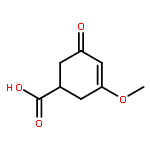
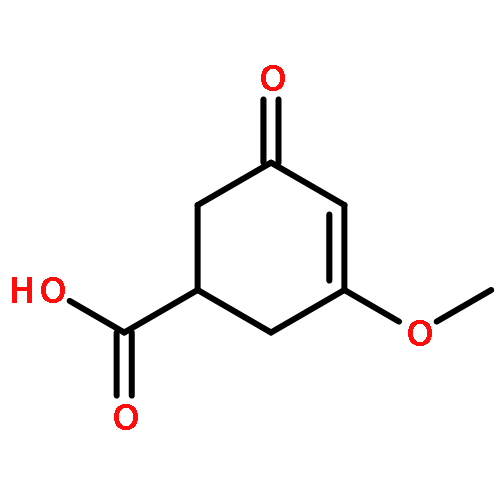
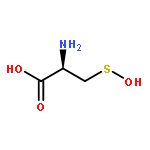
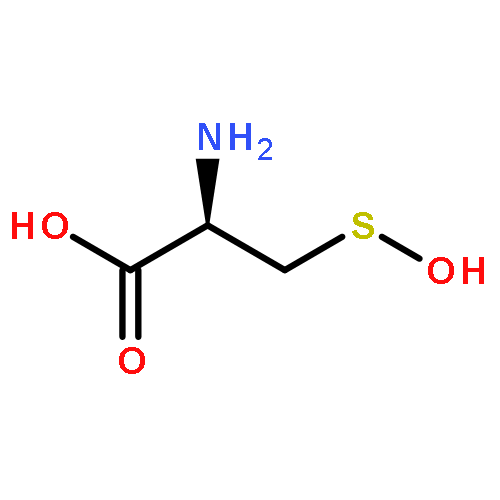
Co-reporter:Vinayak Gupta, Jing Yang, Daniel C. Liebler, and Kate S. Carroll
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5588-5588
Publication Date(Web):March 29, 2017
DOI:10.1021/jacs.7b01791
Targeted covalent inhibitors have emerged as a powerful approach in the drug discovery pipeline. Key to this process is the identification of signaling pathways (or receptors) specific to (or overexpressed in) disease cells. In this context, fragment-based ligand discovery (FBLD) has significantly expanded our view of the ligandable proteome and affords tool compounds for biological inquiry. To date, such covalent ligand discovery has almost exclusively employed cysteine-reactive small-molecule fragments. However, functional cysteine residues in proteins are often redox-sensitive and can undergo oxidation in cells. Such reactions are particularly relevant in diseases, like cancer, which are linked to excessive production of reactive oxygen species. Once oxidized, the sulfur atom of cysteine is much less reactive toward electrophilic groups used in the traditional FBLD paradigm. To address this limitation, we recently developed a novel library of diverse carbon-based nucleophile fragments that react selectively with cysteine sulfenic acid formed in proteins via oxidation or hydrolysis reactions. Here, we report analysis of sulfenic acid-reactive C-nucleophile fragments screened against a colon cancer cell proteome. Covalent ligands were identified for >1280 S-sulfenylated cysteines present in “druggable” proteins and orphan targets, revealing disparate reactivity profiles and target preferences. Among the unique ligand–protein interactions identified was that of a pyrrolidinedione nucleophile that reacted preferentially with protein tyrosine phosphatases. Fragment-based covalent ligand discovery with C-nucleophiles affords an expansive snapshot of the ligandable “redoxome” with significant implications for covalent inhibitor pharmacology and also affords new chemical tools to investigate redox-regulation of protein function.
Co-reporter:Vinayak Gupta and Kate S. Carroll
Chemical Science 2016 vol. 7(Issue 1) pp:400-415
Publication Date(Web):07 Oct 2015
DOI:10.1039/C5SC02569A
Oxidation of a protein cysteine thiol to sulfenic acid, termed S-sulfenylation, is a reversible post-translational modification that plays a crucial role in regulating protein function and is correlated with disease states. The majority of reaction-based small molecule and immunochemical probes used for detecting sulfenic acids are based on the 5,5-dimethyl-1,3-cyclohexanedione (dimedone) scaffold, which is selective, but suffers from low reactivity. In addition, mechanistic details and features that diminish or enhance nucleophile reactivity remain largely unknown. A significant hurdle to resolving the aforementioned issues has been the chemically unstable nature of small-molecule sulfenic acid models. Herein, we report a facile mass spectrometry-based assay and repurposed dipeptide-based model to screen a library of cyclic C-nucleophiles for reactivity with sulfenic acid under aqueous conditions. Observed rate constants for ∼100 cyclic C-nucleophiles were obtained and, from this collection, we have identified novel compounds with more than 200-fold enhanced reactivity, as compared to dimedone. The increase in reactivity and retention of selectivity of these C-nucleophiles were validated in secondary assays, including a protein model for sulfenic acid. Together, this work represents a significant step toward developing new chemical reporters for detecting protein S-sulfenylation with superior kinetic resolution. The enhanced rates and varied composition of the C-nucleophiles should enable more comprehensive analyses of the sulfenome and serve as the foundation for reversible or irreversible nucleophilic covalent inhibitors that target oxidized cysteine residues in therapeutically important proteins.
Co-reporter:Vinayak Gupta and Kate S. Carroll
Chemical Communications 2016 vol. 52(Issue 16) pp:3414-3417
Publication Date(Web):25 Jan 2016
DOI:10.1039/C6CC00228E
Concerns about off-target effects has motivated the development of reversible covalent inhibition strategies for targeting cysteine. However, such strategies have not been reported for the unique cysteine oxoform, sulfenic acid. Herein, we have designed and identified linear C-nucleophiles that react selectively with cysteine sulfenic acid. The resulting thioether adducts exhibit reversibility ranging from minutes to days under reducing conditions, showing the feasibility of tuning C-nucleophile reactivity across a wide range of time scales.
