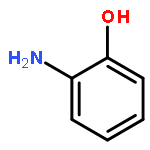
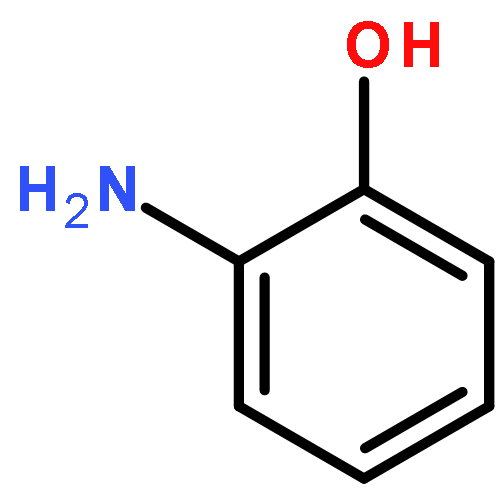
Co-reporter:Lu Zhang;Jing Huang;Weiyi Wang;Jinlong Yang
RSC Advances (2011-Present) 2017 vol. 7(Issue 21) pp:12704-12710
Publication Date(Web):2017/02/21
DOI:10.1039/C7RA01003F
Extensive efforts have been devoted to explore transport behaviors through various molecules and clusters, which are promising building blocks in molecular electronics. Here, we examine the spin-polarized electronic structures and transport properties of a three-shell icosahedral matryoshka cluster, Pb@Mn12@Pb20, by performing density functional theory calculations combining with non-equilibrium Green's function method. Theoretical results clearly reveal that, twelve Mn atoms in the middle layer anti-ferromagnetically couple with the center Pb atom and Pb atoms in the outlayer, while the Pb@Mn12@Pb20 cluster still has a huge magnetic moment of 28.0 bohr magneton, mainly contributed by these Mn atoms. The calculated spin-resolved transmission spectra of the proposed Pb@Mn12@Pb20 junctions exhibit robust spin filtering effect, which is not sensitive to the anchoring distance and the adopted electrode materials, and the conductance through the cluster under the small bias voltage is mainly determined by the spin-up electrons. These findings indicate that this kind of three-shell matryoshka cluster with huge magnetic moment holds potential applications in molecular spintronic devices.
Co-reporter:Zhen Jin, Meng Yang, Shao-Hua Chen, Jin-Huai Liu, Qun-Xiang Li, and Xing-Jiu Huang
Analytical Chemistry 2017 Volume 89(Issue 4) pp:
Publication Date(Web):January 24, 2017
DOI:10.1021/acs.analchem.6b04977
Herein, we revealed that the electrochemical behaviors on the detection of heavy metal ions (HMIs) would largely rely on the exposed facets of SnO2 nanoparticles. Compared to the high-energy {221} facet, the low-energy {110} facet of SnO2 possessed better electrochemical performance. The adsorption/desorption tests, density-functional theory (DFT) calculations, and X-ray absorption fine structure (XAFS) studies showed that the lower barrier energy of surface diffusion on {110} facet was critical for the superior electrochemical property, which was favorable for the ions diffusion on the electrode, and further leading the enhanced electrochemical performance. Through the combination of experiments and theoretical calculations, a reliable interpretation of the mechanism for electroanalysis of HMIs with nanomaterials exposed by different crystal facets has been provided. Furthermore, it provides a deep insight into understanding the key factor to improve the electrochemical performance for HMIs detection, so as to design high-performance electrochemical sensors.
Co-reporter:Jiajun Wang;Ming Zhang;Jie Meng;Jinlong Yang
RSC Advances (2011-Present) 2017 vol. 7(Issue 39) pp:24446-24452
Publication Date(Web):2017/05/03
DOI:10.1039/C7RA01723E
Recently novel two-dimensional materials for solar water splitting have drawn enormous research attention because of their tunable electronic properties for enhancing photocatalytic performance. Here, based on extensive density functional theory calculations, we clearly reveal that single- and few-layer BiOI have favorable band gaps, light electron effective masses, excellent optical absorptions, and nice band edge alignments with the water redox potentials. Moreover, due to the weak interlayer interactions, these remarkable electronic properties are robust, and almost independent of the layer thickness of the BiOI nanosheets. Our theoretical results suggest that the BiOI nanosheets can be efficient photocatalysts for solar water splitting.
