Co-reporter:Cristina Trujillo;Astrid Botte;Stephen J. Connon
Chemical Communications 2017 vol. 53(Issue 63) pp:8874-8877
Publication Date(Web):2017/08/03
DOI:10.1039/C7CC04596D
The first DFT study of the cycloaddition of benzaldehyde with homophthalic anhydride under the influence of a bifunctional organocatalyst is reported. The catalyst first binds and then deprotonates the anhydride, leading to a squaramide-bound enolate, which then adds to the aldehyde with activation of the electrophile by the catalyst's ammonium ion.
Co-reporter:Isabel Rozas
Chem 2017 Volume 2, Issue 1(Volume 2, Issue 1) pp:
Publication Date(Web):12 January 2017
DOI:10.1016/j.chempr.2016.12.012
In this issue of Chem, collaborative efforts from the Cavalli and Steinmetz groups have resulted in a more cogent understanding of the binding of cis- and trans-combretastatin isomers to tubulin by means of structural and computational studies.
Co-reporter:Aoife Flood, Cristina Trujillo, Goar Sanchez-Sanz, Brendan Kelly, Carolina Muguruza, Luis F. Callado, Isabel Rozas
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.06.008
•Phenyl-, thiophenyl- and thiazolylguanidinium derivatives have similar aromaticity.•Thiophenyl- & thiazolylguanidines must act as phenyl-based α2-adrenoceptor ligands.•A high-diversity library of thiophenyl- and thiazolylguanidines was prepared.•Their affinity for α2-adrenoceptor was measured in human prefrontal cortex tissue.•Activity on α2-adrenoceptors was measured: 2 antagonists and 1 agonist were found.Searching for improved antagonists of α2-adrenoceptors, a thorough theoretical study comparing the aromaticity of phenyl-, pyridinyl-, thiophenyl- and thiazolylguanidinium derivatives has been carried out [at M06-2X/6–311++G(p,d) computational level] confirming that thiophene and thiazole will be good ‘ring equivalents’ to benzene in these guanidinium systems. Based on these results, a small but chemically diverse library of guanidine derivatives (15 thiophenes and 2 thiazoles) were synthesised to explore the effect that the bioisosteric change has on affinity and activity at α2-adrenoceptors in comparison with our previously studied phenyl derivatives. All compounds were tested for their α2-adrenoceptor affinity and unsubstituted guanidinothiophenes displayed the strongest affinities in the same range as the phenyl analogues. In the case of cycloakyl systems, thiophenes with 6-membered rings showed the largest affinities, while for the thiazoles the 5-membered analogue presented the strongest affinity. From all the compounds tested for noradrenergic activity, only one compound exhibited agonistic activity, while two compounds showed very promising antagonism of α2-adrenoceptors.Download high-res image (248KB)Download full-size image
Co-reporter:Michela McMullan, Aintzane García-Bea, Patricia Miranda-Azpiazu, Luis F. Callado, Isabel Rozas
European Journal of Medicinal Chemistry 2016 Volume 123() pp:48-57
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.07.011
•Conformationally restricted guanidines allow probing the α2-AR binding site.•Preparation of conformationally restricted guanidines involves novel routes.•Benzodiazepine guanidine derivatives show the highest α2-adrenoceptor affinity.•Substitution at the cyclic N of the guanidine moiety is positive for α2-AR affinity.•Compounds 8b & 18c show high α2-AR affinity + antagonism in vitro human brain tissue.In this paper we report the design, synthesis and pharmacological evaluation of new N-substituted 2-amino-1,4-dihydroquinazolines, 2-amino-1,4-dihydropyridopyrimidines and 2-amino-4,5-dihydro-1,3-benzodiazepines as α2-adrenoceptors ligands. Computational studies show that the proposed substitutions and guanidine-containing ring size will probe an extensive area of the active site. Preparation of these molecules involved novel routes than those previously utilised in our laboratory for the preparation of the acyclic aryl-guanidine counterparts. Compounds 8b and 18c showed the highest affinity and antagonistic activity, within their series, towards the α2-adrenoceptor in human brain tissue in vitro experiments. Structure-activity relationships have been established for the design and biological evaluation of novel α2-adrenoceptor ligands.
Co-reporter:Amila Kahvedžić-Seljubac, Seema-Maria Nathwani, Daniela M. Zisterer, Isabel Rozas
European Journal of Medicinal Chemistry 2016 Volume 117() pp:269-282
Publication Date(Web):19 July 2016
DOI:10.1016/j.ejmech.2016.03.047
•Isouronium and N-hydroxyguanidinium form strong HBs as donors and acceptors.•Presence of a HB acceptor in these cations is detrimental for DNA binding.•Isouroniums and N-hydroxyguanidiniums are strong growth inhibitors in HL-60& Kelly.•No correlation found between DNA binding and cell growth inhibition.•Two isouroniums inhibit growth and are good apoptosis-inducers in both cell lines.Based on the results obtained from a computational study on the suitability of the isouronium and N-hydroxyguanidinium cations as hydrogen bond donors/acceptors, the DNA binding of a series of isouronium derivatives was assessed by DNA thermal denaturation experiments and compared to related N-hydroxyguanidines. Due to the poor DNA binding observed, the nature of the diaromatic linker was explored by preparing the corresponding amide-linked bis-isouronium derivative and measuring its DNA affinity. Next, the inhibitory effects of the isouronium derivatives on cell viability were evaluated in two different cancer cell lines providing IC50 values in the range of 36.9–57.4 μM (HL-60, leukemia), and 17.3–33.9 μM (Kelly, neuroblastoma). These values are comparable to those previously found for the N-hydroxyguanidine series. Compounds with the –S- linker (3, 6, and 10) proved to be considerably active in the HL-60 cells and even more active in the Kelly cell line. No correlation was found between DNA minor groove binding and cell growth inhibition; hence, activity may depend on different modes of action. Further studies into the apoptotic potential of these compounds indicated that, besides inhibiting cell viability and proliferation, derivatives 9 and 10, are significant apoptosis-inducers in both cell lines. Results obtained with HL-60 cells suggest that G2/M arrest and subsequent apoptosis induced by compound 10 are associated with microtubular depolymerisation, loss of mitochondrial membrane potential and activation of the caspase cascade. Moreover, the effects of compound 10 on cell viability and apoptosis in two non-cancereous cell lines (NIH3T3 and MCF-10A) indicate none or minimal toxicity.
Co-reporter:Brendan Kelly; Michela McMullan; Carolina Muguruza; Jorge E. Ortega; J. Javier Meana; Luis F. Callado
Journal of Medicinal Chemistry 2015 Volume 58(Issue 2) pp:963-977
Publication Date(Web):December 9, 2014
DOI:10.1021/jm501635e
We have previously identified phenylguanidine and phenyl-2-aminoimidazoline compounds as high affinity ligands with conflicting functional activity at the α2-adrenoceptor, a G-protein-coupled receptor with relevance in several neuropsychiatric conditions. In this paper we describe the design, synthesis, and pharmacological evaluation of a new series of pyridine derivatives [para substituted 2- and 3-guanidino and 2- and 3-(2-aminoimidazolino)pyridines, disubstituted 2-guanidinopyridines and N-substituted-2-amino-1,4-dihydroquinazolines] that were found to be antagonists/inverse agonists of the α2-adrenoceptor. Furthermore, the compounds exert their effects at the α2-adrenoceptor both in vitro in human prefrontal cortex tissue and in vivo in rat brain as shown by microdialysis experiments. We also provide a docking study at the α2A- and α2C-adrenoceptor subtypes demonstrating the structural features required for high affinity binding to the receptor.
Co-reporter:Elena Diez-Cecilia, Robert Carson, Brendan Kelly, Sandra van Schaeybroeck, James T. Murray, Isabel Rozas
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 19) pp:4287-4292
Publication Date(Web):1 October 2015
DOI:10.1016/j.bmcl.2015.07.082
Mutations in the Ras-pathway occur in 40–45% of colorectal cancer patients and these are refractory to treatment with anti-EGFR-targeted therapies. With this in mind, we have studied novel guanidinium-based compounds with demonstrated ability to inhibit protein kinases. We have performed docking studies with several proteins involved in the Ras-pathway and evaluated 3,4′-bis-guanidinium derivatives as inhibitors of B-Raf. Compound 3, the most potent in this series, demonstrated strong cytotoxicity in WTB-Raf colorectal cancer cells and also cells with V600EB-Raf mutations. Cell death was induced by apoptosis, detected by cleavage of PARP. Compound 3 also potently inhibited ERK1/2 signalling, inhibited EGFR activation, as well as Src, STAT3 and AKT phosphorylation. Mechanistically, compound 3 did not inhibit ATP binding to B-Raf, but direct assay of B-Raf activity was inhibited in vitro. Summarizing, we have identified a novel B-Raf type-III inhibitor that exhibits potent cellular cytotoxicity.
Co-reporter:Julian W. Shaw, Laure Barbance, David H. Grayson, Isabel Rozas
Tetrahedron Letters 2015 Volume 56(Issue 35) pp:4990-4992
Publication Date(Web):26 August 2015
DOI:10.1016/j.tetlet.2015.07.007
The use of 2-chloro-4,6-dimethoxypyrimdine as a tool for the syntheses of substituted guanidines is presented. This method, that we had previously shown to be very useful for aromatic amines, introduces an atom economical, cost effective and environmentally safe method for the installation of the guanidine functionality in aliphatic primary and secondary amines.
Co-reporter:Padraic S. Nagle ; Caitriona McKeever ; Fernando Rodriguez ; Binh Nguyen ; W. David Wilson
Journal of Medicinal Chemistry 2014 Volume 57(Issue 18) pp:7663-7672
Publication Date(Web):August 26, 2014
DOI:10.1021/jm5008006
In this paper we report the design and biophysical evaluation of novel rigid-core symmetric and asymmetric dicationic DNA binders containing 9H-fluorene and 9,10-dihydroanthracene cores as well as the synthesis of one of these fluorene derivatives. First, the affinity toward particular DNA sequences of these compounds and flexible core derivatives was evaluated by means of surface plasmon resonance and thermal denaturation experiments finding that the position of the cations significantly influence the binding strength. Then their affinity and mode of binding were further studied by performing circular dichroism and UV studies and the results obtained were rationalized by means of DFT calculations. We found that the fluorene derivatives prepared have the ability to bind to the minor groove of certain DNA sequences and intercalate to others, whereas the dihydroanthracene compounds bind via intercalation to all the DNA sequences studied here.