Co-reporter:Prakash B. Palde, Ashima Bhaskar, Laura E. Pedró Rosa, Franck Madoux, Peter Chase, Vinayak Gupta, Timothy Spicer, Louis Scampavia, Amit Singh, and Kate S. Carroll
ACS Chemical Biology 2016 Volume 11(Issue 1) pp:172
Publication Date(Web):November 2, 2015
DOI:10.1021/acschembio.5b00517
Development of effective therapies to eradicate persistent, slowly replicating M. tuberculosis (Mtb) represents a significant challenge to controlling the global TB epidemic. To develop such therapies, it is imperative to translate information from metabolome and proteome adaptations of persistent Mtb into the drug discovery screening platforms. To this end, reductive sulfur metabolism is genetically and pharmacologically implicated in survival, pathogenesis, and redox homeostasis of persistent Mtb. Therefore, inhibitors of this pathway are expected to serve as powerful tools in its preclinical and clinical validation as a therapeutic target for eradicating persisters. Here, we establish a first functional HTS platform for identification of APS reductase (APSR) inhibitors, a critical enzyme in the assimilation of sulfate for the biosynthesis of cysteine and other essential sulfur-containing molecules. Our HTS campaign involving 38 350 compounds led to the discovery of three distinct structural classes of APSR inhibitors. A class of bioactive compounds with known pharmacology displayed potent bactericidal activity in wild-type Mtb as well as MDR and XDR clinical isolates. Top compounds showed markedly diminished potency in a conditional ΔAPSR mutant, which could be restored by complementation with Mtb APSR. Furthermore, ITC studies on representative compounds provided evidence for direct engagement of the APSR target. Finally, potent APSR inhibitors significantly decreased the cellular levels of key reduced sulfur-containing metabolites and also induced an oxidative shift in mycothiol redox potential of live Mtb, thus providing functional validation of our screening data. In summary, we have identified first-in-class inhibitors of APSR that can serve as molecular probes in unraveling the links between Mtb persistence, antibiotic tolerance, and sulfate assimilation, in addition to their potential therapeutic value.
Co-reporter:Vinayak Gupta, Hanumantharao Paritala, and Kate S. Carroll
Bioconjugate Chemistry 2016 Volume 27(Issue 5) pp:1411
Publication Date(Web):April 28, 2016
DOI:10.1021/acs.bioconjchem.6b00181
The comparative reaction efficiencies of currently used nucleophilic and electrophilic probes toward cysteine sulfenic acid have been thoroughly evaluated in two different settings—(i) a small molecule dipeptide based model and (ii) a recombinant protein model. We further evaluated the stability of corresponding thioether and sulfoxide adducts under reducing conditions which are commonly encountered during proteomic protocols and in cell analysis. Powered by the development of new cyclic and linear C-nucleophiles, the unsurpassed efficiency in the capture of sulfenic acid under competitive conditions is achieved and thus holds great promise as highly potent tools for activity-based sulfenome profiling.
Co-reporter:Francisco J. Garcia and Kate S. Carroll
Molecular BioSystems 2016 vol. 12(Issue 6) pp:1790-1798
Publication Date(Web):08 Jan 2016
DOI:10.1039/C5MB00847F
Protein tyrosine phosphatases are crucial regulators of signal transduction and function as antagonists towards protein tyrosine kinases to control reversible tyrosine phosphorylation, thereby regulating fundamental physiological processes. Growing evidence has supported the notion that reversible oxidative inactivation of the catalytic cysteine residue in protein tyrosine phosphatases serves as an oxidative post-translational modification that regulates its activity to influence downstream signaling by promoting phosphorylation and induction of the signaling cascade. The oxidation of cysteine to the sulfenic acid is often transient and difficult to detect, thus making it problematic in understanding the role that this oxidative post-translational modification plays in redox-biology and pathogenesis. Several methods to detect cysteine oxidation in biological systems have been developed, though targeted approaches to directly detect oxidized phosphatases are still lacking. Herein we describe the development of a novel immunochemical approach to directly profile oxidized phosphatases. This immunochemical approach consists of an antibody designed to recognize the conserved sequence of the PTP active site (VHCDMDSAG) harboring the catalytic cysteine modified with dimedone (CDMD), a nucleophile that chemoselectively reacts with cysteine sulfenic acids to form a stable thioether adduct. Additionally, we provide biochemical and mass spectrometry workflows to be used in conjugation with this newly developed immunochemical approach to assist in the identification and quantification of basal and oxidized phosphatases.
Co-reporter:Jia Pan and Kate S. Carroll
Organic Letters 2015 Volume 17(Issue 24) pp:6014-6017
Publication Date(Web):December 7, 2015
DOI:10.1021/acs.orglett.5b02981
S-Sulfenylation is a post-translational modification with a crucial role in regulating protein function. However, its analysis has remained challenging due to the lack of facile sulfenic acid models. We report the first photocaged cysteine sulfenic acid with efficient photodeprotection and demonstrate its utility by generating sulfenic acid in a thiol peroxidase after illumination in vitro. These caged sulfoxides should be promising for site-specific incorporation of Cys sulfenic acid in living cells via genetic code expansion.
Co-reporter:Mauro Lo Conte, Jiusheng Lin, Mark A. Wilson, and Kate S. Carroll
ACS Chemical Biology 2015 Volume 10(Issue 8) pp:1825
Publication Date(Web):June 3, 2015
DOI:10.1021/acschembio.5b00124
Protein sulfinic acids are formed by the reaction of reactive oxygen species with protein thiols. Sulfinic acid formation has long been considered an irreversible state of oxidation and is associated with high cellular oxidative stress. Increasing evidence, however, indicates that cysteine is oxidized to sulfinic acid in cells to a greater extent, and is more controlled, than first thought. The discovery of sulfiredoxin has demonstrated that cysteine sulfinic acid can be reversed, pointing to a vast array of potential implications for redox biology. Identification of the site of protein sulfinylation is crucial in clarifying the physiological and pathological effects of post-translational modifications. Currently, the only methods for detection of sulfinic acids involve mass spectroscopy and the use of specific antibodies. However, these methodologies are not suitable for proteomic studies. Herein, we report the first probe for detection of protein sulfinylation, NO-Bio, which combines a C-nitroso warhead for rapid labeling of sulfinic acid with a biotin handle. Based on this new tool, we developed a selective two-step approach. In the first, a sulfhydryl-reactive compound is introduced to selectively block free cysteine residues. Thereafter, the sample is treated with NO-Bio to label sulfinic acids. This new technology represents a rapid, selective, and general technology for sulfinic acid detection in biological samples. As proof of our concept, we also evaluated protein sulfinylation levels in various human lung tumor tissue lysates. Our preliminary results suggest that cancer tissues generally have higher levels of sulfinylation in comparison to matched normal tissues. A new ability to monitor protein sulfinylation directly should greatly expand the impact of sulfinic acid as a post-translational modification.
