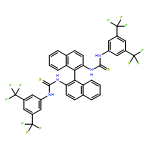
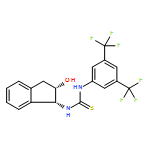
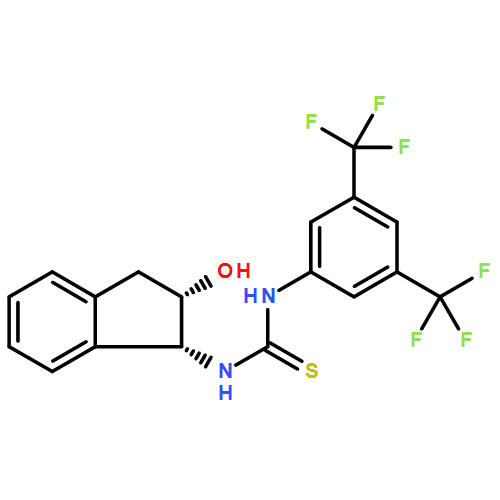
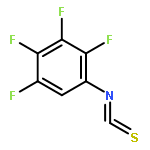
Co-reporter:Elango Kumarasamy, Ramya Raghunathan, Mukund P. Sibi, and J. Sivaguru
Chemical Reviews 2015 Volume 115(Issue 20) pp:11239
Publication Date(Web):September 28, 2015
DOI:10.1021/acs.chemrev.5b00136
Co-reporter:Dr. Saravanakumar Rajendran;Ramya Raghunathan;Dr. Ivan Hevus;Dr. Retheesh Krishnan;Dr. Angel Ugrinov;Dr. Mukund P. Sibi;Dr. Dean C. Webster;Dr. Jayaraman Sivaguru
Angewandte Chemie International Edition 2015 Volume 54( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/anie.201411878
Co-reporter:Dr. Saravanakumar Rajendran;Ramya Raghunathan;Dr. Ivan Hevus;Dr. Retheesh Krishnan;Dr. Angel Ugrinov;Dr. Mukund P. Sibi;Dr. Dean C. Webster;Dr. Jayaraman Sivaguru
Angewandte Chemie International Edition 2015 Volume 54( Issue 4) pp:1159-1163
Publication Date(Web):
DOI:10.1002/anie.201408492
Abstract
Renewable polymeric materials derived from biomass with built-in phototriggers were synthesized and evaluated for degradation under irradiation of UV light. Complete decomposition of the polymeric materials was observed with recovery of the monomer that was used to resynthesize the polymers.
Co-reporter:Nandini Vallavoju and J. Sivaguru
Chemical Society Reviews 2014 vol. 43(Issue 12) pp:4084-4101
Publication Date(Web):07 Apr 2014
DOI:10.1039/C3CS60471C
Using non-bonding interactions to control photochemical reactions requires an understanding of not only thermodynamics and kinetics of ground state and excited state processes but also the intricate interactions that dictate the dynamics within the system of interest. This review is geared towards a conceptual understanding of how one can control the reactivity and selectivity in the excited state by employing confinement and non-covalent interactions. Photochemical reactivity of organic molecules within confined containers and organized assemblies as well as organic templates that interact through H-bonding and/or cation–carbonyl/cation–π interactions is reviewed with an eye towards understanding supramolecular effects and photocatalysis.
Co-reporter:Elango Kumarasamy ; Ramya Raghunathan ; Steffen Jockusch ; Angel Ugrinov ;J. Sivaguru
Journal of the American Chemical Society 2014 Volume 136(Issue 24) pp:8729-8737
Publication Date(Web):June 9, 2014
DOI:10.1021/ja5034638
Atropisomeric maleimides were synthesized and employed for stereospecific [2 + 2] photocycloaddition. Efficient reaction was observed under direct irradiation, triplet-sensitized UV irradiation, and non-metal catalyzed visible-light irradiation, leading to two regioisomeric (exo/endo) photoproducts with complete chemoselectivity (exclusive [2 + 2] photoproduct). High enantioselectivity (ee > 98%) and diastereoselectivity (dr > 99:1%) were observed under the employed reaction conditions and were largely dependent on the substituent on the maleimide double bond but minimally affected by the substituents on the alkenyl tether. On the basis of detailed photophysical studies, the triplet energies of the maleimides were estimated. The triplet lifetimes appeared to be relatively short at room temperature as a result of fast [2 + 2] photocycloaddition. For the visible-light mediated reaction, triplet energy transfer occurred with a rate constant close to the diffusion-limited value. The mechanism was established by generation of singlet oxygen from the excited maleimides. The high selectivity in the photoproduct upon reaction from the triplet excited state was rationalized on the basis of conformational factors as well as the type of diradical intermediate that was preferred during the photoreaction.
