity.Download high-res image (121KB)Download full-size image
Co-reporter:Jianhui Liu, Nanyan Fu, Changyun Wei, Zhucao Song
Tetrahedron Letters 2016 Volume 57(Issue 23) pp:2530-2532
Publication Date(Web):8 June 2016
DOI:10.1016/j.tetlet.2016.04.105
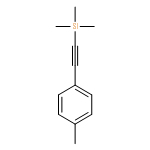
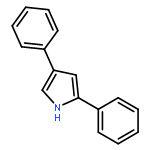
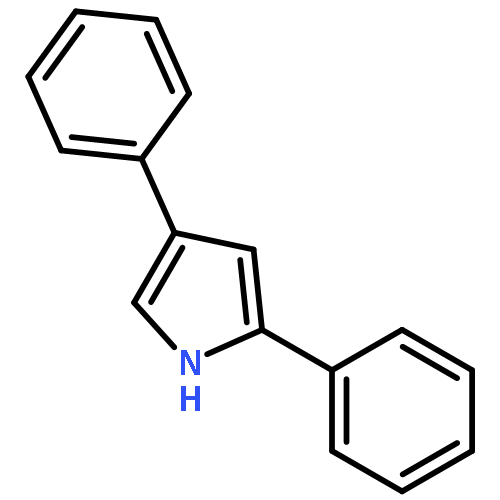
•A new strategy for the synthesis of 2,4-disubstituted pyrroles.•A stepwise iododesilylation and palladium-catalysed Suzuki coupling reaction.•Regioselective iododesilylation can be achieved under simple and mild conditions.In this study, we report a new strategy for the synthesis of 2,4-disubstituted pyrroles using a stepwise iododesilylation and palladium-catalysed Suzuki coupling reaction of 2,4-bis(trimethylsilyl)-1-t-Boc-1H-pyrrole (2). The regioselective iodo-desilylation followed by the Suzuki coupling reaction with concomitant Boc deprotection are critical aspects of the new synthesis method.
Co-reporter:Zhen Cao, Kang Du, Jianhui Liu, Wenjun Tang
Tetrahedron 2016 Volume 72(Issue 14) pp:1782-1786
Publication Date(Web):7 April 2016
DOI:10.1016/j.tet.2016.02.043
Enantioselective syntheses of triptoquinone H and its C-5 epimer were accomplished for the first time in seven linear steps and 33% overall yield from aldehyde 6 and Wittig salt 5 via a key palladium-catalyzed asymmetric dearomative cyclization. The P-chiral biaryl monophosphorus ligand AntPhos was proved to be efficient for the asymmetric dearomative cyclization to provide the tricyclic compound 8 bearing a chiral all-carbon quaternary center in 85% ee and 87% yield.
Co-reporter:Lei Wang, Lele Duan, Beverly Stewart, Maoping Pu, Jianhui Liu, Timofei Privalov, and Licheng Sun
Journal of the American Chemical Society 2012 Volume 134(Issue 45) pp:18868-18880
Publication Date(Web):October 12, 2012
DOI:10.1021/ja309805m
Using the combinations of imidazole and dimethyl sulfoxide (DMSO) as axial ligands and 2,2′-bipyridine-6,6′-dicarboxylate (bda) as the equatorial ligand, we have synthesized six novel ruthenium complexes with noticeably different activity as water oxidation catalysts (WOCs). In four Cs symmetric RuII(κ3-bda)(DMSO)L2 complexes L = imidazole (1), N-methylimidazole (2), 5-methylimidazole (3), and 5-bromo-N-methylimidazole (4). Additionally, in two C2v symmetric RuII(κ4-bda)L2 complexes L = 5-nitroimidazole (5) and 5-bromo-N-methylimidazole (6), that is, fully equivalent axial imidazoles. A detailed characterization of all complexes and the mechanistic investigation of the catalytic water oxidation have been carried out with a number of experimental techniques, that is, kinetics, electrochemistry and high resolution mass spectrometry (HR-MS), and density functional theory (DFT) calculations. We have observed the in situ formation of a RuII-complex with the accessible seventh coordination position. The measured catalytic activities and kinetics of complex 1–6 revealed details about an important structure–activity relation: the connection between the nature of axial ligands in the combination and either the increase or decrease of the catalytic activity. In particular, an axial DMSO group substantially increases the turnover frequency of WOCs reported in the article, with the ruthenium-complex having one axial 5-bromo-N-methyl-imidazole and one axial DMSO (4), we have obtained a high initial turnover frequency of ∼180 s–1. DFT modeling of the binuclear reaction pathway of the O–O bond formation in catalytic water oxidation further corroborated the concept of the mechanistic significance of the axial ligands and rationalized the experimentally observed difference in the activity of complexes with imidazole/DMSO and imidazole/imidazole combinations of axial ligands.