Co-reporter:Elena Diez-Cecilia, Brendan Kelly, Concepcion Perez, Daniela M. Zisterer, Daniel K. Nevin, David G. Lloyd, Isabel Rozas
European Journal of Medicinal Chemistry 2014 Volume 81() pp:427-441
Publication Date(Web):23 June 2014
DOI:10.1016/j.ejmech.2014.05.025
•New families of diaromatic 3,4′-(substituted)guanidinium derivatives were prepared.•Good cytotoxicity and apoptosis effects in HL-60 cancer cell line were observed.•Some compounds show high percentage inhibition on the RAF-1/MEK-1 pathway.•Docking studies on RAF-1 and MEK-1 structures suggest MEK-1 allosteric inhibition.Considering the structural similarities between the kinase inhibitor sorafenib and 4,4′-bis-guanidinium derivatives previously prepared by Rozas and co., which display interesting cytotoxicity in cancer cells, we have studied whether this activity could result from kinase inhibition. Five new families have been prepared consisting of unsubstituted and aryl-substituted 3,4′-bis-guanidiniums, 3,4′-bis-2-aminoimidazolinium and 3-acetamide-4′-(4-chloro-3-trifluoromethylphenyl)guanidinium derivatives. Cytotoxicity (measuring the IC50 values) and apoptosis studies in human HL-60 promyelocytic leukemia cells were carried out for these compounds. Additionally, their potential inhibitory effect was explored on a panel of kinases known to be involved in apoptotic pathways. The previously prepared cytotoxic 4,4′-bis-guanidiniums did not inhibit any of these kinases; however, some of the novel 3,4′-substituted derivatives showed a high percentage inhibition of RAF-1/MEK-1, for which the potential mode of binding was evaluated by docking studies. The interesting antitumour properties showed by these compounds open up new exciting lines of investigation for kinase inhibitors as anticancer agents and also highlights the relevance of the guanidinium moiety for protein kinase inhibitors chemical design.
Co-reporter:Daniel H. O'Donovan, Carolina Muguruza, Luis F. Callado, Isabel Rozas
European Journal of Medicinal Chemistry 2014 Volume 82() pp:242-254
Publication Date(Web):23 July 2014
DOI:10.1016/j.ejmech.2014.05.057
•A pharmacophore model has been developed to design new α2-AR antagonists.•Increased substitution around the guanidinium will selectively lead to antagonists.•Thirty new compounds incorporating a multiply substituted guanidinium were prepared.•Functional assays showed that this feature exclusively yields α2-AR antagonists.Depression has been linked to a selective increase in the high affinity conformation of the α2-adrenergic autoreceptors (α2-ARs) in the human brain as well as to an overexpression of α2-ARs in the hippocampus and cerebral cortex. Thus, the development of novel α2-AR antagonists represents an attractive source of new antidepressants. This paper describes the design, synthesis and pharmacological evaluation of 30 new guanidinium and 2-iminoimidazolidinium as potential α2-AR antagonists. In order to design this new series of α2-AR antagonists, a pharmacophore model was developed using the GALAHAD software. This study suggested that increased substitution in the space surrounding the cationic guanidine moiety might lead selectively to antagonist activity. Following the preparation of compounds incorporating this feature and competitive radioligand binding, [35S]GTPγS functional assays revealed that this structural modification affords exclusively α2-AR antagonists, in contrast with the analogous unsubstituted compounds in which a mixture of antagonist/agonist activities was previously observed.
Co-reporter:Julian W. Shaw;David H. Grayson
European Journal of Organic Chemistry 2014 Volume 2014( Issue 17) pp:3565-3569
Publication Date(Web):
DOI:10.1002/ejoc.201402179
Abstract
A novel method for the synthesis of aryl-substituted guanidines in good overall yields is presented; it consists of the acidic cleavage of 2-(arylamino)-4,6-dimethoxypyrimidines, which were prepared by coupling aryl bromides with 2-amino-4,6-dimethoxypyrimidine. This methodology introduces a new means of protection for the guanidine functionality.
Co-reporter:Marta Marin-Luna, Goar Sanchez-Sanz, Patrick O’Sullivan, and Isabel Rozas
The Journal of Physical Chemistry A 2014 Volume 118(Issue 29) pp:5540-5547
Publication Date(Web):July 2, 2014
DOI:10.1021/jp504483x
We have studied theoretically the complexes of model N-phenylguanidine/ium derivatives with PtCl3– and PtCl2 in different coordinating modes (mono- and bidentate) with different N atoms of the guanidine/ium moiety using the B3LYP/6-31+G** and LANL2DZ mixed basis set. This will aid the understanding of the complexation between platinum and the guanidine or guanidinium moiety in order to design dual anticancer agents that combine a guanidine-based DNA minor groove binder and a cisplatin-like moiety. Calculated interaction and relative energies, analysis of the electron density, and examination of the orbital interactions indicate that the most stable type of complex is that with a monodentate interaction between PtCl3– and guanidinium established through one of the NH2 groups. Next, we optimized the structure of three bis-guanidinium diaromatic systems developed in our group as DNA minor groove binders and their complexation with PtCl3–, finding that the formation of Pt complexes of these minor groove binders is favorable and would produce stable monodentate coordinated systems.
Co-reporter:Patrick O'Sullivan ; Isabel Rozas
ChemMedChem 2014 Volume 9( Issue 9) pp:2065-2073
Publication Date(Web):
DOI:10.1002/cmdc.201402264
Abstract
Based on our previous positive results with bis-guanidine-like diaromatic compounds as DNA minor groove binders, we propose a new family: bis-2-amino-1,4,5,6-tetrahydropyrimidines. According to calculated parameters, these dicationic systems would have a more suitable size and lipophilicity for binding into the minor groove than previous series. Moreover, their DFT-optimised structures and docking into an AT oligomer model show that they would bind in the minor groove with good strength and without energy penalty. Hence, we prepared compounds 4 a–c and evaluated their binding to ssDNA and poly(dA-dT)2 by thermal denaturation experiments. The results showed that 4 a (CO) and 4 d (NH) were the best DNA binders. Compared to the previous series, 4 a–d are better binders than bis-guanidiniums but poorer than bis-2-aminoimidazolinium derivatives. Moreover, circular dichroism experiments using ssDNA and poly(dA-dT)2 confirmed binding into the minor groove. Based on our computational design as well as biophysical studies, we have been able to determine that the optimal interaction of guanidine-like dications in the minor grove occurs with bis-2-aminoimidazolinium systems.
Co-reporter:Amila Kahvedžić ; Seema-Maria Nathwani ; Daniela M. Zisterer
Journal of Medicinal Chemistry 2013 Volume 56(Issue 2) pp:451-459
Publication Date(Web):December 20, 2012
DOI:10.1021/jm301358s
In this paper we report the synthesis of a new family of hydroxyguanidinium aromatic derivatives (4a–g) as potential minor groove binders and cytotoxic agents. Their DNA affinity was evaluated by thermal denaturation experiments using salmon sperm DNA. The antiproliferative effects of derivatives 4a, 4d, and 4f were evaluated in human promyelocytic HL-60, breast carcinoma MCF-7, and neuroblastoma Kelly cell lines using the AlamarBlue viability assay, and IC50 values were obtained. All three compounds were active in the HL-60 cell line. In particular, 4b exhibits antiproliferative effects in all three cell lines while 4d reduced HL-60 and Kelly viability. Both 4b and 4d produced considerable antiproliferative activity in the Kelly cell line. Derivative 4d was chosen for further cell cycle and apoptosis studies using flow cytometric analysis of cellular DNA content.
Co-reporter:Caitriona McKeever ; Marcel Kaiser
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:700-711
Publication Date(Web):January 9, 2013
DOI:10.1021/jm301614w
Considering the strong DNA minor groove binding observed for our previous series of diaromatic symmetric and asymmetric guanidinium and 2-aminoimidazolinium derivatives, we report now the synthesis of new aminoalkyl derivatives of diaromatic guanidines with potential as DNA minor groove binders and antiprotozoal activity. The preparation of these aminoalkyl derivatives (12a–e, 13a–e, 14a–c,e, 15a–e, 16a–e) is presented as well as their affinity for DNA which was evaluated by means of DNA thermal denaturation experiments. Finally, the antiprotozoal activity of most of these aminoalkyl minor groove binders was evaluated in vitro against Trypanosoma brucei rhodesiense (8 compounds) and Plasmodium falciparum (18 compounds). The O-linked derivatives 13c and 14c showed 100 nM activities against P. falciparum, whereas for T. b. rhodesiense all compounds tested showed micromolar activity. Some of the derivatives prepared seem to exert the antimalarial activity by binding to the DNA minor groove whereas other sets of compounds could exert this antimalarial activity by inhibiting the parasite dihydrofolate reductase, for example.
Co-reporter:Daniel H. O'Donovan, Brendan Kelly, Elena Diez-Cecilia, Martin Kitson and Isabel Rozas
New Journal of Chemistry 2013 vol. 37(Issue 8) pp:2408-2418
Publication Date(Web):28 May 2013
DOI:10.1039/C3NJ00285C
The N-aryl- and N-acylguanidine structural motifs are essential for the function of several important classes of molecules, including pharmaceuticals, catalysts and natural products. Compounds combining both motifs can exist as different isomers due to tautomerism within the guanidine subunit, E/Z isomerism with reference to the guanidine double bond, and conformational isomerism. This complex phenomenon results in unresolved broad signal NMR spectra that strongly complicate the characterisation of these derivatives. Hence, the present study examines isomerism in N,N′-bis-aryl-N′′-acylguanidines using low temperature NMR spectroscopy in tandem with Density Functional Theory (DFT), Natural Bond Analysis (NBO) and the Gauge-Invariant Atomic Orbital (GIAO) approach for calculating the NMR chemical shifts associated with each isomer. It was found that the structural preference of these compounds is strongly influenced by intramolecular hydrogen bond (IMHB) effects.
