Co-reporter:Jiao Song, Peng Peng, Jun Chang, Ming-Ming Liu, Jian-Ming Yu, Lu Zhou, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2174-2178
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.064
Novel Ilomastat analogs with substituted benzamide groups, instead of hydroxamic acid groups, were designed, synthesized and evaluated against MMP-2 and MMP-9. Among these analogs, the most potent compound 10a exhibited potent inhibitory activity against MMP-2 with IC50 value of 0.19 nM, which is 5 times more potent than that of Ilomastat (IC50 = 0.94 nM). Importantly, 10a exhibited more than 8300 fold selectivity for MMP-2 versus MMP-9 (IC50 = 1.58 μM). Molecular docking studies showed that 10a bond to the catalytic active pocket of MMP-2 by a non-zinc-chelating mechanism which was different from that of Ilomastat. Furthermore, the invasion assay showed that 10a was effective in reducing HEY cells invasion at 84.6% in 50 μM concentration. For 10a, the pharmacokinetic properties had been improved and especially the more desirable t1/2z was achieved compared with these of the lead compound Ilomastat.
Co-reporter:Wu-Hui Song, Ming-Ming Liu, Dong-Wei Zhong, Ye-lin Zhu, Mike Bosscher, Lu Zhou, De-Yong Ye, Zheng-Hong Yuan
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 16) pp:4528-4531
Publication Date(Web):15 August 2013
DOI:10.1016/j.bmcl.2013.06.045
A series of diketo tetrazoles and diketo triazoles were designed and synthesized as bioisosteres of α,γ-diketo acid, the active site inhibitor of HCV (Hepatitis C virus) polymerase NS5B. Among the synthesized compounds, 4-(4-fluorobenzyloxy)phenyl diketo triazole (30) exhibited anti-HCV activity with an EC50 value of 3.9 μM and an SI value more than 128. The reduction of viral protein and mRNA levels were also validated, supporting the anti-HCV activity of compound 30. These results provide convincing evidence that the diketo tetrazoles and diketo triazoles can be developed as bioisosteres of α,γ-diketo acid to exhibit potent inhibitory activity against HCV.A series of diketo tetrazoles and diketo triazoles were designed and synthesized as bioisosteres of α,γ-diketo acid. Among them, compound 30 exhibited potent anti-HCV activity with an EC50 value of 3.9 μM and an SI value more than 128. The reduction of viral protein and mRNA levels were also validated.
Co-reporter:Xing Gao;Haojun Gong;Peng Men;Deyong Ye
Chinese Journal of Chemistry 2013 Volume 31( Issue 9) pp:1164-1170
Publication Date(Web):
DOI:10.1002/cjoc.201300079
Abstract
A novel series of eight SMS and sPLA2 dual inhibitors containing indole and α-amino cyanide fragments of different length and substitution position was synthesized and evaluated by three different in vitro assays. Biological evaluation showed that all compounds provided inhibitory effects against SMS (about 50% inhibition at 100 µmol/L) and sPLA2 (14–32 µmol/L). All the compounds had the SMS activity better than the positive control compound D609 in SMS2 homogenate, with compounds 5b and 5e ideal for liver homogenate and SMS2 high expression cell homogenate, respectively.



![Benzofuran, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/904400/904325-87-7.png)
![Benzofuran, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/904400/904325-87-7_b.png)


![Acetamide, 2-chloro-N-[2-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/656300/656227-27-9.png)
![Acetamide, 2-chloro-N-[2-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/656300/656227-27-9_b.png)










![2-chloro-n-[(4-fluorophenyl)methyl]acetamide](http://img.cochemist.com/ccimg/257300/257279-75-7.png)
![2-chloro-n-[(4-fluorophenyl)methyl]acetamide](http://img.cochemist.com/ccimg/257300/257279-75-7_b.png)













![Acetamide,2-chloro-N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99585-88-3.png)
![Acetamide,2-chloro-N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99585-88-3_b.png)











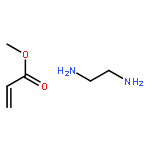
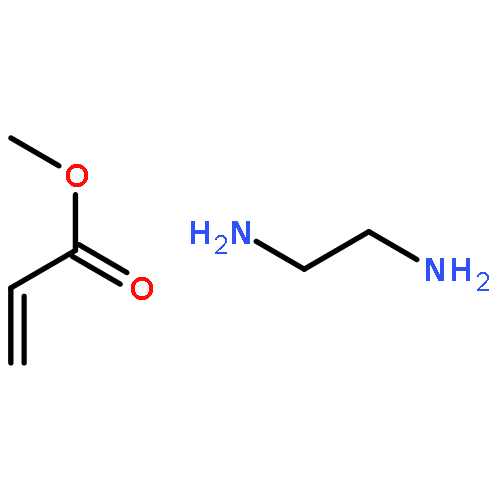


![Acetamide, N-[(4-bromophenyl)methyl]-2-chloro-](http://img.cochemist.com/ccimg/25000/24942-06-1.png)
![Acetamide, N-[(4-bromophenyl)methyl]-2-chloro-](http://img.cochemist.com/ccimg/25000/24942-06-1_b.png)




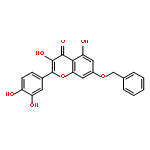
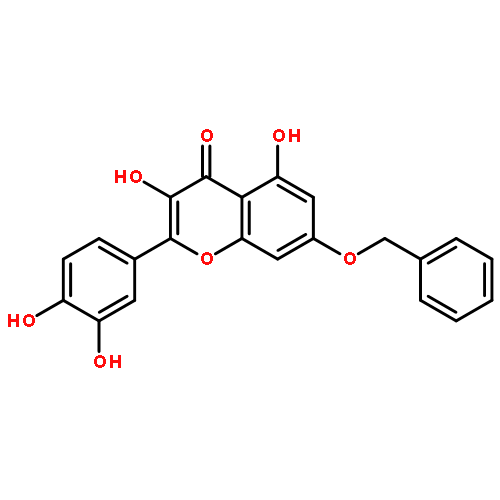








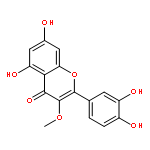

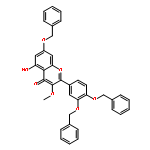
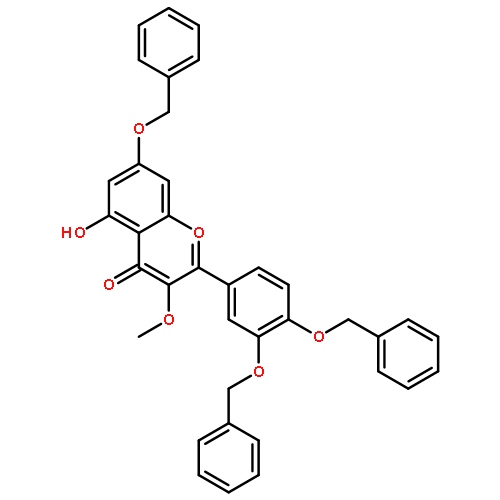
![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8.png)
![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8_b.png)







