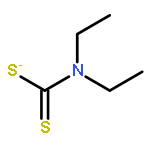
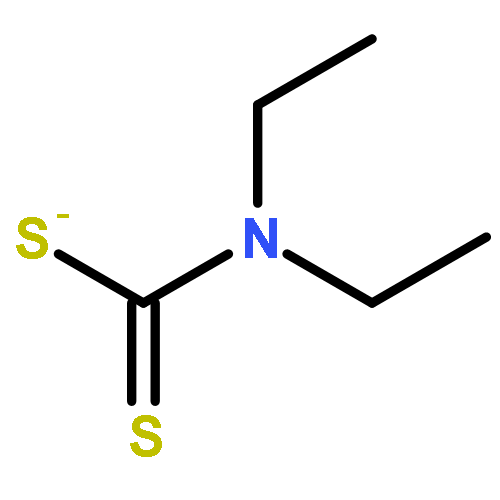
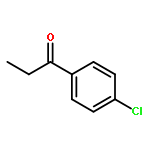
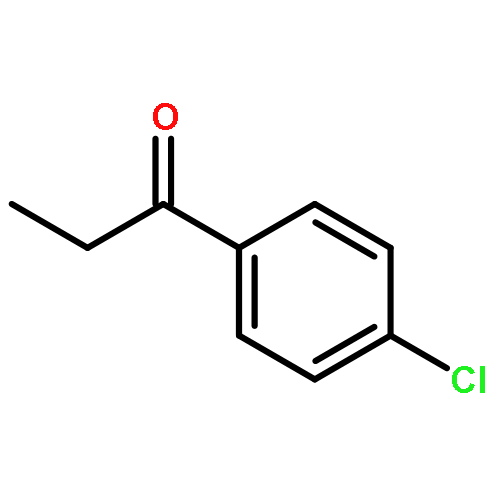
Co-reporter:Thaer M. M. Al-Rammahi and Richard A. Henderson
Dalton Transactions 2016 vol. 45(Issue 1) pp:307-314
Publication Date(Web):20 Nov 2015
DOI:10.1039/C5DT04008F
Kinetic studies on the acid-catalyzed substitution reactions of the teminal chloro-ligands in [Fe4S4Cl4]2− by PhS− in the presence of the acids NHR3+ (R = Me, Prn or Bun) are reported. Although these acids have very similar pKas (17.6–18.4) the reactions show a variety of different kinetics, some of which are inconsistent with a mechanism involving simple protonation of the cluster followed by substitution of a terminal ligand. The observed behaviour is more consistent with the recently proposed mechanism in which Fe–(μ3-SH) bond elongation/cleavage occurs upon protonation of a μ3-S, and suggests that both the acidity and bulk of the acid is important in the protonation step. Other studies have determined the activation parameters (ΔH‡ and ΔS‡) for both the protonation and substitution steps of the acid-catalyzed substitution reactions of [Fe4S4X4]2− (X = Cl or SEt). A significantly negative ΔS‡ is observed for the substitution steps of both clusters indicating associative pathways. This is inconsistent with earlier interpretation of the kinetics of these reactions (based exclusively on the dependence of the rate on the concentration of nucleophile) and indicates that there is no dissociative substitution mechanism and the pathway associated with a zero order dependence on the concentration of PhS− involves associative substitution with the solvent (MeCN) being the nucleophile.
Co-reporter:Thaer M. M. Al-Rammahi and Richard A. Henderson
Dalton Transactions 2016 vol. 45(Issue 4) pp:1373-1381
Publication Date(Web):08 Dec 2015
DOI:10.1039/C5DT04523A
The mechanism of the acid-catalyzed substitution reaction of the terminal chloro-ligands in [Fe4S4Cl4]2− by PhS− in the presence of NHBun3+ involves rate-limiting proton transfer from NHBun3+ to the cluster (k0 = 490 ± 20 dm3 mol−1 s−1). A variety of small molecules and ions (L = substrate = Cl−, Br−, I−, RNHNH2 (R = Me or Ph), Me2NNH2, HCN, NCS−, N3−, ButNC or pyridine) bind to [Fe4S4Cl4]2− and this affects the rate of subsequent protonation of [Fe4S4Cl4(L)]n−. Where the kinetics allow, the equilibrium constants for the substrates binding to [Fe4S4Cl4]2− (KL) and the rates of proton transfer from NHBun3+ to [Fe4S4Cl4(L)]n− (kL0) have been determined. The results indicate the following general features. (i) Bound substrates increase the rate of protonation of the cluster, but the rate increase is modest (kL0/k0 = 1.6 to ≥72). (ii) When KL is small, so is kL0/k0. (iii) Binding substrates which are good σ-donors or good π-acceptors lead to the largest kL0/k0. This behaviour is discussed in terms of the recent proposal that protonation of [Fe4S4Cl4]2− at a μ3-S, is coupled to concomitant Fe–(μ3-SH) bond elongation/cleavage.