Co-reporter:Nini Vallavoju;Sermadurai Selvakumar;Steffen Jockusch;Manoj Thathamkulam Prabhakaran;Mukund P. Sibi
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 13) pp:2763-2768
Publication Date(Web):
DOI:10.1002/adsc.201400677
Co-reporter:Anoklase Jean-Luc Ayitou, Anthony Clay, Elango Kumarasamy, Steffen Jockusch and J. Sivaguru
Photochemical & Photobiological Sciences 2014 vol. 13(Issue 2) pp:141-144
Publication Date(Web):27 Sep 2013
DOI:10.1039/C3PP50278C
Direct irradiation of atropisomeric α-substituted acrylanilides in the presence of alkali metal ions gave high ee values in the 3,4-dihydro-2-quinolin-2-one photoproduct, while in the absence of alkali metal ions, racemic photoproduct was observed. The heavy atom effect leading to enhanced triplet yields alters the reactive pathway leading to the observed enantioselectivity in the photoproduct.
Co-reporter:Akila Iyer, Steffen Jockusch, and J. Sivaguru
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10596-10602
Publication Date(Web):September 8, 2014
DOI:10.1021/jp505678b
Nonbiaryl atropisomeric acrylimides underwent facile [2 + 2] photocycloaddition leading to cross-cyclobutane adducts with very high stereospecificity (enantiomeric excess (ee): 99% and diastereomeric excess (de): 99%). The photoreactions proceeded smoothly in isotropic media for both direct and triplet sensitized irradiations. The reactions were also found to be very efficient in the solid state where the same cross-cyclobutane adduct was observed. Photophysical studies enabled us to understand the excited-state photochemistry of acrylimides. The triplet energy was found to be ∼63 kcal/mol. The reactions proceeded predominantly via a singlet excited state upon direct irradiation with very poor intersystem crossing that was ascertained by quantification of the generated singlet oxygen. The reactions progressed smoothly with triplet sensitization with UV or visible-light irradiations. Laser flash photolysis experiments established the triplet transient of atropisomeric acrylimides with a triplet lifetime at room temperature of ∼40 ns.
Co-reporter:Nini Vallavoju;Dr. Sermadurai Selvakumar;Dr. Steffen Jockusch;Dr. Mukund P. Sibi;Dr. Jayaraman Sivaguru
Angewandte Chemie International Edition 2014 Volume 53( Issue 22) pp:5604-5608
Publication Date(Web):
DOI:10.1002/anie.201310940
Abstract
Can photocatalysis be performed without electron or energy transfer? To address this, organo-photocatalysts that are based on atropisomeric thioureas and display lower excited-state energies than the reactive substrates have been developed. These photocatalysts were found to be efficient in promoting the [2+2] photocycloaddition of 4-alkenyl-substituted coumarins, which led to the corresponding products with high enantioselectivity (77–96 % ee) at low catalyst loading (1–10 mol %). The photocatalytic cycle proceeds by energy sharing via the formation of both static and dynamic complexes (exciplex formation), which is aided by hydrogen bonding.
Co-reporter:Nini Vallavoju;Dr. Sermadurai Selvakumar;Dr. Steffen Jockusch;Dr. Mukund P. Sibi;Dr. Jayaraman Sivaguru
Angewandte Chemie 2014 Volume 126( Issue 22) pp:5710-5714
Publication Date(Web):
DOI:10.1002/ange.201310940
Abstract
Can photocatalysis be performed without electron or energy transfer? To address this, organo-photocatalysts that are based on atropisomeric thioureas and display lower excited-state energies than the reactive substrates have been developed. These photocatalysts were found to be efficient in promoting the [2+2] photocycloaddition of 4-alkenyl-substituted coumarins, which led to the corresponding products with high enantioselectivity (77–96 % ee) at low catalyst loading (1–10 mol %). The photocatalytic cycle proceeds by energy sharing via the formation of both static and dynamic complexes (exciplex formation), which is aided by hydrogen bonding.