Co-reporter:Prakash B. Palde
PNAS 2015 Volume 112 (Issue 26 ) pp:7960-7965
Publication Date(Web):2015-06-30
DOI:10.1073/pnas.1504376112
Cysteine residues in cytosolic proteins are maintained in their reduced state, but can undergo oxidation owing to posttranslational
modification during redox signaling or under conditions of oxidative stress. In large part, the reduction of oxidized protein
cysteines is mediated by a small 12-kDa thiol oxidoreductase, thioredoxin (Trx). Trx provides reducing equivalents for central
metabolic enzymes and is implicated in redox regulation of a wide number of target proteins, including transcription factors.
Despite its importance in cellular redox homeostasis, the precise mechanism by which Trx recognizes target proteins, especially
in the absence of any apparent signature binding sequence or motif, remains unknown. Knowledge of the forces associated with
the molecular recognition that governs Trx–protein interactions is fundamental to our understanding of target specificity.
To gain insight into Trx–target recognition, we have thermodynamically characterized the noncovalent interactions between
Trx and target proteins before S-S reduction using isothermal titration calorimetry (ITC). Our findings indicate that Trx
recognizes the oxidized form of its target proteins with exquisite selectivity, compared with their reduced counterparts.
Furthermore, we show that recognition is dependent on the conformational restriction inherent to oxidized targets. Significantly,
the thermodynamic signatures for multiple Trx targets reveal favorable entropic contributions as the major recognition force
dictating these protein–protein interactions. Taken together, our data afford significant new insight into the molecular forces
responsible for Trx–target recognition and should aid the design of new strategies for thiol oxidoreductase inhibition.
Co-reporter:Francisco J. Garcia, Kate S. Carroll
European Journal of Medicinal Chemistry 2014 Volume 88() pp:28-33
Publication Date(Web):17 December 2014
DOI:10.1016/j.ejmech.2014.06.040
•Designed and synthesized redox-based probes (RBPs) bearing an alkyne handle.•RBPs facilitate cellular investigation of oxidized PTPs in redox cell signaling.•RBPs detect increased levels of oxidized PTP1B in cells upon insulin stimulation.•RBPs trapped oxidized PTP1B and enhanced glucose uptake upon insulin stimulation.•Proof-of-concept for development of nucleophile-based inhibitors for oxidized PTPs.Reversible oxidation of protein tyrosine phosphatases (PTPs) has emerged as an important regulatory mechanism whereby reactive oxygen species (ROS) inactivates the PTP and promotes phosphorylation and induction of the signaling cascade. The lack of sensitive and robust methods to directly detect oxidized PTPs has made it difficult to understand the effects that PTP oxidative inactivation play in redox signaling. We report the use of redox-based probes to directly detect oxidized PTPs in a cellular context, which highlights the importance of direct approaches to assist in the study of physiological and pathophysiological PTP activity in redox regulation. We also demonstrate, as a proof-of-concept, that these redox-based probes serve as prototypes for the design and development of a new class of inhibitors for phosphatases. We envision a nucleophile reacting with the oxidized inactive catalytic cysteine to generate an irreversible thioether adduct which prevents the phosphatase from being reactivated and ultimately fortifies the signaling cascade. Our results reveal the potential of translation of our redox-based probes, which are used to understand redox cell circuitry and disease biology, to small-molecule nucleophile-based inhibitors, which may treat diseases associated with redox stress. This may have implications in the treatment of type 2 diabetes and cancer.
Co-reporter:Jia Pan
Biopolymers 2014 Volume 101( Issue 2) pp:165-172
Publication Date(Web):
DOI:10.1002/bip.22255
ABSTRACT
The oxidation of cysteine thiol side chains by hydrogen peroxide to afford protein sulfenyl modifications is an important mechanism in signal transduction. In addition, aberrant protein sulfenylation contributes to a range of human pathologies, including cancer. Efforts to elucidate the roles of protein sulfenylation in physiology and disease have been hampered by the lack of techniques to probe these modifications in native environments with molecular specificity. In this review, we trace the history of chemical and biological methods that have been developed to detect protein sulfenylation and illustrate how a recent cell-permeable chemical reporter, DYn-2, has been used to detect and identify intracellular targets of endogenous H2O2 during growth factor signaling, including the epidermal growth factor receptor. The array of new tools and methods discussed herein enables the discovery of new biological roles for cysteine sulfenylation in human health and disease. © 2013 Wiley Periodicals, Inc. Biopolymers 101: 165–172, 2014.
Co-reporter:Candice E. Paulsen and Kate S. Carroll
Chemical Reviews 2013 Volume 113(Issue 7) pp:4633
Publication Date(Web):March 20, 2013
DOI:10.1021/cr300163e
Co-reporter:Jia Pan and Kate S. Carroll
ACS Chemical Biology 2013 Volume 8(Issue 6) pp:1110
Publication Date(Web):April 4, 2013
DOI:10.1021/cb4001052
Hydrogen sulfide (H2S) has emerged as a new member of the gaseous transmitter family of signaling molecules and appears to play a regulatory role in the cardiovascular and nervous systems. Recent studies suggest that protein cysteine S-sulfhydration may function as a mechanism for transforming the H2S signal into a biological response. However, selective detection of S-sulfhydryl modifications is challenging since the persulfide group (RSSH) exhibits reactivity akin to other sulfur species, especially thiols. A modification of the biotin switch technique, using S-methyl methanethiosulfonate (MMTS) as an alkylating reagent, was recently used to identify a large number of proteins that may undergo S-sulfhydration, but the underlying mechanism of chemical detection was not fully explored. To address this key issue, we have developed a protein persulfide model and analogue of MMTS, S-4-bromobenzyl methanethiosulfonate (BBMTS). Using these new reagents, we investigated the chemistry in the modified biotin switch method and examined the reactivity of protein persulfides toward different electrophile/nucleophile species. Together, our data affirm the nucleophilic properties of the persulfide sulfane sulfur and afford new insights into protein S-sulfhydryl chemistry, which may be exploited in future detection strategies.
