Co-reporter:Noah L. Wieder, Patrick J. Carroll, and Donald H. Berry
Organometallics 2011 Volume 30(Issue 8) pp:2125-2136
Publication Date(Web):March 31, 2011
DOI:10.1021/om100953g
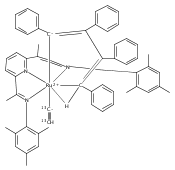
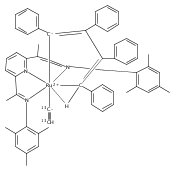
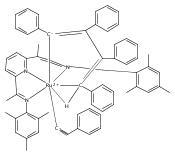
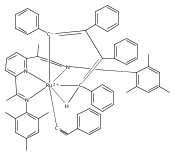
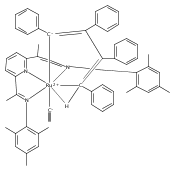
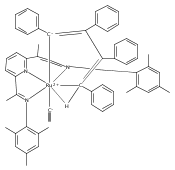
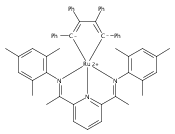
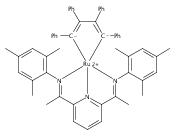
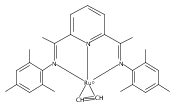
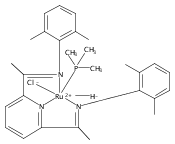
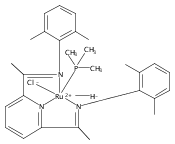
Reaction of the low-valent ruthenium complex [κ2-N3]Ru(η6-MeC6H5) (1), [N3] = 2,6-(MesN═CMe)2C5H3N, with acetylene leads to the displacement of toluene and formation of the monoacetylene adduct, [N3]Ru(C2H2) (2). The short alkyne−metal distances in 2 are consistent with 4 e− donation to the metal; that is, there is some degree of overlap of the perpendicular alkyne π- and metal dxz-orbitals. The NMR data and DFT calculations suggest π⊥ donation is weaker in 2 than in many 4 e− donor alkyne complexes. Further reaction of 2 with acetylene leads to catalytic cyclotrimerization and release of benzene, although the catalyst is short-lived. In the case of diphenylacetylene, two molecules of C2Ph2 react per molecule of 1 to generate the metallacyclic [N3]Ru(C4Ph4) (3), which is best described as a ruthenacyclopentadiene, or ruthenole. Compound 3 does not react further with diphenylacetylene, but does react with terminal alkynes by addition of the acetylenic C−H bond across a ruthenole Ru−C bond. The new complexes, [N3]Ru(C≡C-R)(cis,cis-1,2,3,4-tetraphenylbutadienyl-μ-H) (R = H, 5; Ph, 6), contain terminal acetylide and cis,cis-tetraphenylbutadienyl ligands (Ru-(CPh)4H), where the vinylic C−H bond is weakly bound to the metal through an agostic interaction. This type of ruthenole cleavage by terminal acetylenes may explain the short life of 2 as a catalyst for cyclotrimerization. The order in which HCCH and PhCCPh are introduced into the coordination sphere alters the course of the reaction: whereas isolated Ru(C4Ph4) metallacycle 3 is cleaved by acetylene to give 5, preformed acetylene complex 2 reacts with diphenylacetylene to produce the free cyclization product 1,2,3,4-tetraphenylbenzene (and 3). These observations highlight the key role of five-membered metallacycles in alkyne cyclotrimerization, as well as the importance of steric factors in these reactions. Cyclization is observed in cases where the π-system of the ruthenacyclic intermediate is accessible to an incoming alkyne, but not in cases where steric bulk hinders access.