Co-reporter:Ramya Raghunathan, Elango Kumarasamy, Akila Iyer, Angel Ugrinov and J. Sivaguru
Chemical Communications 2013 vol. 49(Issue 77) pp:8713-8715
Publication Date(Web):20 Aug 2013
DOI:10.1039/C3CC44281K
Atropisomeric α-oxoamides were synthesized and employed for intramolecular Paternò–Büchi reaction leading to very high enantio- and diastereoselectivity in the bicyclic oxetane photoproduct. A reversal of product selectivity was observed in solution and in the solid-state.
Co-reporter:Elango Kumarasamy and J. Sivaguru
Chemical Communications 2013 vol. 49(Issue 39) pp:4346-4348
Publication Date(Web):20 Nov 2012
DOI:10.1039/C2CC37123E
Atropisomeric 3,4-dihydro-2-pyridones undergo stereospecific [2+2]-photocycloaddition in solution with high stereoselectivity (ee > 98% and de > 96%) in the product. The chiral transfer during phototransformation was rationalized based on the stability/reactivity of the biradical.
Co-reporter:Barry C. Pemberton, Angel Ugrinov, J. Sivaguru
Journal of Photochemistry and Photobiology A: Chemistry 2013 Volume 255() pp:10-15
Publication Date(Web):1 March 2013
DOI:10.1016/j.jphotochem.2013.01.005
6-Methylcoumarin (1) upon mechanical grinding with cucurbit[8]uril (CB[8]) or other additives (e.g. thiourea) showed enhanced photodimerization efficiency with exclusive formation of head-to-head photodimer. Mechanical grinding of 1 with CB[8] resulted in host–guest interaction with red-shift fluorescence in the solid-state. This red shift in fluorescence emission is similar to the red-shifted emission observed for CB[8]–1 host–guest complex in solution at 298 K. Phosphorescence emission profile in the solid state at 77 K for mechanically ground 1 with CB[8] was similar to the phosphorescence profile observed for CB[8]–1 host–guest complex at 77 K. Single crystal X-ray determination of 1 from water reveals that the coumarins are oriented in a head-to-head fashion with the distance between the double bonds being 4.695 Å, a distance that is greater than the required Schmidt distance of 4.2 Å for efficient photodimerization in the crystalline state. Enhancement of photoreactivity of 1 upon mechanical grinding with additives appears to be the result of a combination of factors, viz.: (a) crystalline imperfection/defects that are likely formed during mechanical grinding and/or; (b) formation of a new phase due to mechanical grinding (we were not able to observe any noticeable change in crystallinity, while the formation of an amorphous phase could not be ruled out completely).Graphical abstractHighlights► The photochemistry and photophysics of 6-methylcomarin 1 are influenced by mechanical grinding with cucurbit[8]uril. ► Bathochromic shift was observed in the room temperature fluorescence spectra of 1 upon mechanical grinding with CB[8]. ► A red shift in the phosphorescence spectra of 1 was observed at 77 K upon mechanical grinding with CB[8]. ► 6-Methylcoumarin 1 is not photoreactive in the solid-state but the photodimerization efficiency was enhanced upon mechanical grinding of 1 leading to the formation of head-to-head photoproduct.
Co-reporter:Anoklase J.-L. Ayitou;Dr. Gaku Fukuhara;Elango Kumarasamy;Dr. Yoshihisa Inoue;Dr. J. Sivaguru
Chemistry - A European Journal 2013 Volume 19( Issue 13) pp:4327-4334
Publication Date(Web):
DOI:10.1002/chem.201203665
Abstract
Enantiospecific axial-to-point chiral transfer in light-induced transformations was efficient under elevated pressure at high temperatures. Model photoreactions with atropisomeric compounds showed higher enantioselectivity in the photoproducts under elevated pressure. The ee values in the photoproducts were rationalized based on the increased stability of optically pure atropisomeric compounds at elevated pressure, even at high temperatures.