Co-reporter:Jianhui Liu and Weina Jiang
Dalton Transactions 2012 vol. 41(Issue 32) pp:9700-9707
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2DT30468F
Coordination of the pyridyl-attached diiron azadithiolate hexacarbonyl complexes (2 and 3) through the pyridyl nitrogen to the Re on 10-phenanthroline rhenium (5a) and 2,9-diphenyl-1,10-phenanthroline rhenium (5b) forms novel [Re–Fe] complexes 7a, 7b and 8 respectively. Under visible light illumination using triethylamine as a sacrificial electron donor and [Re–Fe] type complexes (7a, 7b or 8) as catalysts, remarkably increased efficiency was observed for photoinduced hydrogen production with a turnover number reaching 11.8 from complex 7a and 8.75 from 7b. To the best of our knowledge, these are the best values compared to other [Re–Fe] photocatalysts reported so far. In contrast to the parent molecules, the turnover number by the intermolecular combination of complexes 6a and 2 showed a value of 5.23, and that from 6b and 2 is 3.8, while no H2 was detected from 8a and 3 under the same experimental conditions. Obviously, the intramolecular combination of rhenium(I) and [2Fe2S] as a catalyst is promising for efficient H2 evolution, and it is better than the intermolecular multi-component system.
Co-reporter:Weina Jiang, Jianhui Liu, Cheng Li
Inorganic Chemistry Communications 2012 Volume 16() pp:81-85
Publication Date(Web):February 2012
DOI:10.1016/j.inoche.2011.11.038
In order to conduct photoactive catalysts for hydrogen production, a novel trimetallic [Re-Fe] complex 1, consisting of the phenanthroline rhenium photosensitizer and the [2Fe2S] complex (connected by the axial coordination of a pyridyl group), was prepared and spectroscopically characterized. The apparent fluorescence quenching of the complex 1 was observed in comparison with the reference complex 1b, suggesting the possibility for an electron transfer from the excited state of the rhenium moiety to the [2Fe2S] moiety. Visible light-driven H2 generation was achieved by using triethylamine (sacrificial electron donor) in the presence of the complex 1 (catalyst) in CH3CN/H2O, with a turnover number reaching to 1.5. This is by far as we know the highest for the [Re-Fe] photocatalysts. In contrast to the molecular device, the multicomponent catalyst of complexes 1b and 1c, no H2 was detected by GC analysis in the same experimental condition. The plausible mechanism for the photochemical H2-evolution with this molecular device is discussed.A novel trimetallic [Re–Fe] complex 1 was prepared and spectroscopically characterized. Visible light-driven H2 generation was achieved by using triethylamine in the presence of complex 1 in CH3CN/H2O, with a turnover number reaching to 1.5, so far as we know the highest for the [Re–Fe] photocatalysts.Highlights► A novel [Re–Fe] photocatalyst and its reference complexes were synthesized and characterized. ► Fluorescence quenching suggested a possibility for an electron transfer from the rhenium moiety to the [2Fe2S] moiety. ► The [Re–Fe] photocatalyst can catalyze the hydrogen evolution with the TEA as the electron donor. ► Its turnover number reach to 1.5, so far the highest for the [Re–Fe] photocatalysts.