Co-reporter:Hanumantharao Paritala, Kate S. Carroll
Analytical Biochemistry 2013 Volume 440(Issue 1) pp:32-39
Publication Date(Web):1 September 2013
DOI:10.1016/j.ab.2013.05.007
Abstract
Mycobacterium tuberculosis (Mtb) adenosine 5′-phosphosulfate (APS) reductase (APR) catalyzes the first committed step in sulfate reduction for the biosynthesis of essential reduced sulfur-containing biomolecules, such as cysteine, and is essential for survival in the latent phase of tuberculosis (TB) infection. Despite the importance of APR to Mtb and other bacterial pathogens, current assay methods depend on the use of 35S-labeled APS or shunt adenosine 5′-monophosphate (AMP) to a coupled-enzyme system. Both methods are cumbersome and require the use of expensive reagents. Here, we report the development of a continuous spectrophotometric method for measuring APR activity by using novel sulfite-selective colorimetric or “off–on” fluorescent levulinate-based probes. Thus, the APR activity can be followed by monitoring the increase in absorbance or fluorescence of the resulting phenolate product. Using this assay, we determined Michaelis–Menten kinetic constants (Km, kcat, and kcat/Km) and the apparent inhibition constant (Ki) for adenosine 5′-diphosphate (ADP), which compared favorably with values obtained in the “gold standard” radioactive assay. The newly developed assay is robust and easy to perform with a simple spectrophotometer.
Co-reporter:Hanumantharao Paritala, Yuta Suzuki, Kate S. Carroll
Tetrahedron Letters 2013 Volume 54(Issue 14) pp:1869-1872
Publication Date(Web):3 April 2013
DOI:10.1016/j.tetlet.2013.01.109
Nucleosides are essential bio-molecules that participate in a wide array of biological processes involved in maintaining physiologic homeostasis. Recent efforts geared toward the synthesis of nucleoside analogues and development of nucleoside combinatorial libraries using solid phase synthesis have contributed invaluable information toward drug design and development. These studies have provided information concerning the structural requirements of substrate binding pockets of enzymes and evaluation of enzyme kinetics. However, the synthesis of nucleosides and its corresponding analogues remains a challenging and time consuming process. Herein, we report an efficient, microwave assisted solid phase coupling of nucleosides, combinatorial chemistry on the coupled nucleosides to generate small library, and mild cleavage conditions to release nucleoside derivatives from its solid support. We anticipate these findings will accelerate the development of synthetic methods or combinatorial library design of nucleoside analogues in similar settings.A methodology for an efficient, microwave assisted solid phase coupling of nucleosides, small library generation, and a mild cleavage condition to release nucleoside derivatives is developed.
Co-reporter:Devayani P. Bhave, Jiyoung A. Hong, Rebecca L. Keller, Carsten Krebs, and Kate S. Carroll
ACS Chemical Biology 2012 Volume 7(Issue 2) pp:306
Publication Date(Web):October 24, 2011
DOI:10.1021/cb200261n
Assimilatory sulfate reduction supplies prototrophic organisms with reduced sulfur that is required for the biosynthesis of all sulfur-containing metabolites, including cysteine and methionine. The reduction of sulfate requires its activation via an ATP-dependent activation to form adenosine-5′-phosphosulfate (APS). Depending on the species, APS can be reduced directly to sulfite by APS reductase (APR) or undergo a second phosphorylation to yield 3′-phosphoadenosine-5′-phosphosulfate (PAPS), the substrate for PAPS reductase (PAPR). These essential enzymes have no human homologue, rendering them attractive targets for the development of novel antibacterial drugs. APR and PAPR share sequence and structure homology as well as a common catalytic mechanism, but the enzymes are distinguished by two features, namely, the amino acid sequence of the phosphate-binding loop (P-loop) and an iron–sulfur cofactor in APRs. On the basis of the crystal structures of APR and PAPR, two P-loop residues are proposed to determine substrate specificity; however, this hypothesis has not been tested. In contrast to this prevailing view, we report here that the P-loop motif has a modest effect on substrate discrimination. Instead, by means of metalloprotein engineering, spectroscopic, and kinetic analyses, we demonstrate that the iron–sulfur cluster cofactor enhances APS reduction by nearly 1000-fold, thereby playing a pivotal role in substrate specificity and catalysis. These findings offer new insights into the evolution of this enzyme family and extend the known functions of protein-bound iron–sulfur clusters.
Co-reporter:Thu H. Truong and Kate S. Carroll
Biochemistry 2012 Volume 51(Issue 50) pp:
Publication Date(Web):November 27, 2012
DOI:10.1021/bi301441e
Epidermal growth factor receptor (EGFR) exemplifies the family of receptor tyrosine kinases that mediate numerous cellular processes, including growth, proliferation, and differentiation. Moreover, gene amplification and EGFR mutations have been identified in a number of human malignancies, making this receptor an important target for the development of anticancer drugs. In addition to ligand-dependent activation and concomitant tyrosine phosphorylation, EGFR stimulation results in the localized generation of H2O2 by NADPH-dependent oxidases. In turn, H2O2 functions as a secondary messenger to regulate intracellular signaling cascades, largely through the modification of specific cysteine residues within redox-sensitive protein targets, including Cys797 in the EGFR active site. In this review, we highlight recent advances in our understanding of the mechanisms that underlie redox regulation of EGFR signaling and how these discoveries may form the basis for the development of new therapeutic strategies for targeting this and other H2O2-modulated pathways.
Co-reporter:Dr. Mauro LoConte ;Dr. Kate S. Carroll
Angewandte Chemie 2012 Volume 124( Issue 26) pp:6608-6611
Publication Date(Web):
DOI:10.1002/ange.201201812
Co-reporter:Dr. Mauro LoConte ;Dr. Kate S. Carroll
Angewandte Chemie International Edition 2012 Volume 51( Issue 26) pp:6502-6505
Publication Date(Web):
DOI:10.1002/anie.201201812
Co-reporter:Stephen E Leonard, Kate S Carroll
Current Opinion in Chemical Biology 2011 Volume 15(Issue 1) pp:88-102
Publication Date(Web):February 2011
DOI:10.1016/j.cbpa.2010.11.012
Oxidative cysteine modifications have emerged as a central mechanism for dynamic post-translational regulation of all major protein classes and correlate with many disease states. Elucidating the precise roles of cysteine oxidation in physiology and pathology presents a major challenge. This article reviews the current, targeted proteomic strategies that are available to detect and quantify cysteine oxidation. A number of indirect methods have been developed to monitor changes in the redox state of cysteines, with the majority relying on the loss of reactivity with thiol-modifying reagents or restoration of labeling by reducing agents. Recent advances in chemical biology allow for the direct detection of specific cysteine oxoforms based on their distinct chemical attributes. In addition, new chemical reporters of cysteine oxidation have enabled in situ detection of labile modifications and improved proteomic analysis of redox-regulated proteins. Progress in the field of redox proteomics should advance our knowledge of regulatory mechanisms that involve oxidation of cysteine residues and lead to a better understanding of oxidative biochemistry in health and disease.