Co-reporter:Isabel Rozas;Goar Sánchez-Sanz;Ibon Alkorta;José Elguero
Journal of Physical Organic Chemistry 2013 Volume 26( Issue 5) pp:378-385
Publication Date(Web):
DOI:10.1002/poc.3099
We have calculated the complexes formed by guanidine/guanidinium and HCl/Cl−, HNO3/NO3− and H2SO4/HSO4− both in the gas and aqueous Polarizable Continuum Model (PCM) phase to understand the effect that solvation has on their interaction energies. In the gas phase, the cation–anion complexes are much more stable than the rest; however, when PCM-water is considered, this energetic difference is not as large due to the extra stabilization that the ions suffer when in aqueous solution. All the complexes were analyzed in terms of their AIM and NBO properties. In all cases, water solvation seems to “dampen” those properties observed in the gas phase. The values of Nucleus Independent Chemical Shift (NICS)(1) and NICS(2) indicate a huge influence of the proximity of the carbon atom for short distances; thus, the 3D NICS values on the van der Waal isosurfaces have been used to evaluate the possible Y-aromaticity of the guanidinium system. The isosurface in this system is more similar to cyclohexane than to benzene as indication of poor aromaticity. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Brendan Kelly, Isabel Rozas
Tetrahedron Letters 2013 Volume 54(Issue 30) pp:3982-3984
Publication Date(Web):24 July 2013
DOI:10.1016/j.tetlet.2013.05.070
We present a concise, less-toxic and broadly applicable method for coupling weakly nucleophilic amines with N,N′-di-(tert-butoxycarbonyl)thiourea, N-(tert-butxoycarbonyl), N′-alkyl/arylsubstituted-thioureas and N,N′-di-(tert-butoxycarbonyl)imidazolidine-2-thione in the presence of copper(II) chloride. Subsequent removal of Boc protecting groups affords guanidines, di-substituted guanidines and 2-aminoimidazolines in modest to excellent overall yields.
Co-reporter:Fernando Blanco, Brendan Kelly, Goar Sánchez-Sanz, Cristina Trujillo, Ibon Alkorta, Jose Elguero, and Isabel Rozas
The Journal of Physical Chemistry B 2013 Volume 117(Issue 39) pp:11608-11616
Publication Date(Web):September 2, 2013
DOI:10.1021/jp407339v
Considering that guanidine-based derivatives are good DNA minor groove binders, we have theoretically studied, using the Polarizable Continuum model mimicking water solvation, the complexes formed by the biologically relevant guanidinium cation and the DNA and RNA nucleobases (adenine, guanine, cytosine, thymine, and uracil). The interactions established within these complexes both by hydrogen bonds and by cation−π interactions have been analyzed by means of the Atoms in Molecules and Natural Bond Orbital approaches. Moreover, maps of electron density difference have been produced to understand the cation−π complexes. Finally, the NICS and three-dimensional NICS maps of the cation−π complexes have been studied to understand the effect of the guanidinium cation on the aromaticity of the nucleobases.
Co-reporter:Padraic S. Nagle ; Fernando Rodriguez ; Binh Nguyen ; W. David Wilson
Journal of Medicinal Chemistry 2012 Volume 55(Issue 9) pp:4397-4406
Publication Date(Web):April 12, 2012
DOI:10.1021/jm300296f
In this paper we report the design and synthesis of a new family of asymmetric peptide linked diaromatic dications as potent DNA minor groove binders. These peptide-linked compounds, with a linear core, displayed a much larger affinity than other guanidinium-like derivatives from the same series with curved cores. As a first screening, the DNA affinity of these structures was evaluated by means of thermal denaturation experiments, finding that the nature of the cation (guanidinium vs 2-aminoimidazolinium) significantly influenced the binding strength. Their binding affinity was assessed by implementing further biophysical measurements such as surface plasmon resonance and circular dichroism. In particular, it was observed that compounds 6, 7, and 8 displayed both a strong binding affinity and significant selectivity for AT oligonucleotides. In addition, the thermodynamics of their binding was evaluated using isothermal titration calorimetry, indicating that the binding is derived from favorable enthalpic and entropic contributions.
Co-reporter:C. Matijssen;G. K. Kinsella;G. W. Watson;I. Rozas
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 4) pp:351-360
Publication Date(Web):
DOI:10.1002/poc.1940
The α1-adrenoceptor is a target for the treatment of several conditions from hypertension to benign prostatic hyperplasia. In this paper, we describe a new analysis approach to explore the conformational space of several ligands of the α1-adrenoceptor and we also present the calculation of their proton affinity and basicity. For each compound a conformational search followed by a semi-empirical optimisation was performed and a selection of conformations for each ligand was subjected to further optimisation using density functional theory methods. Different positions were explored to determine the favoured site of protonation, and then, the proton affinity (in the gas phase) and basicity (using the polarisable continuum model for the aqueous solution) were calculated for each of them. In addition, an alternative method using one explicit water molecule in combination with the polarisable continuum model for aqueous solvent was explored. Moreover, the acid dissociation constant (pKa) in water of these 26 compounds was calculated because this is an important parameter for a ligand when binding to its receptor. The experimental pKa values of six of these ligands and those of two compounds with a very low and a very large pKa were used to validate the theoretical methodology. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Daniel H. O’Donovan, Isabel Rozas
Tetrahedron Letters 2012 Volume 53(Issue 34) pp:4532-4535
Publication Date(Web):22 August 2012
DOI:10.1016/j.tetlet.2012.06.042
A divergent strategy for the synthesis of 1-aryl- and 2-aryl-2-iminoimidazolidines is presented. Cyclization of N-Boc-N′-aryl-N′′-(2-hydroxyethyl)guanidines in the presence of methanesulfonyl chloride and triethylamine or sodium hydride at 0 °C affords the corresponding 2-iminoimidazolidines in good yields.
Co-reporter:Brendan Kelly, Goar Sánchez-Sanz, Fernando Blanco, Isabel Rozas
Computational and Theoretical Chemistry 2012 Volume 998() pp:64-73
Publication Date(Web):15 October 2012
DOI:10.1016/j.comptc.2012.06.004
We have theoretically studied, using PCM–water solvation, the cation–π and π–π complexes established by the biologically relevant 5-substituted 2-pyridinylguanidinium derivatives and electron-rich and electron-depleted aromatic systems (benzene and (hexafluoro)benzene). In condensed phase using PCM–water and M06-2X/6-311++G(d,p) different cation–π and π–π complexes were found. The interactions established within these complexes have been analyzed by means of the Atoms in Molecules and Natural Bond Orbital approaches and electron density difference maps have been calculated. Finally, the effect of the cation–π and π–π complexation on the aromaticity of pyridine, benzene and hexafluorobenzene was evaluated by calculating the corresponding aromaticity indexes, NICS0, 1 and 2 as well as the NICS on the 0.001 a.u. isodensity surface.Graphical abstractHighlights► 2-Pyridinylguanidiniums form cation–π and π–π complexes with (hexafluoro)benzene. ► Interactions in complexes studied by AIM, NBO and electron-density difference maps. ► Cation–π and π–π effect on aromaticity studied by NICS and NICS isodensity surface.
Co-reporter:Padraic S. Nagle;Amila Kahvedžić;Thomas McCabe
Structural Chemistry 2012 Volume 23( Issue 2) pp:315-323
Publication Date(Web):2012 April
DOI:10.1007/s11224-011-9861-5
We present the crystal structures of the chloride salts of the mono-guanidinium 1 (–CH2CH2– linker) and the bis-isouronium 2 (–O– linker) that have been resolved by us indicating that these compounds are diprotonated in the solid state as informed by the counterions positions. To determine the pKa values of these compounds as well as those of their analogues 3 (mono-2-aminoimidazolinium with a –CH2CH2– linker) and 4 (mono-guanidinium with a –O– linker), the corresponding UV–Vis titrations were carried out. Thus, in aqueous solution compounds 1, 3 and 4 were present as mono-cationic species while derivative 2 was a bis-cation.
Co-reporter:Sergio Lopez, Geoffrey P. Margison, R. Stanley McElhinney, Alessandra Cordeiro, T. Brian H. McMurry, Isabel Rozas
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 5) pp:1658-1665
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmc.2011.01.038
Searching for a novel family of inactivators of the human DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) which is known to bind to the DNA minor groove, we have computationally modelled and synthesised two series of 2-amino-6-aryloxy-5-nitropyrimidines with morpholino or aminodiaryl substituents (potential minor groove binders) at the 4-position. Synthesis of these compounds was achieved by successive substitution of each of the two Cl atoms of 2-amino-4,6-dichloro-5-nitropyrimidine by the corresponding amino and aryloxy derivatives. Biochemical evaluation of these compounds as MGMT inactivators showed poor activities, but in general the 4-bromothenyloxy derivatives showed better inactivation than the benzyloxy versions. DNA binding assessment was not possible due to insolubility problems.
Co-reporter:Alessra Cordeiro;Julian Shaw;John O'Brien;Ferno Blanco
European Journal of Organic Chemistry 2011 Volume 2011( Issue 8) pp:1504-1513
Publication Date(Web):
DOI:10.1002/ejoc.201001459
Abstract