Co-reporter:Barry C. Pemberton;Ramya Raghunathan;Sabine Volla
Chemistry - A European Journal 2012 Volume 18( Issue 39) pp:12178-12190
Publication Date(Web):
DOI:10.1002/chem.201202083
Abstract
Cucurbiturils are a family of molecular container compounds with superior molecular recognition properties. The use of cucurbiturils for supramolecular catalysis is highlighted in this concept. Both photochemical reactions as well as thermal transformations are reviewed with an eye towards tailoring substrates for supramolecular catalysis mediated by cucurbiturils.
Co-reporter:Elango Kumarasamy ; Josepha L. Jesuraj ; Joseph N. Omlid ; Angel Ugrinov
Journal of the American Chemical Society 2011 Volume 133(Issue 43) pp:17106-17109
Publication Date(Web):October 7, 2011
DOI:10.1021/ja203087a
Nonbiaryl axially chiral 2-pyridones were synthesized and employed for light-induced electrocyclic 4π ring closure leading to bicyclo-β-lactam photoproducts in solution. The enantioselectivity in the photoproducts varied from 22 to 95% depending on the reaction temperature and the ability of the axially chiral chromophore to form intramolecular and/or intermolecular H-bonds with the solvent. On the basis of the differential activation parameters, entropic control of the enantiospecificity was observed for 2-pyridones lacking the ability to form H-bonds. Conversely, enthalpy played a significant role for 2-pyridones having the ability to form H-bonds.
Co-reporter:Barry C. Pemberton, Raushan K. Singh, Alexander C. Johnson, Steffen Jockusch, José P. Da Silva, Angel Ugrinov, Nicholas J. Turro, D. K. Srivastava and J. Sivaguru
Chemical Communications 2011 vol. 47(Issue 22) pp:6323-6325
Publication Date(Web):04 May 2011
DOI:10.1039/C1CC11164G
Guest induced shape change of the cucurbit[8]uril cavity is likely rate limiting in the supramolecular photocatalytic cycle for CB8 mediated photodimerization of 6-methylcoumarin.
Co-reporter:Anoklase Jean-Luc Ayitou and J. Sivaguru
Chemical Communications 2011 vol. 47(Issue 9) pp:2568-2570
Publication Date(Web):23 Dec 2010
DOI:10.1039/C0CC04416D
The enantiomeric ratio (e.r.) in the 3,4-dihydroquinolin-2-one photoproduct during 6π-photocyclization of α-substituted axially chiral ortho-tert-butyl-acrylanilides depends on the nature of the reactive spin state (singlet or triplet), where the singlet-spin state reactivity gives a racemic mixture and the triplet reactivity gives an e.r. value >95∶5.
Co-reporter:Josepha L. Jesuraj and J. Sivaguru
Chemical Communications 2010 vol. 46(Issue 26) pp:4791-4793
Publication Date(Web):25 May 2010
DOI:10.1039/C0CC00470G
Atropisomeric benzoylformamides 1 undergo Type II reaction leading to cis-2 and trans-2 oxazolidin-4-one photoproducts. The N–C(Aryl) chiral axis is maintained during the course of the phototransformation in spite the reaction proceeding through a near planar intermediate(s). As the rotational barrier of the N–C(Aryl) chiral axis in the cis-2 and trans-2 photoproducts is lowered when compared with the reactant 1, the isolated optically pure trans-2 isomer is converted to the ent-cis-2 isomer without affecting the C-5 stereogenic center, resulting in resolution of the cis-2 enantiomers.
Co-reporter:Barry C. Pemberton, Nilotpal Barooah, D. K. Srivatsava and J. Sivaguru
Chemical Communications 2010 vol. 46(Issue 2) pp:225-227
Publication Date(Web):17 Nov 2009
DOI:10.1039/B920605A
Cucurbit[8]uril (as low as 10 mol%) acts as a supramolecular catalytic nanoreaction vessel and facilitates the photodimerization of coumarins in water leading to syn dimers. Saturation kinetics shows a sigmoidal dependence with a turnover number of 3.4 min−1 and a Hill constant of 1.8 indicating a co-operative mechanism in the catalytic process.