Co-reporter:Devayani P. Bhave, Wen-Ge Han, Samuel Pazicni, James E. Penner-Hahn, Kate S. Carroll, and Louis Noodleman
Inorganic Chemistry 2011 Volume 50(Issue 14) pp:6610-6625
Publication Date(Web):June 16, 2011
DOI:10.1021/ic200446c
Adenosine-5′-phosphosulfate reductase (APSR) is an iron–sulfur protein that catalyzes the reduction of adenosine-5′-phosphosulfate (APS) to sulfite. APSR coordinates to a [4Fe-4S] cluster via a conserved CC-X∼80-CXXC motif, and the cluster is essential for catalysis. Despite extensive functional, structural, and spectroscopic studies, the exact role of the iron–sulfur cluster in APS reduction remains unknown. To gain an understanding into the role of the cluster, density functional theory (DFT) analysis and extended X-ray fine structure spectroscopy (EXAFS) have been performed to reveal insights into the coordination, geometry, and electrostatics of the [4Fe-4S] cluster. X-ray absorption near-edge structure (XANES) data confirms that the cluster is in the [4Fe-4S]2+ state in both native and substrate-bound APSR while EXAFS data recorded at ∼0.1 Å resolution indicates that there is no significant change in the structure of the [4Fe-4S] cluster between the native and substrate-bound forms of the protein. On the other hand, DFT calculations provide an insight into the subtle differences between the geometry of the cluster in the native and APS-bound forms of APSR. A comparison between models with and without the tandem cysteine pair coordination of the cluster suggests a role for the unique coordination in facilitating a compact geometric structure and “fine-tuning” the electronic structure to prevent reduction of the cluster. Further, calculations using models in which residue Lys144 is mutated to Ala confirm the finding that Lys144 serves as a crucial link in the interactions involving the [4Fe-4S] cluster and APS.
Co-reporter:Thu H. Truong, Francisco J. Garcia, Young Ho Seo, Kate S. Carroll
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 17) pp:5015-5020
Publication Date(Web):1 September 2011
DOI:10.1016/j.bmcl.2011.04.115
We have developed an approach that allows relative quantification of protein sulfenic acids using a pair of light and heavy isotope labled probes, DAz-2 and d6-DAz-2. In conjunction with a new complementary acid-cleavable linker, Yn-ACL, we demonstrate that tagged peptides are successfully labeled, enriched, and fully characterized by LC–MS/MS analysis. Overall, this method can be applied to map sites of cysteine oxidation and compare protein sulfenylation in normal and disease states.
Co-reporter:Dr. Young Ho Seo;Dr. Kate S. Carroll
Angewandte Chemie 2011 Volume 123( Issue 6) pp:1378-1381
Publication Date(Web):
DOI:10.1002/ange.201007175
Co-reporter:Dr. Young Ho Seo;Dr. Kate S. Carroll
Angewandte Chemie International Edition 2011 Volume 50( Issue 6) pp:1342-1345
Publication Date(Web):
DOI:10.1002/anie.201007175
Co-reporter:Stephen E. Leonard;Francisco J. Garcia;Dr. David S. Goodsell;Dr. Kate S. Carroll
Angewandte Chemie International Edition 2011 Volume 50( Issue 19) pp:4423-4427
Publication Date(Web):
DOI:10.1002/anie.201007871
Co-reporter:Stephen E. Leonard;Francisco J. Garcia;Dr. David S. Goodsell;Dr. Kate S. Carroll
Angewandte Chemie 2011 Volume 123( Issue 19) pp:4515-4519
Publication Date(Web):
DOI:10.1002/ange.201007871
Co-reporter:Vinayak Gupta and Kate S. Carroll
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN415-415
Publication Date(Web):2015/10/07
DOI:10.1039/C5SC02569A
Oxidation of a protein cysteine thiol to sulfenic acid, termed S-sulfenylation, is a reversible post-translational modification that plays a crucial role in regulating protein function and is correlated with disease states. The majority of reaction-based small molecule and immunochemical probes used for detecting sulfenic acids are based on the 5,5-dimethyl-1,3-cyclohexanedione (dimedone) scaffold, which is selective, but suffers from low reactivity. In addition, mechanistic details and features that diminish or enhance nucleophile reactivity remain largely unknown. A significant hurdle to resolving the aforementioned issues has been the chemically unstable nature of small-molecule sulfenic acid models. Herein, we report a facile mass spectrometry-based assay and repurposed dipeptide-based model to screen a library of cyclic C-nucleophiles for reactivity with sulfenic acid under aqueous conditions. Observed rate constants for ∼100 cyclic C-nucleophiles were obtained and, from this collection, we have identified novel compounds with more than 200-fold enhanced reactivity, as compared to dimedone. The increase in reactivity and retention of selectivity of these C-nucleophiles were validated in secondary assays, including a protein model for sulfenic acid. Together, this work represents a significant step toward developing new chemical reporters for detecting protein S-sulfenylation with superior kinetic resolution. The enhanced rates and varied composition of the C-nucleophiles should enable more comprehensive analyses of the sulfenome and serve as the foundation for reversible or irreversible nucleophilic covalent inhibitors that target oxidized cysteine residues in therapeutically important proteins.
Co-reporter:Vinayak Gupta and Kate S. Carroll
Chemical Communications 2016 - vol. 52(Issue 16) pp:NaN3417-3417
Publication Date(Web):2016/01/25
DOI:10.1039/C6CC00228E
Concerns about off-target effects has motivated the development of reversible covalent inhibition strategies for targeting cysteine. However, such strategies have not been reported for the unique cysteine oxoform, sulfenic acid. Herein, we have designed and identified linear C-nucleophiles that react selectively with cysteine sulfenic acid. The resulting thioether adducts exhibit reversibility ranging from minutes to days under reducing conditions, showing the feasibility of tuning C-nucleophile reactivity across a wide range of time scales.