Co-reporter:Jiajun Wang;Jing Huang;Jie Meng;Jinlong Yang
RSC Advances (2011-Present) 2017 vol. 7(Issue 63) pp:39877-39884
Publication Date(Web):2017/08/11
DOI:10.1039/C7RA03175K
Codoping can effectively engineer the band structures of photocatalysts (e.g. TiO2) to enhance their photoelectrochemical performance, however, previous investigations mainly focused on codoped bulk materials. In this work, we explore the (Rh + F) surface codoping effect on anatase TiO2 (101) and (001) facets for solar water splitting by performing extensive density functional theory calculations. According to the calculated defect formation energies, we find that the noble metal (Rh) atoms can be stably doped at the anatase TiO2 (101) surface with the aid of the codoped F atoms, thus can act as active sites for photocatalytic H2 evolution, which also provides the possibility of single-atom Rh catalysis on the (Rh + F) codoped anatase TiO2 (101) surface. The band gap of the codoped system is narrowed to about 2.14 eV through introducing several occupied and delocalized intermediate states which prevent the recombination of photogenerated carriers. Remarkably, the valence band maximum and conduction band minimum of the (Rh + F) codoped anatase TiO2 (101) surface match well with the water redox potentials and the visible light absorption is significantly enhanced. These findings imply that this kind of surface codoping is an effective approach to obtain visible light photocatalysts for water splitting.
Co-reporter:Jing Huang, Rong Xie, Weiyi Wang, Qunxiang Li and Jinlong Yang
Nanoscale 2016 vol. 8(Issue 1) pp:609-616
Publication Date(Web):17 Nov 2015
DOI:10.1039/C5NR05601B
As one of the most promising building blocks in molecular spintronics, spin crossover (SCO) complexes have attracted increasing attention due to their magnetic bistability between the high-spin (HS) and low-spin (LS) states. Here, we explore the electronic structures and transport properties of SCO magnet Fe2 complexes with three different spin-pair configurations, namely [LS–LS], [LS–HS], and [HS–HS], by performing extensive density functional theory calculations combined with the non-equilibrium Green's function technique. Our calculations clearly reveal that the SCO magnet Fe2 complexes should display two-step spin transitions triggered by external stimuli, i.e. temperature or light, which confirm the previous phenomenological model and agree well with previous experimental measurements. Based on the calculated transport results, we observe a nearly perfect spin-filtering effect and negative differential resistance (NDR) behavior integrated in the SCO magnet Fe2 junction with the [HS–HS] configuration. The current through the [HS–HS] SCO magnet Fe2 complex under a small bias voltage is mainly contributed by the spin-down electrons, which is significantly larger than those of the [LS–LS] and [LS–HS] cases. The bias-dependent transmissions are responsible for the observed NDR effect. These theoretical findings suggest that SCO Fe2 complexes hold potential applications in molecular spintronic devices.
Co-reporter:Jiajun Wang, Jie Meng, Qunxiang Li and Jinlong Yang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 25) pp:17029-17036
Publication Date(Web):17 May 2016
DOI:10.1039/C6CP01001F
Recently, various single-layer materials have been explored as desirable photocatalyts for water splitting. In this work, based on extensive density functional theory calculations, we examine the geometric, electronic, optical, and potential photocatalytic properties of single-layer cadmium chalcogenides (CdX sheets, X = S, Se, and Te), which are cleaved from the (001) plane of the bulk wurtzite structure. The predicted formation energies have relatively low values and a suitable substrate (i.e. graphene) that can effectively stabilize CdX sheets, which imply that the fabrication and application of CdX sheets are highly possible in experiments. The calculated band gaps, band edge positions and optical absorptions clearly reveal that CdSe and CdTe sheets are promising photocatalysts for water splitting driven by visible light. Moreover, the band gaps and band edge positions of three CdX sheets can be effectively tuned by applying biaxial strain, which then can enhance their photocatalytic performance. These theoretical findings imply that CdX sheets are promising candidates for photocatalytic water splitting.