A revision of the literature on the nitration of tetrahydroquinolines yielded a number of inconsistencies. Thus, we have carried out a thorough study on the nitration of tetrahydroquinoline and several of its N-protected derivatives both experimentally and at theoretical level. Usually, nitration is carried out in acidic conditions and, thus, tetrahydroquinoline would be N-protonated; however, if the amino group is protected, the neutral system will be the one undergoing nitration. Different protecting groups have been explored varying, not only electronic and steric effects, but also deprotection conditions. Additionally, different reagents and reaction conditions have been investigated. From this study we have been able to achieve total regioselectivity for nitration at the 6-position. A very detailed NMR study has been carried out to unequivocally characterise the four nitro isomers. In parallel, a computational study has been performed that is in agreement with the experimental results obtained. With this purpose, all the σ complexes of the four nitro isomers neutral and N-protonated have been optimized both in gas and water condensed phases by using the B3LYP/6-31++G** level of computation.
Co-reporter:Fernando Blanco, Brendan Kelly, Ibon Alkorta, Isabel Rozas, José Elguero
Chemical Physics Letters 2011 Volume 511(1–3) pp:129-134
Publication Date(Web):26 July 2011
DOI:10.1016/j.cplett.2011.06.012
Abstract
We have theoretically studied, in gas phase and using PCM-water solvation, the complexes established by the biologically relevant guanidinium cation and simple aromatic systems (benzene, naphthalene and pyridine). In gas phase only hydrogen bonded complexes were obtained, whereas using PCM-water different cation–π complexes for the three aromatic systems were found. The interactions established within these complexes have been analyzed by means of the atoms in molecules and natural bond orbital approaches. Finally, experimental evidence of the cation–π interactions created by guanidinium was found in the crystal structure of N-(5-methylpyridin-2-yl)guanidinium chloride, which was also theoretically analyzed.
Co-reporter:Elena Diez-Cecilia, Brendan Kelly, Isabel Rozas
Tetrahedron Letters 2011 Volume 52(Issue 50) pp:6702-6704
Publication Date(Web):14 December 2011
DOI:10.1016/j.tetlet.2011.09.112
Single-step reduction of aryl nitro and carbonyl groups to the corresponding synthetically useful alkyl-anilines occurs with excellent yields by treatment with hydrazine and a base in a solvent-free reaction. The method has been applied to a broad range of compounds with different properties. Investigations into the mechanism of the reduction reveal that each group is reduced independently. A mechanism is proposed for this novel reduction of aromatic nitro groups.
Co-reporter:Daniel H. O’Donovan, Isabel Rozas
Tetrahedron Letters 2011 Volume 52(Issue 32) pp:4117-4119
Publication Date(Web):10 August 2011
DOI:10.1016/j.tetlet.2011.05.132
We present a new and concise method for the preparation of asymmetrical N,N′-disubstituted guanidines starting from thiourea via the reaction of N-Boc-protected N′-alkyl/aryl substituted thioureas with an amine in the presence of mercury(II) chloride and triethylamine.
Co-reporter:Brendan Kelly, Daniel H. O'Donovan, John O'Brien, Thomas McCabe, Fernando Blanco, and Isabel Rozas
The Journal of Organic Chemistry 2011 Volume 76(Issue 22) pp:9216-9227
Publication Date(Web):October 6, 2011
DOI:10.1021/jo200954c
The synthesis and conformational analysis of a series of pyridin-2-yl guanidine derivatives using NMR, X-ray crystallography, and B3LYP/6-31+G** theoretical studies are reported. A remarkable difference was observed in the 1H NMR spectra of the guanidinium salts as compared with their N,N′-di-Boc protected and neutral analogues. This difference corresponds to a 180° change in the dihedral angle between the guanidine/ium moiety and the pyridine ring in the salts as compared to the Boc-protected derivatives, a conclusion that was supported by theoretical studies, X-ray data, and NMR analysis. Moreover, our data sustain the existence of two intramolecular hydrogen-bonding systems: (i) between the pyridine N1 atom and the guanidinium protons in the salts and (ii) within the tert-butyl carbamate groups of the Boc-protected derivatives. To verify that the observed conformational control arises from these intramolecular interactions, a new series of N-Boc-N′-propyl-substituted pyridin-2-yl guanidines were also prepared and studied.
Co-reporter:Padraic S. Nagle, Susan J. Quinn, John M. Kelly, Daniel H. O'Donovan, Amir R. Khan, Fernando Rodriguez, Binh Nguyen, W. David Wilson and Isabel Rozas
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 24) pp:5558-5567
Publication Date(Web):15 Oct 2010
DOI:10.1039/C0OB00428F
Biophysical studies have been carried out on a family of asymmetric guanidinium-based diaromatic derivatives to assess their potential as DNA minor groove binding agents. To experimentally assess the binding of these compounds to DNA, solution phase biophysical studies have been performed. Thus, surface plasmon resonance, UV-visible spectroscopy and circular and linear dichroism have been utilized to evaluate binding constants, stoichiometry and mode of binding. In addition, the thermodynamics of the binding process have been determined by using isothermal titration calorimetry. These results show significant DNA binding affinity that correlates with the expected 1:1 binding ratio usually observed for minor groove binders. Moreover, a simple computational approach has been devised to assess the potential as DNA binders of this family of compounds.
Co-reporter:Fernando Rodriguez ; Isabel Rozas ; Jorge E. Ortega ; Amaia M. Erdozain ; J. Javier Meana ;Luis F. Callado
Journal of Medicinal Chemistry 2009 Volume 52(Issue 3) pp:601-609
Publication Date(Web):January 9, 2009
DOI:10.1021/jm800838r
In this paper, we report the synthesis of three new 2-aminoimidazoline (compounds 4b, 5b, and 6b) and three new guanidine derivatives (compounds 7b, 8b, and 9b) as potential α2-adrenoceptor antagonists for the treatment of depression. Their pharmacological profile was evaluated in vitro in human brain tissue and compared to the potential antidepressant 1 and the agonists 2 and 3. All new substrates were evaluated by in vitro functional [35S]GTPγS binding assays in human prefrontal cortex to determine their agonistic or antagonistic activity. Compound 8b was found to be an antagonist in vitro and was subjected to in vivo microdialysis experiments in rats. Moreover, a new synthesis of the precursor amines for compounds 4b−9b is presented.
Co-reporter:Padraic S. Nagle ; Fernando Rodriguez ; Amila Kahvedžić ; Susan J. Quinn
Journal of Medicinal Chemistry 2009 Volume 52(Issue 22) pp:7113-7121
Publication Date(Web):October 29, 2009
DOI:10.1021/jm901017t
In this paper we report the synthesis of three families of new amidine-based aromatic derivatives as potential DNA minor groove binding agents for the treatment of cancer. The preparation of monoguanidine, mono-2-aminoimidazoline, and asymmetric diphenylguanidine/2-aminoimidazoline derivatives (compounds 1a−c to 8a−c) is presented. The affinity of these substrates and of a family of mono- and bis-isoureas (previously prepared in Rozas’ laboratory) for DNA was evaluated by means of DNA thermal denaturation measurements. In particular, compounds 2c, 5c, 6c, 7c, and 8c were found to bind strongly both to natural DNA and to adenine−thymine oligonucleotides, showing a preference for the adenine−thymine base pair sequences.
Co-reporter:Sergio Lopez, Thomas McCabe, R. Stanley McElhinney, T. Brian H. McMurry, Isabel Rozas
Tetrahedron Letters 2009 50(44) pp: 6022-6024
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.08.029
Co-reporter:Fernando Rodriguez ; Isabel Rozas ; Jorge E. Ortega ; Amaia M. Erdozain ; J. Javier Meana ;Luis F. Callado
Journal of Medicinal Chemistry 2008 Volume 51(Issue 11) pp:3304-3312
Publication Date(Web):May 7, 2008
DOI:10.1021/jm800026x
The preparation of a number of (bis)guanidine and (bis)2-aminoimidazoline derivatives as potential α2-adrenoceptor antagonists for the treatment of depression is presented. Human brain tissue was used to measure their affinity toward the α2-adrenoceptors in vitro. Compounds 6b, 8b, 9b, 10b, 15b, 17b, 18b, 20b, and 21b displayed a good affinity (pKi > 7) and were evaluated in in vitro functional [35S]GTPγS binding assays in human prefrontal cortex to determine their agonistic or antagonistic activity. Among these compounds, 17b and 20b showed the expected behavior for an antagonist and were subject to in vivo microdialysis experiments in rats. Significantly, these experiments confirmed the antagonistic properties of 17b and 20b, and therefore both compounds can be considered as potential antidepressants.
Co-reporter:Áine Goonan, Amila Kahvedžić, Fernando Rodriguez, Padraic S. Nagle, Thomas McCabe, Isabel Rozas, Amaia M. Erdozain, J. Javier Meana, Luis F. Callado
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 17) pp:8210-8217
Publication Date(Web):1 September 2008
DOI:10.1016/j.bmc.2008.07.033
The synthesis of nine new mono- and bis-O-phenylisouronium compounds (2, 6b–10b and 12b–14b) and their Boc-protected isourea precursors (2a, 6a–10a and 12a–14a) is described. The carbodiimide 4, which was formed, had been suggested as the reactive intermediate species and driving force of the reaction. All final substrates were tested as potential α2-ARs ligands in human brain tissue by means of radioligand binding experiments and were compared to the potential antidepressant 1, as well as other related guanidine containing derivatives.
Co-reporter:Isabel Rozas;Ibon Alkorta;José Elguero
Structural Chemistry 2008 Volume 19( Issue 6) pp:923-933
Publication Date(Web):2008/12/01
DOI:10.1007/s11224-008-9377-9
It is frequently said that hydrogen bonds (HBs) are enhanced by ionic interactions and in this article we intend to determine the degree at which this reinforcement happens. Considering our interest in the Guanidine(neutral)/Guanidinium(cation) system and its particular nature, all the possible 1:1 complexes with the Chloride(anion)/Hydrochloric acid(neutral) system have been studied at different levels of computation (B3LYP with 6-31+G* and TZVP basis sets; MP2 with 6-31+G*, 6-311++G** and aug-cc-pVDZ basis sets; CBS-QB3 and G3MP2). The nature of these interactions established in all the systems and, when possible, at all the levels of computation used in this study, has been analyzed using Atoms in Molecules and Natural Bond Orbital methodologies. By examining the interaction energy, the electron density at the bond critical bonds, the atomic energy, the charge transfer, the orbital energy, and the deformation energy we can conclude that HBs are stronger when the ionic interaction is stronger. Thus, both interactions do not work in an independent manner but one reinforces the other to different degrees depending on the nature of the charges present. Several correlations with the interaction energy have been found and a partition of the contributions of both the HB and ionic forces to the total interactions is proposed.