Co-reporter:Yu Shi;Xiaoyu Li;Wenfeng Jiang;Licheng Sun
Applied Organometallic Chemistry 2011 Volume 25( Issue 7) pp:514-520
Publication Date(Web):
DOI:10.1002/aoc.1795
Abstract
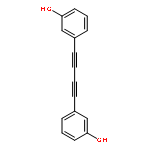
A PdCl2-catalyzed direct alkynylation of arylboronic acids to give diarylacetylenes is described. The optimal conditions using PdCl2 as catalyst, MeOHPhMeH2O as solvent and K2CO3 as base effectively suppressed the formation of homo-coupling product and afforded moderate to good yield of the desired unsymmetrical coupling product. This reaction represents a Suzuki-type sp2(CB)–sp(CX) cross-coupling. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Yu Shi, Xiaoyu Li, Jianhui Liu, Wenfeng Jiang, Licheng Sun
Tetrahedron Letters 2010 Volume 51(Issue 28) pp:3626-3628
Publication Date(Web):14 July 2010
DOI:10.1016/j.tetlet.2010.05.011
A new Suzuki-type cross-coupling reaction between 1-iodo-2-arylalkynes and arylboronic acids to afford a wide variety of functionalized diarylacetylenes in a mild reaction condition was developed. The reaction was catalyzed by a small amount of a structurally simple, commercially available, and stable PdCl2. This unique sp–sp2 carbon–carbon bond formation provides a new protocol for the synthesis of diarylacetylenes, which is a new addition to the Suzuki cross-coupling reaction.
Co-reporter:Shi Jiang, Jianhui Liu, Yu Shi, Zhen Wang, Björn Åkermark and Licheng Sun
Dalton Transactions 2007 (Issue 8) pp:896-902
Publication Date(Web):24 Jan 2007
DOI:10.1039/B615037C
Three biomimetic 2Fe2S complexes [{(µ-SCH2)2NCH2(2-C4H3O)}](Fe2(CO)6) (6a), [{(µ-SCH2)2 NCH2(2-C4H3S)}](Fe2(CO)6) (6b) and [{(µ-SCH2)2NCH2(5-Br-2-C4H2S)}Fe2(CO)6] (6c) were prepared as models for the active site of Fe-only hydrogenase by the convergent process from [(µ-S2)Fe2(CO)6] and N,N-bis(hydromethyl)-2-furan and thiophene. The structures of these complexes were identified spectroscopically and crystallographically. The electrochemical behavior of the complexes 6a and 6c was unique as they showed catalytic proton reduction with a low reduction potential at −1.13 and −1.09 V vs Fc/Fc+, respectively, in the presence of HClO4.
Co-reporter:Zhen Wang, Jianhui Liu, Chengjiang He, Shi Jiang, Björn Åkermark, Licheng Sun
Inorganica Chimica Acta 2007 Volume 360(Issue 7) pp:2411-2419
Publication Date(Web):2 May 2007
DOI:10.1016/j.ica.2006.12.008
Three novel complexes (μ-adt)[Fe2(CO)5PTA] (2-PTA), (μ-adt)[Fe2(CO)4PTA2] (2-PTA2) and (μ-adt)[Fe2(CO)5DAPTA] (2-DAPTA), where adt is SCH2N(CH2CH2CH3)CH2S, PTA stands for 1,3,5-triaza-7-phosphaadamantane and DAPTA is 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane, were prepared as the models of the iron hydrogenase active site through controlled CO displacement of (μ-adt)[Fe2(CO)6] with PTA and DAPTA. The coordination configurations of 2-PTA and 2-PTA2 were characterized by X-ray crystallography. The disubstituted diiron complex 2-PTA2 features a basal/apical coordination mode, instead of the typical transoid basal/basal configuration. Protonation of three complexes only occurred at the bridging-N atom, rather than at the tertiary nitrogen atom on the PTA or DAPTA ligands. Electrochemical properties of the complexes were studied in acetonitrile or a mixture of acetonitrile and water in the presence of acetic acid, by cyclic voltammetry. The current sensitivity of the reduced species to acid concentration in the presence of H2O is greater than in the pure CH3CN solution.Three diiron azadithiolates with hydrophilic phosphatriazaadamantane ligand were prepared. Their structures are characterized by 1H NMR, IR spectra and two of them further are determined by X-ray analysis. Protonation occurred only at the bridging-N atom, rather than the N atom on the ligands. Their electrochemical properties are studied in acetonitrile or a mixture of acetonitrile and water.