Co-reporter:Jiajun Wang, Jing Huang, Jie Meng, Qunxiang Li and Jinlong Yang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 26) pp:17517-17524
Publication Date(Web):01 Jun 2016
DOI:10.1039/C6CP02047J
Double-hole doping is an effective approach to engineer the band structures of semiconductors for enhancing the photoelectrochemical performance. Here, we explore the anionic monodoping (i.e. N, C, and P) and codoping (i.e. N + N, C + S, and N + P pairs) effects on the electronic structures and photocatalytic activities of ZrO2 by performing extensive density functional theory calculations. Upon anionic monodoping, several unoccupied impurity states appear within the band gap, which may trap the photogenerated carriers and then reduce the photocatalytic efficiency. Remarkably, double-hole doping via introducing three anionic (N + N), (C + S), and (N + P) codoping pairs in ZrO2 can not only effectively narrow the band gap, but can also create several fully filled delocalized intermediate bands for preventing the recombination of the photogenerated electron–hole pairs. Moreover, the band edge positions matching well with the redox potentials of water and the improved visible light absorption ability indicate that the three examined codoped ZrO2 systems are promising photocatalysts for visible light water splitting. In short, double-hole doping via anionic pairs provides an effective path to tune the huge-gap semiconductor band structures and to develop high efficient catalysts for solar-driven water splitting.
Co-reporter:Jiajun Wang, Shaohua Chen, Qunxiang Li, Jinlong Yang
International Journal of Hydrogen Energy 2016 Volume 41(Issue 30) pp:13050-13057
Publication Date(Web):10 August 2016
DOI:10.1016/j.ijhydene.2016.05.073
•The acceptor IIB metal (Zn or Cd) and sulfur codoping effect on the electronic structures of anatase TiO2 is explored.•The metal-assisted SO coupling can not only effectively reduce the band gap of anatase TiO2, improve visible-light harvesting, and but also can prevent the recombination of the photogenerated electron–hole pairs.•The band edge alignments in the (Zn + S) and (Cd + S) codoped anatase TiO2 are desirable for photocatalytic visible light water-splitting.Codoping is an effective method to improve the photoelectrochemical performance of anatase TiO2. In this work, through performing extensive hybrid density functional theory calculations, we explore the (Zn + S) and (Cd + S) codoping effects on the electronic structures and photocatalytic activities of anatase TiO2. Theoretical results clearly reveal that the coupling of the incorporated S dopant with the second-nearest neighboring O atom assisted by the acceptor metals leads to the fully occupied and delocalized intermediate bands within the band gap, which originate from the SO antibonding states (π∗). This kind of metal-assisted SO coupling can not only effectively reduce the band gap of TiO2, but also prevent the recombination of the photogenerated carriers. Moreover, the visible light absorption of anatase TiO2 is significantly enhanced via codoping, and both the reduction and oxidation potentials of water lie within the band gap of the (Zn + S) and (Cd + S) codoped anatase TiO2, which are desirable for photocatalytic water splitting. These theoretical findings imply that the metal-assisted SO coupling is an effectively approach to engineer the band structures and the visible light photoelectrochemical performance of semiconductors photocatalysts.
Co-reporter:Jiajun Wang, Zhaoyong Guan, Jing Huang, Qunxiang Li and Jinlong Yang
Journal of Materials Chemistry A 2014 vol. 2(Issue 21) pp:7960-7966
Publication Date(Web):03 Mar 2014
DOI:10.1039/C4TA00275J
Here, we explore the enhanced photocatalytic mechanism for the hybrid g-C3N4/MoS2 nanocomposites for the first time by performing extensive density functional theory calculations. The calculated band alignment between the g-C3N4 monolayer and MoS2 sheets clearly reveals that the conduction band minimum and valence band maximum of the g-C3N4 monolayer are higher by about 0.83 eV and 0.15 eV respectively than those of the MoS2 sheet. This predicted type-II band alignment ensures the photogenerated electrons easily migrate from the g-C3N4 monolayer to the MoS2 sheet, and leads to the high hydrogen-evolution reaction activity. The charge transfer between MoS2 and g-C3N4 results in a polarized field within the interface region, which will benefit the separation of photogenerated carriers. The calculated optical absorption curves verify that this proposed layered nanocomposite is a good light-harvesting semiconductor. Moreover, a g-C3N4 bilayer covering a MoS2 sheet also displays desirable properties. These findings indicate that the MoS2 sheet is a promising candidate as a non-noble metal co-catalyst for g-C3N4 photocatalysts, and also provide useful information for understanding the observed enhanced photocatalytic mechanism in experiments.