Co-reporter:Isabel Rozas
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 22) pp:2782-2790
Publication Date(Web):23 Feb 2007
DOI:10.1039/B618225A
The nature of hydrogen bond interactions (HB) is still today the subject of many discussions. We present an overview of computational methods and parameters (interaction energy, HB distance and radii, electron density topological parameters or orbital energies) required for an accurate description of HB systems. As well, we present the different correlations that have been found between these descriptors providing a global view of HB interactions. A synopsis of the different HBs reported in terms of their strength was presented. Considering the definitions of covalent and ionic bonds, HB interactions could occur between these two extremes. Thus, we look into some of the very strong HBs (LBHB, CAHB, RAHB) and some of the weak HBs (weak donors: C–H or weak acceptors: π systems). Subsequently, aspects such as cooperativity or solvation are examined. Finally, we present a study on multiple “parallel” and “bifurcated” HB systems. Our results indicate that HB pattern and electron density determine the strength of the interaction and that “parallel” HB interactions are more stable than the “bifurcated” ones.
Co-reporter:Gemma K. Kinsella, Fernando Rodriguez, Graeme W. Watson, Isabel Rozas
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 8) pp:2850-2855
Publication Date(Web):15 April 2007
DOI:10.1016/j.bmc.2007.02.026
In order to design any new potential drug, it is crucial to know their corresponding pKa since their protonation state will be critical in the ligand–receptor interaction and it will play an essential role in their pharmacokinetic profile. Several authors have developed approaches for the computational determination of pKa which involve the use of a thermodynamic cycle relating pKa to the gas-phase proton basicity via the solvation energies of the products and the reactants. Such methods are very dependent on the solvation model used and the nature of the system. The theoretical pKa of a number of agonists and antagonists of the α1A-adrenoceptor has been computed and the performance of this approach has been tested through comparison with the available and/or measured experimental pKa values.The figure illustrates some of the α1-adrenoceptor ligands object of this study. Their pKa has been theoretically computed at B3LYP/6-31G∗ level and the experimental pKa for some of them has been measured by us.
Co-reporter:Jonathan Corcoran, Fernando Rodriguez, Isabel Rozas, J. Javier Meana, Luis F. Callado
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 21) pp:6009-6012
Publication Date(Web):1 November 2007
DOI:10.1016/j.bmcl.2007.07.093
Continuing with our search of aliphatic dicationic derivatives as I2-IBS ligands and looking at Amiloride, a known ligand of I2-IBS, we have incorporated the guanidinocarbonyl moiety into our aliphatic compounds with the intention of improving the binding to I2-IBS. Thus, we present the different approaches to the preparation and pharmacological evaluation (in human brain tissue) as I2-IBS ligands of a new series of aliphatic derivatives incorporating the guanidinocarbonyl group and with different chain length (n = 8–12, and 14 methylene groups).
Co-reporter:Ibon Alkorta;José Elguero
Theoretical Chemistry Accounts 2007 Volume 118( Issue 3) pp:533-539
Publication Date(Web):2007 September
DOI:10.1007/s00214-007-0340-4
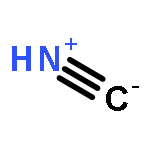
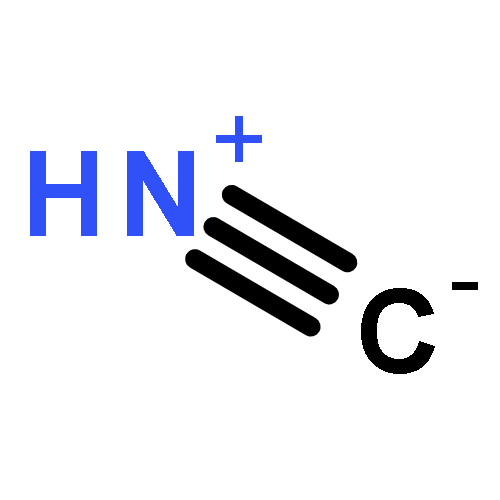
A theoretical study of the possible protonation sites of simple molecules formed by C, N, Si, P, B and Al that present a triple bond between those atoms has been carried out. The calculations performed include MP2 and CCSD(T) methods with the aug-cc-pVTZ basis set. The nature of the protonated species has been analyzed with the Atoms In Molecules methodology.
Co-reporter:Gemma K. Kinsella, Graeme W. Watson, Isabel Rozas
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 5) pp:1580-1587
Publication Date(Web):1 March 2006
DOI:10.1016/j.bmc.2005.10.007
A systematic study has been performed of the proton affinity of a large family of agonists and antagonists of the α1-adrenoceptor at the B3LYP/6-31G* level of theory. After a conformational search, all the N atoms were considered as protonation sites and protonation energy values were determined. The inclusion of solvation by means of the Onsager model yielded stabilization in the proton affinity values obtained. In addition, a good correlation was found between the proton affinity values corresponding to the first protonation in gas phase of some of the compounds and their corresponding experimental affinity constants Ki for the α1A adrenergic receptor.The figure illustrates some of the α1-adrenoceptor ligand objects of this study. Their proton affinity have been theoretically computed at the B3LYP/6-31G* level.
Co-reporter:Isabel Rozas, Ibon Alkorta and José Elguero
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 2) pp:366-371
Publication Date(Web):15 Dec 2004
DOI:10.1039/B415768K
The complexes formed by the double interaction established between RNA bases and guanidinium and formate ions, as a model for the interacting groups of arginine and glutamic or aspartic amino acid side chains, have been theoretically studied. A density functional theory method (B3LYP/6-31 + G**) has been used for this study. The range of interaction energies obtained allowed for a distinction between bidentate and bifurcate hydrogen bond interactions. The analysis of the electron density and the natural bond orbital analysis shows that these complexes are bound by double hydrogen bonds established between the donor and acceptor groups of guanidinium and formate respectively and those of the RNA bases. Comparisons are made with the results obtained in some previous theoretical and experimental studies.
Co-reporter:Christophe Dardonville, Isabel Rozas, Luis F Callado, J.Javier Meana
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 5) pp:1525-1533
Publication Date(Web):May 2002
DOI:10.1016/S0968-0896(01)00420-5
Two families of compounds with affinity towards the I2 imidazoline binding sites are reported. The first is a family of compounds structurally related to agmatine with two guanidine or 2-aminoimidazoline groups at each end of an aliphatic chain of six, eight, nine or 12 methylene groups. Second, and following the model of clonidine, we propose another family of compounds also with two guanidine or 2-aminoimidazoline groups at each end of a chain consisting of two phenyl rings connected by groups such as CH2, CO, NH and SO2. The affinity of the compounds towards the I2 imidazoline binding sites was then evaluated in human brain tissues. In order to determine their pharmacological selectivity versus α2-adrenoceptors, the affinity for these receptors was also evaluated for the compounds with the highest affinities at I2 imidazoline binding sites. The results obtained show that many of the compounds exhibit a considerable affinity towards the I2 imidazoline binding sites. The aliphatic derivatives, in particular, present a very interesting selectivity for the I2 imidazoline binding sites versus the α2 adrenoceptors. To better understand these findings, mono-guanidinium analogues of the aliphatic derivatives were synthesised and tested showing poor affinity for I2 imidazoline binding sites. The importance of these results lies in the novelty of the chemical structures studied (dicationic aliphatic compounds particularly) because they are significantly different to those of the I2 imidazoline binding site ligands reported to date.Graphic
Co-reporter:Padraic S. Nagle, Susan J. Quinn, John M. Kelly, Daniel H. O'Donovan, Amir R. Khan, Fernando Rodriguez, Binh Nguyen, W. David Wilson and Isabel Rozas
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 24) pp:NaN5567-5567
Publication Date(Web):2010/10/15
DOI:10.1039/C0OB00428F
Biophysical studies have been carried out on a family of asymmetric guanidinium-based diaromatic derivatives to assess their potential as DNA minor groove binding agents. To experimentally assess the binding of these compounds to DNA, solution phase biophysical studies have been performed. Thus, surface plasmon resonance, UV-visible spectroscopy and circular and linear dichroism have been utilized to evaluate binding constants, stoichiometry and mode of binding. In addition, the thermodynamics of the binding process have been determined by using isothermal titration calorimetry. These results show significant DNA binding affinity that correlates with the expected 1:1 binding ratio usually observed for minor groove binders. Moreover, a simple computational approach has been devised to assess the potential as DNA binders of this family of compounds.
Co-reporter:Isabel Rozas
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 22) pp:NaN2790-2790
Publication Date(Web):2007/02/23
DOI:10.1039/B618225A
The nature of hydrogen bond interactions (HB) is still today the subject of many discussions. We present an overview of computational methods and parameters (interaction energy, HB distance and radii, electron density topological parameters or orbital energies) required for an accurate description of HB systems. As well, we present the different correlations that have been found between these descriptors providing a global view of HB interactions. A synopsis of the different HBs reported in terms of their strength was presented. Considering the definitions of covalent and ionic bonds, HB interactions could occur between these two extremes. Thus, we look into some of the very strong HBs (LBHB, CAHB, RAHB) and some of the weak HBs (weak donors: C–H or weak acceptors: π systems). Subsequently, aspects such as cooperativity or solvation are examined. Finally, we present a study on multiple “parallel” and “bifurcated” HB systems. Our results indicate that HB pattern and electron density determine the strength of the interaction and that “parallel” HB interactions are more stable than the “bifurcated” ones.