Co-reporter:Ming Xia;Yan Gao;Björn Åkermark;Licheng Sun
Helvetica Chimica Acta 2007 Volume 90(Issue 3) pp:553-561
Publication Date(Web):19 MAR 2007
DOI:10.1002/hlca.200790056
As bio-inspired chemical model of the oxygen-evolving complex (OEC) in photosystem II, a new tyrosine-modified corrole ligand 3 and its high-valent copper and manganese complexes 3a and 3b were synthesized and characterized. The copper complexes 1a and 2a of corrole 1 and 2 were also prepared for comparison. The emission property indicates that the emission of ligands 2 and 3 is located at 670 nm, but no emission is observed for their metal complexes due to its suppression by the metal center. The electrochemical study shows that 3a might dimerize at the first two reversible oxidations, a behavior which was not observed in the case of 1a and 2a. The corrolato manganese(IV) complex 3b shows one reversible reduction and one quasireversible oxidation at −0.17 and 0.77 V vs. Ag/Ag+, respectively.
Co-reporter:Weiming Gao, Jianhui Liu, Chengbing Ma, Linhong Weng, Kun Jin, Changneng Chen, Björn Åkermark, Licheng Sun
Inorganica Chimica Acta 2006 Volume 359(Issue 4) pp:1071-1080
Publication Date(Web):1 March 2006
DOI:10.1016/j.ica.2005.10.053
The preparation and characterization of two novel amino-incorporated sulfur-bridged dinuclear iron (I) complexes of the type [NR(μ-SCH2)2]Fe2(CO)6, one being amino protected [N(CH2CH2NHTs)(μ-SCH2)2]Fe2(CO)6 (8) and the other [(μ- SCH2)2Fe2(CO)6NCH2CH2N (μ-SCH2)2]Fe2(CO)6 (9) are described. These two complexes are readily prepared in a SN2 manner between double lithium anion and bis(chloromethyl) amine derivatives. The structures of 8 and 9 were characterized by IR, 1H, 13C NMR, MS and HRMS spectra and further determined by X-ray analyses. Protonation of complex 8 gave the mono N-protonated product, while for 9 the protonation occurred in both of the N atoms. The redox properties were evaluated by cyclic voltammograms. It was shown that these two complexes can catalyze electrochemical reduction of proton to molecular hydrogen.This paper describes the preparation of two novel amino-incorporated FeS complexes. Their structures are characterized by 1H, 13C NMR, MS, IR, and HRMS spectra and further determined by X-ray analysis. Their protonations and electrochemical properties showed that they are better catalyst for the reduction of proton to hydrogen than aromatic nitrogen atom bridged complexes.
Co-reporter:Jianhui Liu and Weina Jiang
Dalton Transactions 2012 - vol. 41(Issue 32) pp:NaN9707-9707
Publication Date(Web):2012/06/13
DOI:10.1039/C2DT30468F
Coordination of the pyridyl-attached diiron azadithiolate hexacarbonyl complexes (2 and 3) through the pyridyl nitrogen to the Re on 10-phenanthroline rhenium (5a) and 2,9-diphenyl-1,10-phenanthroline rhenium (5b) forms novel [Re–Fe] complexes 7a, 7b and 8 respectively. Under visible light illumination using triethylamine as a sacrificial electron donor and [Re–Fe] type complexes (7a, 7b or 8) as catalysts, remarkably increased efficiency was observed for photoinduced hydrogen production with a turnover number reaching 11.8 from complex 7a and 8.75 from 7b. To the best of our knowledge, these are the best values compared to other [Re–Fe] photocatalysts reported so far. In contrast to the parent molecules, the turnover number by the intermolecular combination of complexes 6a and 2 showed a value of 5.23, and that from 6b and 2 is 3.8, while no H2 was detected from 8a and 3 under the same experimental conditions. Obviously, the intramolecular combination of rhenium(I) and [2Fe2S] as a catalyst is promising for efficient H2 evolution, and it is better than the intermolecular multi-component system.