Co-reporter:Tao Luo, Qiang-Qiang Meng, Chao Gao, Xin-Yao Yu, Yong Jia, Bai Sun, Zhen Jin, Qun-Xiang Li, Jin-Huai Liu and Xing-Jiu Huang
Chemical Communications 2014 vol. 50(Issue 100) pp:15952-15955
Publication Date(Web):31 Oct 2014
DOI:10.1039/C4CC06064D
We present an effective method to synthesize 15 nm magnetite nanocrystals with the morphology of square and circular nanoplates, which expose (001) facet and (111) facet, respectively. The magnetic property and electrochemical behavior towards As(III) exhibit strong facet-dependent characteristics. Theoretical calculations confirm the facet-dependent characteristics and provide the corresponding explanations.
Co-reporter:Wei-Hong Xu, Qiang-Qiang Meng, Chao Gao, Jing Wang, Qun-Xiang Li, Jin-Huai Liu and Xing-Jiu Huang
Chemical Communications 2014 vol. 50(Issue 39) pp:5011-5013
Publication Date(Web):31 Mar 2014
DOI:10.1039/C4CC01029A
We find for the first time that the electrochemical performances of the α-Fe2O3 nanostructures depend on their exposed facets. Density functional theory calculations are carried out to better and scientifically understand the effect of different exposed facets at the atomic-scale level.
Co-reporter:Qiangqiang Meng, Zhaoyong Guan, Jing Huang, Qunxiang Li and Jinlong Yang
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 23) pp:11519-11526
Publication Date(Web):22 Apr 2014
DOI:10.1039/C4CP01077A
Recently, the synthesis, properties, modifications, and applications of TiO2 nanomaterials have attracted much research attention. Here, based on extensive density functional theory calculations, we explored the stability, electronic structures and optical absorption properties of single-walled TiO2 nanotubes (SWTONTs) and TiO2 nanotube arrays (TONTAs), which are constructed from anatase TiO2(101) monolayers and bilayers, respectively. We obtained the stable Dnd (n = 3–5) and S2n(−n, n) (n = 3–9) SWTONTs, and found that SWTONTs energetically prefer S2n symmetry. Compared with S2n(−n, n) SWTONTs, the calculated Young's moduli of Dnd(−n, n) SWTONTs are more stiff due to their relatively large strain energies. The band gaps of hexagonal TONTAs are not sensitive to their apertures, which are less than that of TiO2 bilayers. The narrow band gaps of TONTAs originate from the edge states mainly contributed by the Ti and O atoms at the core region. The calculated optical absorptions of both SWTONTs and TONTAs display anisotropic features. These results clearly reveal that the electronic and optical properties of TiO2 nanostructures are strongly associated with their symmetry, dimensions and morphology, which provide useful insights into the understanding of the related experimental observations.
Co-reporter:Zhaoyong Guan ; Jiajun Wang ; Jing Huang ; Xiaojun Wu ; Qunxiang Li ;Jinlong Yang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 39) pp:22491-22498
Publication Date(Web):September 8, 2014
DOI:10.1021/jp508617k
Recently transition-metal-free magnetism and half-metallicity have drawn enormous attention due to their potential applications in spintronic devices. In this work, we examine the stability, electronic structures, and magnetic properties of single-walled C4N3 nanotubes (SWCNNTs) by performing extensive spin-polarized density functional theory calculations and molecular dynamics simulations. The theoretical results clearly reveal that all examined SWCNNTs are stable at room temperature. Armchair (n, n) (n = 4–10), zigzag (n, 0) (n = 7–10), and two helical SWCNNTs have ferromagnetic ground states. All armchair SWCNNTs are metal-free half-metals, helical SWCNNTs with small radii are bipolar magnetic semiconductors, and zigzag SWCNNTs show size dependency, in which the semiconductor-to-metal transition can be realized by increasing their radii. Moreover, the total magnetic moments of SWCNNTs can be tuned by changing the number of primary C4N3 unit cells. These findings bring us the possibility of building functional electronic/spintronic devices with SWCNNTs due to the tunable metal-free magnetism and half-metallicity.