![Guanidine, [4-(1-piperidinyl)phenyl]-](http://img.cochemist.com/ccimg/870800/870782-65-3.png)
![Guanidine, [4-(1-piperidinyl)phenyl]-](http://img.cochemist.com/ccimg/870800/870782-65-3_b.png)
![Methanone, bis[4-[(4,5-dihydro-1H-imidazol-2-yl)amino]phenyl]-](http://img.cochemist.com/ccimg/450400/450381-83-6.png)
![Methanone, bis[4-[(4,5-dihydro-1H-imidazol-2-yl)amino]phenyl]-](http://img.cochemist.com/ccimg/450400/450381-83-6_b.png)
![Guanidine, [4-(phenylmethyl)phenyl]-](http://img.cochemist.com/ccimg/666800/666723-37-1.png)
![Guanidine, [4-(phenylmethyl)phenyl]-](http://img.cochemist.com/ccimg/666800/666723-37-1_b.png)


![CARBAMODITHIOIC ACID, [2-(ACETYLOXY)ETHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/50800/50782-67-7.png)
![CARBAMODITHIOIC ACID, [2-(ACETYLOXY)ETHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/50800/50782-67-7_b.png)

![Imidazo[1,2-c]quinazolin-5(3H)-one,2,6-dihydro-2-[[4-(2-methoxyphenyl)-1-piperazinyl]methyl]-](http://img.cochemist.com/ccimg/146000/145938-37-0.png)
![Imidazo[1,2-c]quinazolin-5(3H)-one,2,6-dihydro-2-[[4-(2-methoxyphenyl)-1-piperazinyl]methyl]-](http://img.cochemist.com/ccimg/146000/145938-37-0_b.png)
![2-[4-(dimethylamino)phenyl]guanidine](http://img.cochemist.com/ccimg/67500/67453-82-1.png)
![2-[4-(dimethylamino)phenyl]guanidine](http://img.cochemist.com/ccimg/67500/67453-82-1_b.png)