.jpg)




![ACETONITRILE, [(3-METHOXYPHENYL)THIO]-](http://img.cochemist.com/ccimg/79600/79506-65-3.png)
![ACETONITRILE, [(3-METHOXYPHENYL)THIO]-](http://img.cochemist.com/ccimg/79600/79506-65-3_b.png)
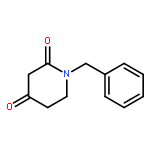
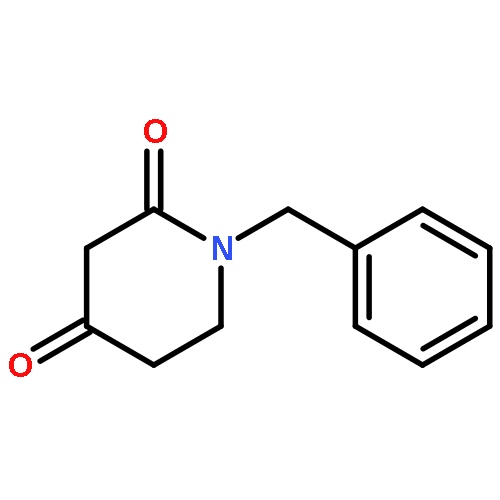
![L-Cysteine, S-[(2-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/61600/61543-53-1.png)
![L-Cysteine, S-[(2-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/61600/61543-53-1_b.png)

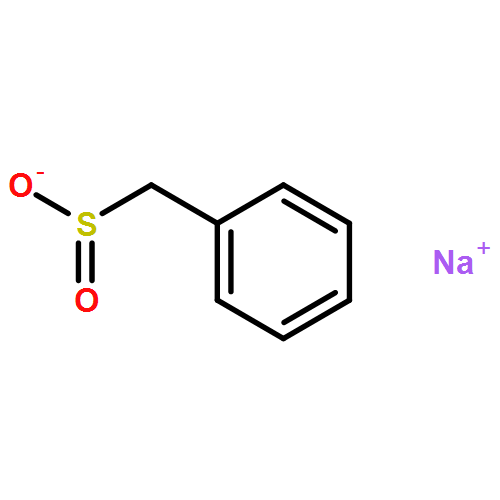
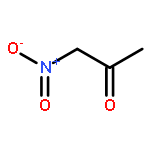
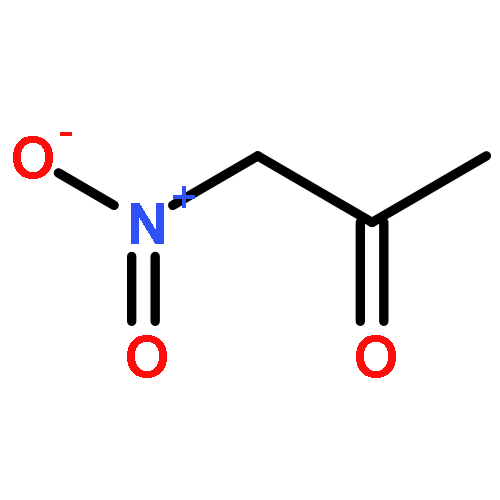
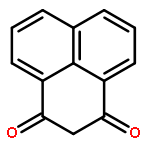
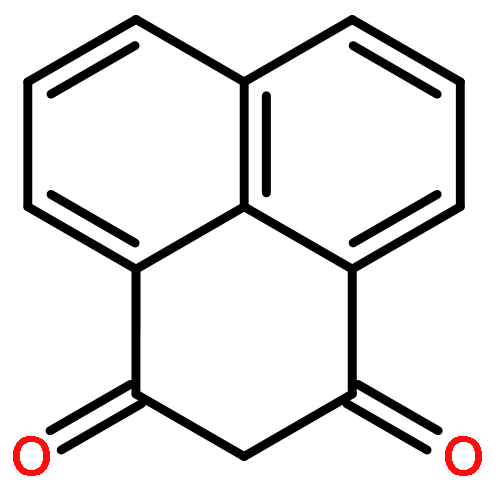
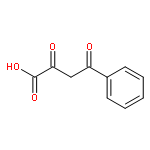








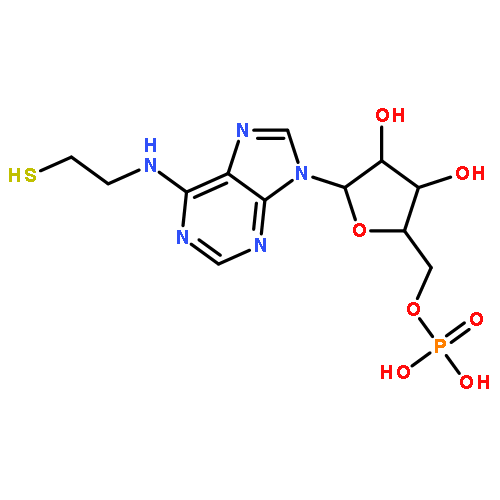
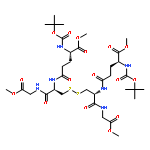
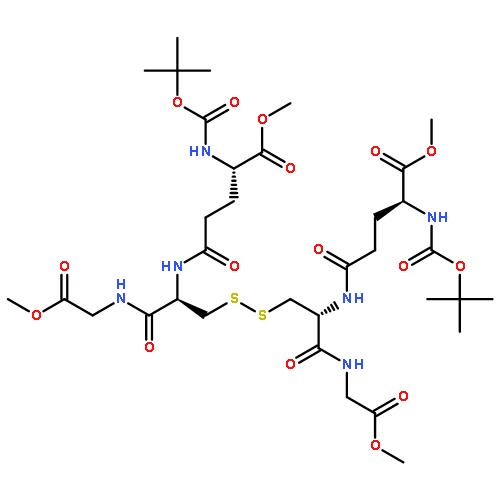
![HEXANOIC ACID, 6-[4-(DIMETHOXYMETHYL)PHENOXY]-, ETHYL ESTER](http://img.cochemist.com/ccimg/518400/518315-74-7.png)
![HEXANOIC ACID, 6-[4-(DIMETHOXYMETHYL)PHENOXY]-, ETHYL ESTER](http://img.cochemist.com/ccimg/518400/518315-74-7_b.png)


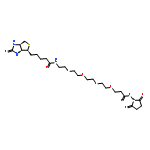

![Benzenesulfenic acid, 2-[(diethylamino)carbonyl]-](http://img.cochemist.com/ccimg/195000/194983-49-8.png)