Co-reporter:Jiajun Wang ; Haifeng Sun ; Jing Huang ; Qunxiang Li ;Jinlong Yang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 14) pp:7451-7457
Publication Date(Web):March 21, 2014
DOI:10.1021/jp5004775
Doping with anion and cation impurities is an effective approach to tune the photoelectrochemical properties of TiO2. Here, we explore the Rh monodoping and (Rh + F) codoping effect on electronic structures and photocatalytic activities of anatase TiO2 by performing extensive density functional theory calculations. Upon Rh monodoping, the band gap of TiO2 can be effectively reduced. But this cationic dopant creates an unoccupied intermediate localized state within the band gap, which will act as photogenerated carrier recombination center, which reduces the photocatalytic efficiency. Fortunately, we find that the stable charge-compensated donor–acceptor pair (Rh + F) codoping in TiO2 can effectively reduce the band gap by forming a delocalized intermediate band within the band gap. Moreover, the band edge alignment in the (Rh + F) codoped TiO2 is desirable for water splitting. The calculated optical absorption curve of (Rh + F) codoped TiO2 verifies that it has significantly improved visible light absorption. These findings imply that the (Rh + F) codoped TiO2 is a promising visible light photocatalyst for water splitting.
Co-reporter:Zhaoyong Guan ; Weiyi Wang ; Jing Huang ; Xiaojun Wu ; Qunxiang Li ;Jinlong Yang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 49) pp:28616-28624
Publication Date(Web):November 18, 2014
DOI:10.1021/jp5089349
Carbon-doped boron nitride nanostructures including nanosheets, nanoribbons, and nanotubes have drawn enormous research attention because of their tunable electronic properties and widespread applications. In this work, we explore the electronic and magnetic properties of graphene flake-doped single-walled boron nitride nanotubes (BNNTs) on the basis of first-principles calculations. Theoretical results reveal that the band structures of these doped BNNTs can be effectively engineered by embedding graphene flakes with different sizes and shapes. Moreover, the Lieb theorem works for the triangle graphene flake-doped BNNTs, and the corresponding doped systems are ferromagnetic, originating from the spin-polarized interface states. All BNNTs embedded with the triangular graphene flakes with relatively small sizes are typical bipolar magnetic semiconductors, which can be easily tuned into half-metals by carrier doping, opening the door to their promising applications in spintronic devices.
Co-reporter:Jing Huang, Weiyi Wang, Shangfeng Yang, Qunxiang Li, Jinlong Yang
Chemical Physics Letters 2013 590() pp: 111-115
Publication Date(Web):
DOI:10.1016/j.cplett.2013.10.049
Co-reporter:Ke Xu, Jing Huang, Zhaoyong Guan, Qunxiang Li, Jinlong Yang
Chemical Physics Letters 2012 Volume 535() pp:111-115
Publication Date(Web):11 May 2012
DOI:10.1016/j.cplett.2012.03.066
Abstract
We present a theoretical study of spin transport through a magnetic C28 molecule sandwiched between two Au (1 1 1) electrodes. The ab initio modeling is performed by spin density functional theory and nonequilibrium Green’s function technique. The results clearly show that the spin-resolved transmission spectra of C28 molecular junctions exhibit robust transport spin polarization (TSP) characteristics, which depends on the contact configuration. At the small bias voltage, the conductance of C28 is mainly determined by the spin-down electrons. The TSP behavior can be effectively tuned by the gate. Our results indicate that C28 molecule holds promise in future molecular spintronics applications.