Co-reporter:Anoklase Jean-Luc Ayitou ; Josepha L. Jesuraj ; Nilotpal Barooah ; Angel Ugrinov ;J. Sivaguru
Journal of the American Chemical Society 2009 Volume 131(Issue 32) pp:11314-11315
Publication Date(Web):July 24, 2009
DOI:10.1021/ja9050586
α-Oxoamides 1 with o-tert-butyl substitution on the N-phenyl moiety were found to be stable axially chiral atropisomers. These axially chiral α-oxoamides undergo enantiospecific photochemical γ-Hydrogen abstraction in CHCl3 to yield β-lactams with high enantioselectivity (e.r. ∼90:10) in solution. The extent of enantioselectivity was found to be dependent on the reaction temperature.
Co-reporter:Anoklase Jean-Luc Ayitou, Angel Ugrinov and J. Sivaguru
Photochemical & Photobiological Sciences 2009 vol. 8(Issue 6) pp:751-754
Publication Date(Web):25 Mar 2009
DOI:10.1039/B903861B
O-tert-Butylacrylanilides with N–H substitution undergo 6π-photocyclization at the unsubstituted ortho carbon, whereas the corresponding N-methyl derivatives cyclize at the ortho carbon bearing the tert-butyl with the eventual loss of 2-methylpropene.
Co-reporter:J. Sivaguru, Marissa R. Solomon, Thomas Poon, Steffen Jockusch, Sara G. Bosio, Waldemar Adam and Nicholas J. Turro
Accounts of Chemical Research 2008 Volume 41(Issue 3) pp:387
Publication Date(Web):February 13, 2008
DOI:10.1021/ar7001254
Photochirogenesis, the control of chirality in photoreactions, is one of the most challenging problems in stereocontrolled photochemistry, in which the stereodifferentiation has to be imprinted within the short lifetime of the electronically excited state. Singlet oxygen (1O2), an electronically excited molecule that is known to be sensitive to vibrational deactivation, has been selected as a model case for testing stereoselective control by vibrational deactivation. The stereoselectivity in the reaction of 1O2 with E/Z enecarbamates 1, equipped with the oxazolidinone chiral auxiliary, has been examined for the mode selectivity ([2 + 2]-cycloaddition versus ene-reaction) and the stereoselectivity in the oxidative cleavage of the alkenyl functionality to the methyldesoxybenzoin (MDB) product. Through the appropriate choice of substituents in the enecarbamate, the mode selectivity (ene versus [2 + 2]), which depends on the alkene geometry (E or Z), the steric bulk of the oxazolidinone substituent at the C-4 position, and the C-3′ configuration on the side chain, may be manipulated. Phenethyl substitution gives exclusively the [2 + 2]-cycloaddition product, irrespective of the alkene geometry. The stereoselection in the resulting methyldesoxybenzoin (MDB) product is examined in a variety of solvents as a function of temperature by using chiral GC analysis. The extent (% ee) as well as the sense (R versus S) of the stereoselectivity in the MDB formation for the E isomer depends significantly on solvent and temperature, whereas the corresponding Z isomers are not affected by such variations. The complex temperature and solvent effects are scrutinized in terms of the differential activation parameters (ΔΔS⧧, ΔΔH⧧) for the photooxygenation of E/Z-enecarbamates in various solvents at different temperatures. The enthalpy−entropy compensations provide a mechanistic understanding of the temperature dependence of the ee values for the MDB product and the difference in the behavior between the Z and E enecarbamates. The E enecarbamates show a relatively high contribution from the entropy term and an appreciable contribution from the enthalpy term; both terms possess the same sign. In contrast, the corresponding relative insensitivity of Z enecarbamates to temperature and solvent variation is convincingly explained by the near-zero ΔΔS‡ and ΔΔH‡. Such effects, associated with temperature- and solvent-dependent conformational factors, are most likely dictated by the stereogenic center at the C-3′ phenethyl substituent. The high stereocontrol during the photooxygenation of the chiral enecarbamates is shown to be independent of the steric demand of the oxazolidinone substituent at the C-4 position. In view of the reduced stereocontrol on deuteration of the oxazolidinone substituent at the C-4 position, we propose that the unusual stereoselective vibrational quenching of the attacking singlet oxygen (excited-state reactivity), a novel mechanistic concept, works in concert with the usual steric impositions (ground-state reactivity) exercised by the substituents to afford the high stereoselectivity observed in the dioxetane product during the [2 + 2] cycloaddition. Such synergistic interplay is held responsible for the highly stereoselective photooxidative cleavage of the chiral enecarbamates. The efficacy of stereocontrol in this photooxidation is demonstrated by kinetically resolving the epimers of the enecarbamate cleavage product (MDB) in essentially perfect stereoselectivity, a new methodology that we coin “photo-Pasteur-type kinetic resolution”.