Co-reporter:Noah L. Wieder ; Michelle Gallagher ; Patrick J. Carroll
Journal of the American Chemical Society 2010 Volume 132(Issue 12) pp:4107-4109
Publication Date(Web):March 3, 2010
DOI:10.1021/ja100894h
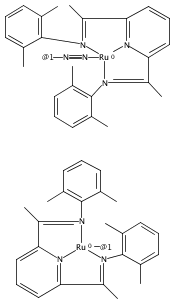
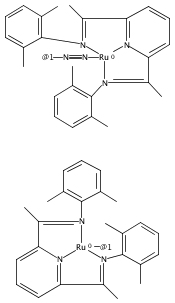
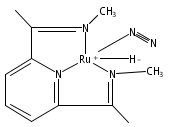
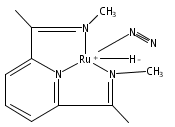
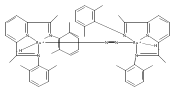
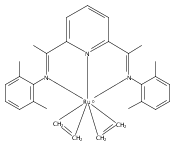
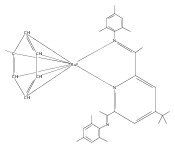
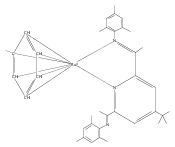
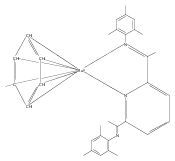
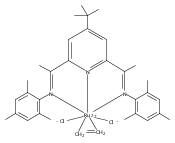
Formally zerovalent, dinitrogen-bridged ruthenium complex, {[N3Xyl]Ru}2(μ-η1:η1-N2) (1), where [N3Xyl] = 2,6-(XylN═CMe)2C5H3N, reacts with excess H2 to give the binuclear hydride species, {[N3]Ru(H)}2(μ-η1:η1-N2) (2) bearing a single hydrogen per ruthenium. Complex 2 is an unusual example of a structurally characterized paramagnetic transition metal hydride, and the first such example for ruthenium. Structural data and DFT calculations suggest unpaired electron density is strongly delocalized onto the non-innocent [N3] ligand, with a relatively small degree of the metalloradical character implied by the Ru(I) formal oxidation state, and that the [N3]−/Ru(II) formalism may be more informative. Consistent with an effective oxidation state greater than Ru(I), further reaction of 2 with excess H2 to give metal dihydride species ([N3]RuH2(L)) is not observed. The magnetic moment of 2 (3.50 μB) in solution is consistent with one unpaired electron per [N3]Ru moiety; however, 2 is diamagnetic in the solid due to close (3.26 Å) head-to-tail contact between Ru pyridine planes of neighboring molecules. Although the geometry is reminiscent of the weak “π-stacking” observed for closed-shell aromatic ring systems, DFT calculations indicate the structure and associated spin pairing result from in-phase overlap of the delocalized SOMOs on neighboring molecules—that is the interaction is best viewed as a weak covalent bond delocalized over 22 atoms.
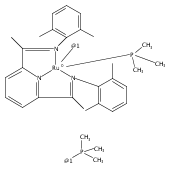
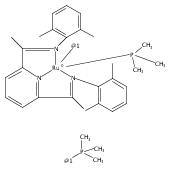
Co-reporter:Michelle Gallagher, Noah L. Wieder, Vladimir K. Dioumaev, Patrick J. Carroll and Donald H. Berry
Organometallics 2010 Volume 29(Issue 3) pp:591-603
Publication Date(Web):January 13, 2010
DOI:10.1021/om9009075
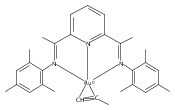
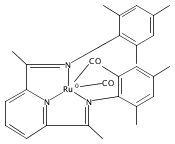
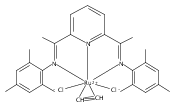
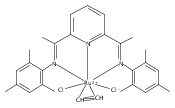
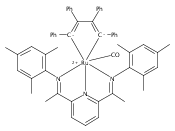
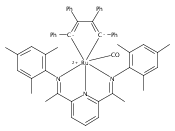
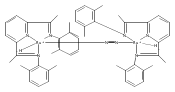
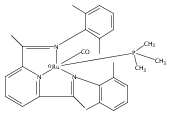
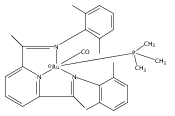
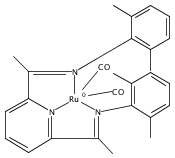