Co-reporter:Jing Huang, Weiyi Wang, Shangfeng Yang, Haibin Su, Qunxiang Li, Jinlong Yang
Chemical Physics Letters 2012 s 539–540() pp: 102-106
Publication Date(Web):
DOI:10.1016/j.cplett.2012.05.002
Co-reporter:Jing Huang, Qunxiang Li, Ke Xu, Haibin Su and Jinlong Yang
The Journal of Physical Chemistry C 2010 Volume 114(Issue 27) pp:11946-11950
Publication Date(Web):June 17, 2010
DOI:10.1021/jp101554c
Using density functional theory calculations combined with nonequilibrium Green’s function method, we report the electronic, magnetic, and transport properties of iron-cyclooctatetraene (Fe-COT) sandwich clusters. Four Fen COTn+1 (n = 1−4) clusters with linear sandwich structure are highly stable because of the strong Fe-COT coupling. The ground state of Fe-COT clusters is ferromagnetic, Fe atoms couple ferromagnetically to the neighboring COT rings, and the large total magnetic moments increase with the number of Fe atoms. The spin-polarized transport calculations indicate that Fe-COT clusters coupled to gold electrodes act as nearly perfect spin-filters. The revealed properties indicate that the Fe-COT clusters would be ideal materials for promising molecular spintronics.
Co-reporter:Jing Huang, Qunxiang Li, Haibin Su, Jinlong Yang
Chemical Physics Letters 2009 Volume 479(1–3) pp:120-124
Publication Date(Web):7 September 2009
DOI:10.1016/j.cplett.2009.08.006
Abstract
We report first-principles calculations of the electronic and transport properties of diarylethene molecules sandwiched between metallic armchair (5, 5) carbon nanotubes with open and closed configurations. The calculated transmission spectra of two configurations are distinctively different near the Fermi level, and their profiles can be tuned by the gate bias voltage. The on–off ratio of currents between the closed and open configurations under a gate bias voltage is predicted to be more than two orders of magnitude, which reproduces the essential features of the experimental results. Moreover, we find that the switching property of diarylethene molecular junctions is very robust to the anchoring configuration, substituent of cyclopentene, and choice of electrodes.
Co-reporter:Jiajun Wang, Zhaoyong Guan, Jing Huang, Qunxiang Li and Jinlong Yang
Journal of Materials Chemistry A 2014 - vol. 2(Issue 21) pp:NaN7966-7966
Publication Date(Web):2014/03/03
DOI:10.1039/C4TA00275J
Here, we explore the enhanced photocatalytic mechanism for the hybrid g-C3N4/MoS2 nanocomposites for the first time by performing extensive density functional theory calculations. The calculated band alignment between the g-C3N4 monolayer and MoS2 sheets clearly reveals that the conduction band minimum and valence band maximum of the g-C3N4 monolayer are higher by about 0.83 eV and 0.15 eV respectively than those of the MoS2 sheet. This predicted type-II band alignment ensures the photogenerated electrons easily migrate from the g-C3N4 monolayer to the MoS2 sheet, and leads to the high hydrogen-evolution reaction activity. The charge transfer between MoS2 and g-C3N4 results in a polarized field within the interface region, which will benefit the separation of photogenerated carriers. The calculated optical absorption curves verify that this proposed layered nanocomposite is a good light-harvesting semiconductor. Moreover, a g-C3N4 bilayer covering a MoS2 sheet also displays desirable properties. These findings indicate that the MoS2 sheet is a promising candidate as a non-noble metal co-catalyst for g-C3N4 photocatalysts, and also provide useful information for understanding the observed enhanced photocatalytic mechanism in experiments.
Co-reporter:Jiajun Wang, Jie Meng, Qunxiang Li and Jinlong Yang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 25) pp:NaN17036-17036
Publication Date(Web):2016/05/17
DOI:10.1039/C6CP01001F
Recently, various single-layer materials have been explored as desirable photocatalyts for water splitting. In this work, based on extensive density functional theory calculations, we examine the geometric, electronic, optical, and potential photocatalytic properties of single-layer cadmium chalcogenides (CdX sheets, X = S, Se, and Te), which are cleaved from the (001) plane of the bulk wurtzite structure. The predicted formation energies have relatively low values and a suitable substrate (i.e. graphene) that can effectively stabilize CdX sheets, which imply that the fabrication and application of CdX sheets are highly possible in experiments. The calculated band gaps, band edge positions and optical absorptions clearly reveal that CdSe and CdTe sheets are promising photocatalysts for water splitting driven by visible light. Moreover, the band gaps and band edge positions of three CdX sheets can be effectively tuned by applying biaxial strain, which then can enhance their photocatalytic performance. These theoretical findings imply that CdX sheets are promising candidates for photocatalytic water splitting.