Co-reporter:Nilotpal Barooah, Barry C. Pemberton, Alexander C. Johnson and J. Sivaguru
Photochemical & Photobiological Sciences 2008 vol. 7(Issue 12) pp:1473-1479
Publication Date(Web):23 Oct 2008
DOI:10.1039/B814230K
Coumarin derivatives with non-polar substituents at the 6 or 7 position undergoes photodimerization in the presence of CB[8] in water to give the syn dimer as the major product. It is postulated that these neutral coumarins form dynamic complexes in the presence of CB[8] and the product selectivity is reflective of the type of complex, available volume in the CB[8] cavity and relative rate of photodimerization inside and outside the CB[8] cavity.
Co-reporter:Ramya Raghunathan, Elango Kumarasamy, Akila Iyer, Angel Ugrinov and J. Sivaguru
Chemical Communications 2013 - vol. 49(Issue 77) pp:NaN8715-8715
Publication Date(Web):2013/08/20
DOI:10.1039/C3CC44281K
Atropisomeric α-oxoamides were synthesized and employed for intramolecular Paternò–Büchi reaction leading to very high enantio- and diastereoselectivity in the bicyclic oxetane photoproduct. A reversal of product selectivity was observed in solution and in the solid-state.
Co-reporter:Barry C. Pemberton, Raushan K. Singh, Alexander C. Johnson, Steffen Jockusch, José P. Da Silva, Angel Ugrinov, Nicholas J. Turro, D. K. Srivastava and J. Sivaguru
Chemical Communications 2011 - vol. 47(Issue 22) pp:NaN6325-6325
Publication Date(Web):2011/05/04
DOI:10.1039/C1CC11164G
Guest induced shape change of the cucurbit[8]uril cavity is likely rate limiting in the supramolecular photocatalytic cycle for CB8 mediated photodimerization of 6-methylcoumarin.
Co-reporter:Josepha L. Jesuraj and J. Sivaguru
Chemical Communications 2010 - vol. 46(Issue 26) pp:NaN4793-4793
Publication Date(Web):2010/05/25
DOI:10.1039/C0CC00470G
Atropisomeric benzoylformamides 1 undergo Type II reaction leading to cis-2 and trans-2 oxazolidin-4-one photoproducts. The N–C(Aryl) chiral axis is maintained during the course of the phototransformation in spite the reaction proceeding through a near planar intermediate(s). As the rotational barrier of the N–C(Aryl) chiral axis in the cis-2 and trans-2 photoproducts is lowered when compared with the reactant 1, the isolated optically pure trans-2 isomer is converted to the ent-cis-2 isomer without affecting the C-5 stereogenic center, resulting in resolution of the cis-2 enantiomers.
Co-reporter:Nandini Vallavoju and J. Sivaguru
Chemical Society Reviews 2014 - vol. 43(Issue 12) pp:NaN4101-4101
Publication Date(Web):2014/04/07
DOI:10.1039/C3CS60471C
Using non-bonding interactions to control photochemical reactions requires an understanding of not only thermodynamics and kinetics of ground state and excited state processes but also the intricate interactions that dictate the dynamics within the system of interest. This review is geared towards a conceptual understanding of how one can control the reactivity and selectivity in the excited state by employing confinement and non-covalent interactions. Photochemical reactivity of organic molecules within confined containers and organized assemblies as well as organic templates that interact through H-bonding and/or cation–carbonyl/cation–π interactions is reviewed with an eye towards understanding supramolecular effects and photocatalysis.