![Benzenesulfenic acid, 2-[(diethylamino)carbonyl]-](http://img.cochemist.com/ccimg/195000/194983-49-8_b.png)




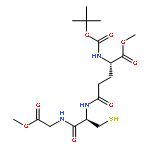
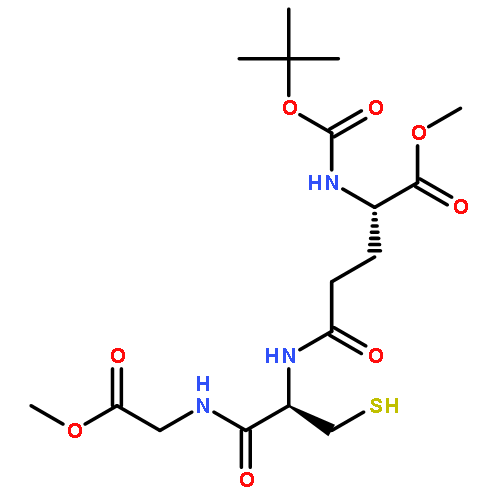
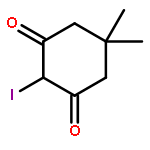
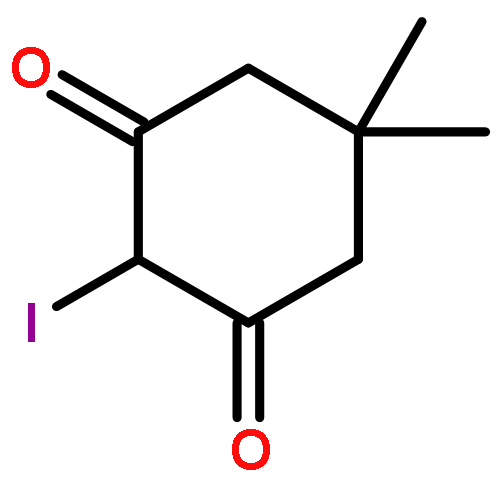
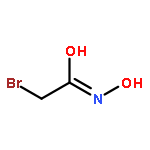
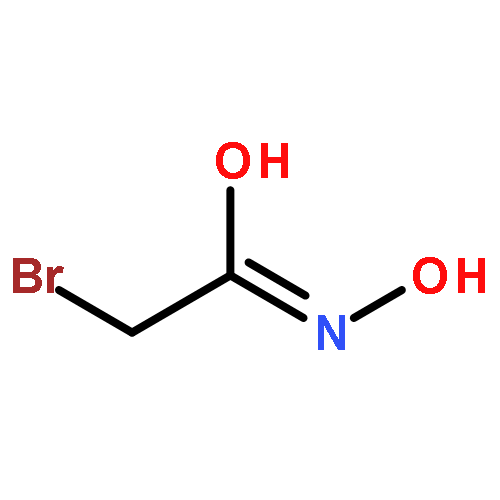
![6H-Pyrido[4,3-b]carbazolium,9-methoxy-2,5,11-trimethyl-, iodide (1:1)](http://img.cochemist.com/ccimg/93900/93841-50-0.png)
![6H-Pyrido[4,3-b]carbazolium,9-methoxy-2,5,11-trimethyl-, iodide (1:1)](http://img.cochemist.com/ccimg/93900/93841-50-0_b.png)
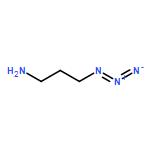
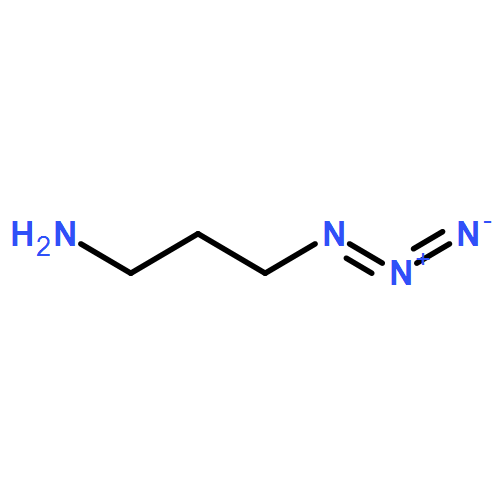
dimethyl-](http://img.cochemist.com/ccimg/85600/85514-43-8.png)
dimethyl-](http://img.cochemist.com/ccimg/85600/85514-43-8_b.png)
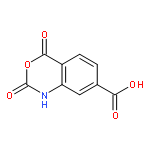
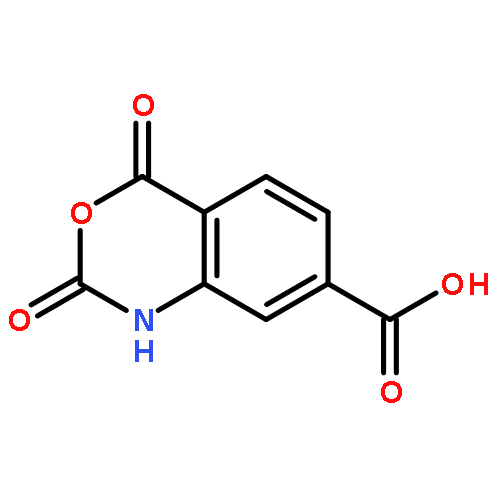
![Glycine,N-[9-(5-O-phosphono-b-D-ribofuranosyl)-9H-purin-6-yl]-](http://img.cochemist.com/ccimg/77200/77133-59-6.png)
![Glycine,N-[9-(5-O-phosphono-b-D-ribofuranosyl)-9H-purin-6-yl]-](http://img.cochemist.com/ccimg/77200/77133-59-6_b.png)
![Adenosine, 5'-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/69600/69530-93-4.png)