Co-reporter:Shi Jiang, Jianhui Liu, Yu Shi, Zhen Wang, Björn Åkermark and Licheng Sun
Dalton Transactions 2007(Issue 8) pp:NaN902-902
Publication Date(Web):2007/01/24
DOI:10.1039/B615037C
Three biomimetic 2Fe2S complexes [{(µ-SCH2)2NCH2(2-C4H3O)}](Fe2(CO)6) (6a), [{(µ-SCH2)2 NCH2(2-C4H3S)}](Fe2(CO)6) (6b) and [{(µ-SCH2)2NCH2(5-Br-2-C4H2S)}Fe2(CO)6] (6c) were prepared as models for the active site of Fe-only hydrogenase by the convergent process from [(µ-S2)Fe2(CO)6] and N,N-bis(hydromethyl)-2-furan and thiophene. The structures of these complexes were identified spectroscopically and crystallographically. The electrochemical behavior of the complexes 6a and 6c was unique as they showed catalytic proton reduction with a low reduction potential at −1.13 and −1.09 V vs Fc/Fc+, respectively, in the presence of HClO4.

![Ruthenate(2-), bis(2,2'-bipyridine-κN1,κN1')[[([2,2'-bipyridine]-4,4'-diyl-κN1,κN1')bis[phosphonato]](4-)]-, hydrogen bromide (1:4:2), (OC-6-22)-](/data/chemimg/133900/239472-59-4.png)
![Ruthenate(2-), bis(2,2'-bipyridine-κN1,κN1')[[([2,2'-bipyridine]-4,4'-diyl-κN1,κN1')bis[phosphonato]](4-)]-, hydrogen bromide (1:4:2), (OC-6-22)-](/data/chemimg/133900/239472-59-4_b.png)











![(+)-1,2-BIS[(2S,5S)-2,5-DIMETHYLPHOSPHOLANO]BENZENE](http://img.cochemist.com/ccimg/136800/136735-95-0.png)
![(+)-1,2-BIS[(2S,5S)-2,5-DIMETHYLPHOSPHOLANO]BENZENE](http://img.cochemist.com/ccimg/136800/136735-95-0_b.png)






![Ethanone, 1-(4-methoxyphenyl)-2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/99300/99236-19-8.png)
![Ethanone, 1-(4-methoxyphenyl)-2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/99300/99236-19-8_b.png)




![[(E)-3,3,3-TRIFLUORO-1-PHENYLPROP-1-EN-2-YL]BENZENE](http://img.cochemist.com/ccimg/76300/76280-44-9.png)
![[(E)-3,3,3-TRIFLUORO-1-PHENYLPROP-1-EN-2-YL]BENZENE](http://img.cochemist.com/ccimg/76300/76280-44-9_b.png)




![1-FLUORO-4-[4-(4-FLUOROPHENYL)BUTA-1,3-DIYNYL]BENZENE](http://img.cochemist.com/ccimg/55700/55606-94-5.png)
![1-FLUORO-4-[4-(4-FLUOROPHENYL)BUTA-1,3-DIYNYL]BENZENE](http://img.cochemist.com/ccimg/55700/55606-94-5_b.png)



















![Di-μ-chlorobis[(1,2,3-η)-1-phenyl-2-propenyl]dipalladium(Ⅱ)](http://img.cochemist.com/ccimg/12200/12131-44-1.png)
![Di-μ-chlorobis[(1,2,3-η)-1-phenyl-2-propenyl]dipalladium(Ⅱ)](http://img.cochemist.com/ccimg/12200/12131-44-1_b.png)