![N-BENZO[1,3]DIOXOL-5-YL-GUANIDINE](http://img.cochemist.com/ccimg/366500/366498-47-7.png)
![N-BENZO[1,3]DIOXOL-5-YL-GUANIDINE](http://img.cochemist.com/ccimg/366500/366498-47-7_b.png)


![3-[3-[4-[4-Fluoro-2-(2,2,2-trifluoroethoxy)phenyl]piperazin-1-yl]propyl]-5-methylpyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/186400/186386-21-0.png)
![3-[3-[4-[4-Fluoro-2-(2,2,2-trifluoroethoxy)phenyl]piperazin-1-yl]propyl]-5-methylpyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/186400/186386-21-0_b.png)
![4H-1-Benzopyran-4-one,3-[[[2-(2,6-dimethoxyphenoxy)ethyl]amino]methyl]-2,3-dihydro-2-phenyl-](http://img.cochemist.com/ccimg/146300/146234-67-5.png)
![4H-1-Benzopyran-4-one,3-[[[2-(2,6-dimethoxyphenoxy)ethyl]amino]methyl]-2,3-dihydro-2-phenyl-](http://img.cochemist.com/ccimg/146300/146234-67-5_b.png)
![Carbamic acid, [4-[(4-aminophenyl)methyl]phenyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/135700/135680-03-4.png)
![Carbamic acid, [4-[(4-aminophenyl)methyl]phenyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/135700/135680-03-4_b.png)
![1H-Imidazol-2-amine, 4,5-dihydro-N-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/677400/677343-27-0.png)
![1H-Imidazol-2-amine, 4,5-dihydro-N-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/677400/677343-27-0_b.png)