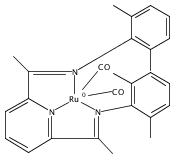
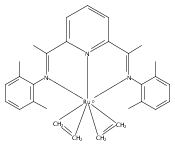
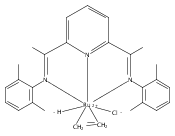
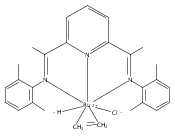
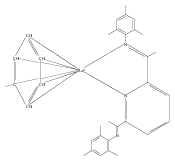
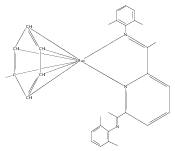
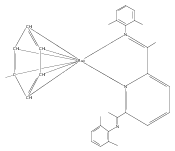
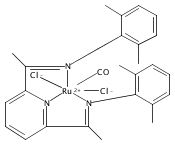
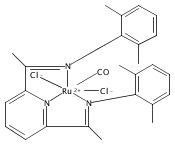
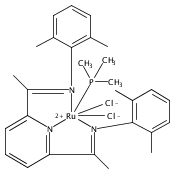
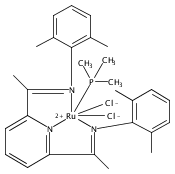
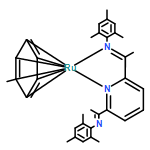
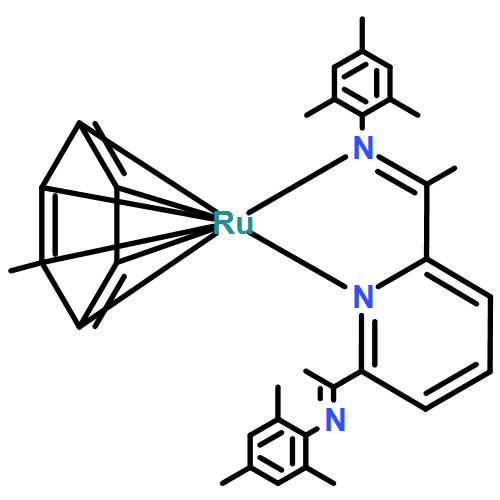
A series of low-valent ruthenium complexes bearing 2,6-bis(imino)pyridyl (“[N3]”) ligands has been synthesized and characterized. Reduction of [N3]RuCl2(C2H4) ([N3xyl] = 2,6-(XylNCMe)2C5H3N, 1a; [N3mes] = 2,6-(MesNCMe)2C5H2N, 1b; [tBu-N3mes] = 2,6-(MesNCMe)2p-tBuC5H2N, 1c) with hydridosilanes in an arene solvent such as toluene yields new 18e− η6-arene complexes [κ2-N3]Ru(η6-MeC6H5), 2a,b,c, in which the [N3] ligand is bidentate and only one imine group is coordinated to the metal. The arene ligand can be displaced with dinitrogen in non-arene solvents to yield the binuclear, four-coordinate, formally Ru(0) complexes {[N3]Ru}2(μ-N2), 3a,b,c. Pyrophoric complex 3c is a rare example of a structurally characterized Ru(0) dinitrogen complex. Treatment of low-valent complexes 2 or 3 with donor ligands generates five-coordinate complexes [N3xyl]RuL1,2 (L1,2 = C2H4, 4a; L1,2 = PMe3, 5a; L1,2 = CO, 6a; L1 = PMe3, L2 = CO, 7a). Complexes 2a, 3c, 5a, 6a, and 7a are diamagnetic and have been structurally characterized by single-crystal X-ray diffraction methods. New six-coordinate Ru(II) complexes [N3xyl]RuCl2(L) (L = PMe3, CO) were also isolated and structurally characterized. The infrared data, observed geometrical parameters, and reactivity patterns of the formally Ru(0) centers suggest varying degrees of electron delocalization to the “non-innocent” bis(imino)pyridyl, but probably not to the extent implied by the valence tautomeric [N3]2−/Ru(II) canonical form. Although the [N3]−/Ru(I) representation may portray the electron distribution more accurately than “Ru(0)”, the inherent odd electron counts on both ligand and metal—and requisite antiferromagnetic coupling—provides little in the way of “useful” distinctions or predictive value for the low-valent [N3]Ru(L)2 complexes with strong-field co-ligands such as CO and PMe3. These five-coordinate adducts seem to be adequately described as Ru(0) complexes of the neutral [N3] ligand. However, “non-innocent” valence tautomeric canonical forms such as [N3]−/Ru+ may be more applicable to the four-coordinate dinitrogen complexes {[N3]Ru}2(μ-N2).
.jpg)












![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1.png)
![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1_b.png)