![Adenosine, 5'-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/69600/69530-93-4_b.png)
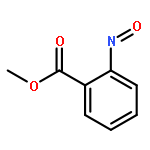
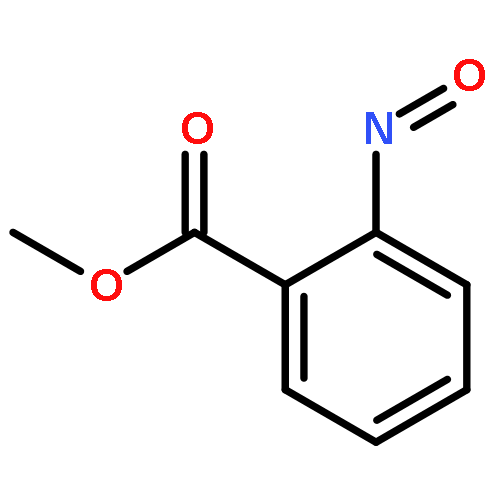
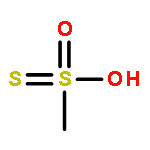



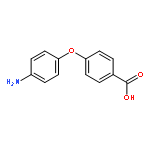
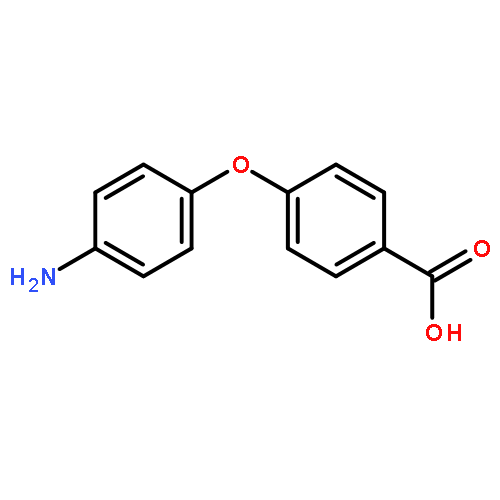


![9-bromo-5,11-dimethyl-6H-pyrido[4,3-b]carbazole](http://img.cochemist.com/ccimg/18100/18073-34-2.png)
![9-bromo-5,11-dimethyl-6H-pyrido[4,3-b]carbazole](http://img.cochemist.com/ccimg/18100/18073-34-2_b.png)
![5,9,11-trimethyl-6h-pyrido[4,3-b]carbazole](http://img.cochemist.com/ccimg/18100/18073-31-9.png)
![5,9,11-trimethyl-6h-pyrido[4,3-b]carbazole](http://img.cochemist.com/ccimg/18100/18073-31-9_b.png)
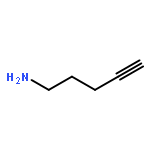

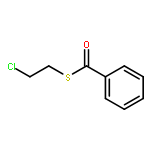
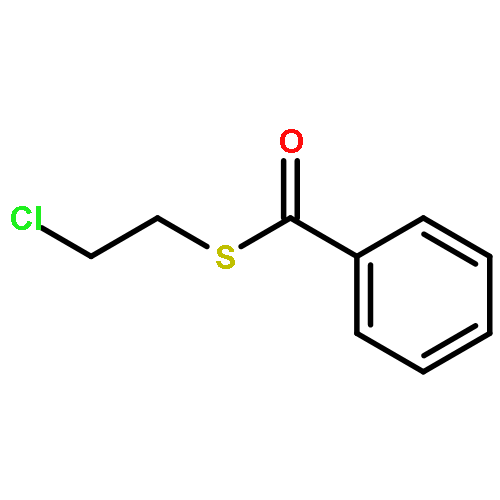
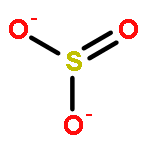
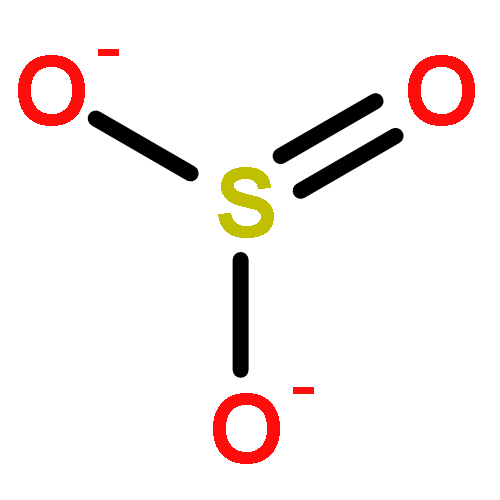


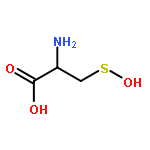
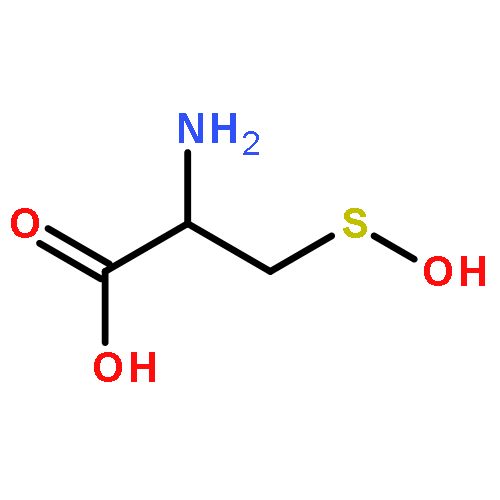
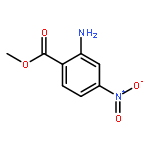
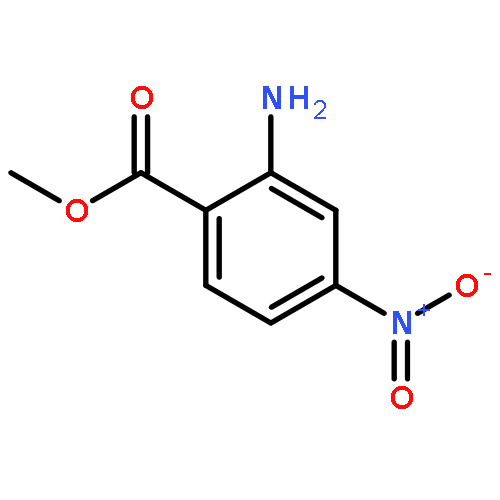







![6H-Pyrido[4,3-b]carbazole,5,11-dimethyl-](http://img.cochemist.com/ccimg/600/519-23-3.png)
![6H-Pyrido[4,3-b]carbazole,5,11-dimethyl-](http://img.cochemist.com/ccimg/600/519-23-3_b.png)