Co-reporter:Jiajun Wang, Jing Huang, Jie Meng, Qunxiang Li and Jinlong Yang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 26) pp:NaN17524-17524
Publication Date(Web):2016/06/01
DOI:10.1039/C6CP02047J
Double-hole doping is an effective approach to engineer the band structures of semiconductors for enhancing the photoelectrochemical performance. Here, we explore the anionic monodoping (i.e. N, C, and P) and codoping (i.e. N + N, C + S, and N + P pairs) effects on the electronic structures and photocatalytic activities of ZrO2 by performing extensive density functional theory calculations. Upon anionic monodoping, several unoccupied impurity states appear within the band gap, which may trap the photogenerated carriers and then reduce the photocatalytic efficiency. Remarkably, double-hole doping via introducing three anionic (N + N), (C + S), and (N + P) codoping pairs in ZrO2 can not only effectively narrow the band gap, but can also create several fully filled delocalized intermediate bands for preventing the recombination of the photogenerated electron–hole pairs. Moreover, the band edge positions matching well with the redox potentials of water and the improved visible light absorption ability indicate that the three examined codoped ZrO2 systems are promising photocatalysts for visible light water splitting. In short, double-hole doping via anionic pairs provides an effective path to tune the huge-gap semiconductor band structures and to develop high efficient catalysts for solar-driven water splitting.
Co-reporter:Qiangqiang Meng, Zhaoyong Guan, Jing Huang, Qunxiang Li and Jinlong Yang
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 23) pp:NaN11526-11526
Publication Date(Web):2014/04/22
DOI:10.1039/C4CP01077A
Recently, the synthesis, properties, modifications, and applications of TiO2 nanomaterials have attracted much research attention. Here, based on extensive density functional theory calculations, we explored the stability, electronic structures and optical absorption properties of single-walled TiO2 nanotubes (SWTONTs) and TiO2 nanotube arrays (TONTAs), which are constructed from anatase TiO2(101) monolayers and bilayers, respectively. We obtained the stable Dnd (n = 3–5) and S2n(−n, n) (n = 3–9) SWTONTs, and found that SWTONTs energetically prefer S2n symmetry. Compared with S2n(−n, n) SWTONTs, the calculated Young's moduli of Dnd(−n, n) SWTONTs are more stiff due to their relatively large strain energies. The band gaps of hexagonal TONTAs are not sensitive to their apertures, which are less than that of TiO2 bilayers. The narrow band gaps of TONTAs originate from the edge states mainly contributed by the Ti and O atoms at the core region. The calculated optical absorptions of both SWTONTs and TONTAs display anisotropic features. These results clearly reveal that the electronic and optical properties of TiO2 nanostructures are strongly associated with their symmetry, dimensions and morphology, which provide useful insights into the understanding of the related experimental observations.
Co-reporter:Tao Luo, Qiang-Qiang Meng, Chao Gao, Xin-Yao Yu, Yong Jia, Bai Sun, Zhen Jin, Qun-Xiang Li, Jin-Huai Liu and Xing-Jiu Huang
Chemical Communications 2014 - vol. 50(Issue 100) pp:NaN15955-15955
Publication Date(Web):2014/10/31
DOI:10.1039/C4CC06064D
We present an effective method to synthesize 15 nm magnetite nanocrystals with the morphology of square and circular nanoplates, which expose (001) facet and (111) facet, respectively. The magnetic property and electrochemical behavior towards As(III) exhibit strong facet-dependent characteristics. Theoretical calculations confirm the facet-dependent characteristics and provide the corresponding explanations.
Co-reporter:Wei-Hong Xu, Qiang-Qiang Meng, Chao Gao, Jing Wang, Qun-Xiang Li, Jin-Huai Liu and Xing-Jiu Huang
Chemical Communications 2014 - vol. 50(Issue 39) pp:NaN5013-5013
Publication Date(Web):2014/03/31
DOI:10.1039/C4CC01029A
We find for the first time that the electrochemical performances of the α-Fe2O3 nanostructures depend on their exposed facets. Density functional theory calculations are carried out to better and scientifically understand the effect of different exposed facets at the atomic-scale level.