![1H-Imidazol-2-amine, 4,5-dihydro-N-[4-(phenylthio)phenyl]-](http://img.cochemist.com/ccimg/677400/677343-39-4.png)
![1H-Imidazol-2-amine, 4,5-dihydro-N-[4-(phenylthio)phenyl]-](http://img.cochemist.com/ccimg/677400/677343-39-4_b.png)


![[4-[4-(cyanoamino)phenoxy]phenyl]cyanamide](http://img.cochemist.com/ccimg/30100/30070-40-7.png)
![[4-[4-(cyanoamino)phenoxy]phenyl]cyanamide](http://img.cochemist.com/ccimg/30100/30070-40-7_b.png)
![2-[4-[[4-(DIAMINOMETHYLIDENEAMINO)PHENYL]METHYL]PHENYL]GUANIDINE](http://img.cochemist.com/ccimg/7800/7773-73-1.png)
![2-[4-[[4-(DIAMINOMETHYLIDENEAMINO)PHENYL]METHYL]PHENYL]GUANIDINE](http://img.cochemist.com/ccimg/7800/7773-73-1_b.png)





![2-[(1,3-Benzodioxol-5-ylmethyl)amino]-nicotinonitrile](http://img.cochemist.com/ccimg/890100/890094-35-6.png)
![2-[(1,3-Benzodioxol-5-ylmethyl)amino]-nicotinonitrile](http://img.cochemist.com/ccimg/890100/890094-35-6_b.png)








![2,4(1H,3H)-Pyrimidinedione,6-[[3-[4-(2-methoxyphenyl)-1-piperazinyl]propyl]amino]-1,3,5-trimethyl-](http://img.cochemist.com/ccimg/34700/34661-85-3.png)
![2,4(1H,3H)-Pyrimidinedione,6-[[3-[4-(2-methoxyphenyl)-1-piperazinyl]propyl]amino]-1,3,5-trimethyl-](http://img.cochemist.com/ccimg/34700/34661-85-3_b.png)







![1,4-Benzodioxin-2-methanamine,N-[2-(2,6-dimethoxyphenoxy)ethyl]-2,3-dihydro-](http://img.cochemist.com/ccimg/700/613-67-2.png)
![1,4-Benzodioxin-2-methanamine,N-[2-(2,6-dimethoxyphenoxy)ethyl]-2,3-dihydro-](http://img.cochemist.com/ccimg/700/613-67-2_b.png)


![Carbamic acid,[(propylamino)thioxomethyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/145100/145012-93-7.png)
![Carbamic acid,[(propylamino)thioxomethyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/145100/145012-93-7_b.png)















![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)




![[4-(4-AMINO-6,7-DIMETHOXY-QUINAZOLIN-2- YL)PIPERAZIN-1-YL]-(2,5-DIOXABI CYCLO[4.4.0]DECA-6,8,10-TRIEN-4-YL)METHANONE](http://img.cochemist.com/ccimg/74200/74191-85-8.png)
![[4-(4-AMINO-6,7-DIMETHOXY-QUINAZOLIN-2- YL)PIPERAZIN-1-YL]-(2,5-DIOXABI CYCLO[4.4.0]DECA-6,8,10-TRIEN-4-YL)METHANONE](http://img.cochemist.com/ccimg/74200/74191-85-8_b.png)


![[4-[[4-(CYANOAMINO)PHENYL]METHYL]PHENYL]CYANAMIDE](http://img.cochemist.com/ccimg/30100/30070-39-4.png)
![[4-[[4-(CYANOAMINO)PHENYL]METHYL]PHENYL]CYANAMIDE](http://img.cochemist.com/ccimg/30100/30070-39-4_b.png)
![Benzeneethanol, β-[(2-methylphenyl)amino]-](/data/chemimg/132000/851660-17-8.png)
![Benzeneethanol, β-[(2-methylphenyl)amino]-](/data/chemimg/132000/851660-17-8_b.png)




![[2,2'-Binaphthalene]-1,1'-diol, (1R)-](http://img.cochemist.com/ccimg/122600/122531-87-7.png)
![[2,2'-Binaphthalene]-1,1'-diol, (1R)-](http://img.cochemist.com/ccimg/122600/122531-87-7_b.png)

![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)

![(E)-amino[(2-chlorobenzyl)imino]methanaminium chloride](http://img.cochemist.com/ccimg/1200/1128-91-2.png)
![(E)-amino[(2-chlorobenzyl)imino]methanaminium chloride](http://img.cochemist.com/ccimg/1200/1128-91-2_b.png)
![5-[(2R)-2-[2-(2-ETHOXYPHENOXY)ETHYLAMINO]PROPYL]-2-METHOXYBENZENESULFONAMIDE](/data/chemimg/307000/106133-20-4.png)
![5-[(2R)-2-[2-(2-ETHOXYPHENOXY)ETHYLAMINO]PROPYL]-2-METHOXYBENZENESULFONAMIDE](/data/chemimg/307000/106133-20-4_b.png)