Co-reporter:Barry C. Pemberton, Nilotpal Barooah, D. K. Srivatsava and J. Sivaguru
Chemical Communications 2010 - vol. 46(Issue 2) pp:NaN227-227
Publication Date(Web):2009/11/17
DOI:10.1039/B920605A
Cucurbit[8]uril (as low as 10 mol%) acts as a supramolecular catalytic nanoreaction vessel and facilitates the photodimerization of coumarins in water leading to syn dimers. Saturation kinetics shows a sigmoidal dependence with a turnover number of 3.4 min−1 and a Hill constant of 1.8 indicating a co-operative mechanism in the catalytic process.
Co-reporter:Anoklase Jean-Luc Ayitou and J. Sivaguru
Chemical Communications 2011 - vol. 47(Issue 9) pp:NaN2570-2570
Publication Date(Web):2010/12/23
DOI:10.1039/C0CC04416D
The enantiomeric ratio (e.r.) in the 3,4-dihydroquinolin-2-one photoproduct during 6π-photocyclization of α-substituted axially chiral ortho-tert-butyl-acrylanilides depends on the nature of the reactive spin state (singlet or triplet), where the singlet-spin state reactivity gives a racemic mixture and the triplet reactivity gives an e.r. value >95∶5.
Co-reporter:Elango Kumarasamy and J. Sivaguru
Chemical Communications 2013 - vol. 49(Issue 39) pp:NaN4348-4348
Publication Date(Web):2012/11/20
DOI:10.1039/C2CC37123E
Atropisomeric 3,4-dihydro-2-pyridones undergo stereospecific [2+2]-photocycloaddition in solution with high stereoselectivity (ee > 98% and de > 96%) in the product. The chiral transfer during phototransformation was rationalized based on the stability/reactivity of the biradical.



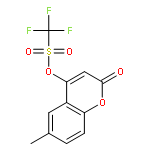
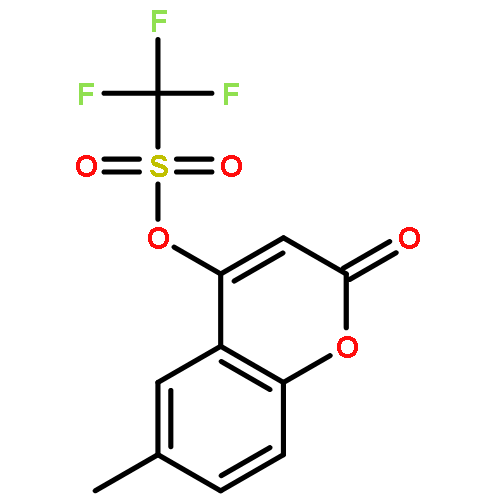
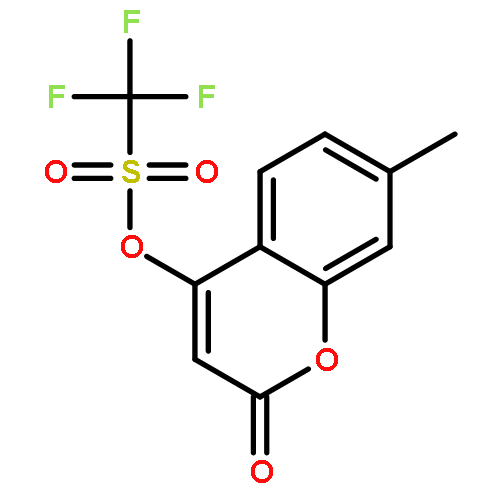
![Benzenemethanamine, N-[2-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/205300/205242-33-7.png)
![Benzenemethanamine, N-[2-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/205300/205242-33-7_b.png)
![1H-Benzotriazole-1-methanamine, N-[2-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/202300/202276-33-3.png)
![1H-Benzotriazole-1-methanamine, N-[2-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/202300/202276-33-3_b.png)



![2-Furancarboxaldehyde, 5-[(benzoyloxy)methyl]-](http://img.cochemist.com/ccimg/42900/42889-91-8.png)
![2-Furancarboxaldehyde, 5-[(benzoyloxy)methyl]-](http://img.cochemist.com/ccimg/42900/42889-91-8_b.png)






![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)