Co-reporter:Thaer M. M. Al-Rammahi;Paul G. Waddell;Richard A. Henderson
Transition Metal Chemistry 2016 Volume 41( Issue 5) pp:555-561
Publication Date(Web):2016 August
DOI:10.1007/s11243-016-0052-5
The X-ray crystal structures of [NHR3]2[Fe4S4X4] (X = PhS, R = Et or nBu; X = Cl, R = nBu), reported in this paper, show NH…X interactions between the cation and the cubanoid cluster. Comparison of the cluster dimensions in [NHR3]2[Fe4S4X4] with those reported earlier for [NR′4]2[Fe4S4X4] (R′ = Me, X = PhS; R′ = Et, X = Cl) indicates that N–H…X interactions have a negligible effect on the dimensions of the cluster. The relevance of these structures to understanding where on [Fe4S4X4]2− protonation labilises the cluster to substitution is discussed.
Co-reporter:Ahmed Alwaaly, William Clegg, Richard A. Henderson, Michael R. Probert and Paul G. Waddell
Dalton Transactions 2015 vol. 44(Issue 7) pp:3307-3317
Publication Date(Web):06 Jan 2015
DOI:10.1039/C4DT03543G
The complexes [Ni(S2CR)(triphos)]BPh4 (R = Me, Et, Bun or Ph; triphos = PhP{CH2CH2PPh2}2) have been prepared and characterised. X-ray crystallography (for R = Et, Ph, C6H4Me-4, C6H4OMe-4 and C6H4Cl-4) shows that the geometry of the five-coordinate nickel in the cation is best described as distorted trigonal bipyramidal, containing a bidentate carboxydithioate ligand with the two sulfur atoms spanning axial and equatorial sites, the other axial site being occupied by the central phosphorus of triphos. The reactions of [Ni(S2CR)(triphos)]+ with mixtures of HCl and Cl− in MeCN to form equilibrium solutions containing [Ni(SH(S)CR)(triphos)]2+ have been studied using stopped-flow spectrophotometry. The kinetics show that proton transfer is slower than the diffusion-controlled limit and involves at least two coupled equilibria. The first step involves the rapid association between [Ni(S2CR)(triphos)]+ and HCl to form the hydrogen-bonded precursor, {[Ni(S2CR)(triphos)]+⋯HCl} (KR1) and this is followed by the intramolecular proton transfer (kR2) to produce [Ni(SH(S)CR)(triphos)]2+. In the reaction of [Ni(S2CMe)(triphos)]+ the rate law is consistent with the carboxydithioate ligand undergoing chelate ring-opening after protonation. It seems likely that chelate ring-opening occurs for all [Ni(S2CR)(triphos)]+, but only with [Ni(S2CMe)(triphos)]+ is the protonation step sufficiently fast that chelate ring-opening is rate-limiting. With all other systems, proton transfer is rate-limiting. DFT calculations indicate that protonation can occur at either sulfur atom, but only protonation at the equatorial sulfur results in chelate ring-opening. The ways in which protonation of either sulfur atom complicates the analyses and interpretation of the kinetics are discussed.
Co-reporter:Ahmed Alwaaly, William Clegg, Ross W. Harrington, Athinoula L. Petrou and Richard A. Henderson
Dalton Transactions 2015 vol. 44(Issue 26) pp:11977-11983
Publication Date(Web):08 Jun 2015
DOI:10.1039/C5DT01716E
Earlier kinetic studies on the protonation of the coordinated thiolate in the square-planar [Ni(SC6H4R′-4)(triphos)]+ (R′ = NO2, Cl, H, Me or MeO) by lutH+ (lut = 2,6-dimethylpyridine) indicate a two-step mechanism involving initial formation of a (kinetically detectable) precursor intermediate, {[Ni(SC6H4R′-4)(triphos)]⋯Hlut}2+ (KR1), followed by an intramolecular proton transfer step (kR2). The analogous [Ni(SR)(triphos)]BPh4 {R = Et, But or Cy; triphos = PhP(CH2CH2PPh2)2} have been prepared and characterized by spectroscopy and X-ray crystallography. Similar to the aryl thiolate complexes, [Ni(SR)(triphos)]+ are protonated by lutH+ in an equilibrium reaction but the observed rate law is simpler. Analysis of the kinetic data for both [Ni(SR)(triphos)]+ and [Ni(SC6H4R′-4)(triphos)]+ shows that both react by the same mechanism, but that KR1 is largest when the thiolate is poorly basic, or the 4-R′ substituent in the aryl thiolates is electron-withdrawing. These results indicate that it is both NH⋯S hydrogen bonding and encapsulation of the bound lutH+ (by the phenyl groups on triphos) which stabilize the precursor intermediate.
Co-reporter:Ahmed Alwaaly, Ian Dance and Richard A. Henderson
Chemical Communications 2014 vol. 50(Issue 37) pp:4799-4802
Publication Date(Web):10 Mar 2014
DOI:10.1039/C4CC00922C
Density functional calculations show that Fe–S clusters undergo unexpected large structural changes when protonated at S. Protonation of prototypical cubanoid [Fe4S4X4]2− to [Fe4S3(SH)X4]− (X = Cl, SR, OR) results in formation of doubly-bridging SH, severance of one Fe–S bond, and creation of a three-coordinate Fe. These findings explain previously enigmatic results concerning the reactivity of these clusters, including the rates of protonation, pKa data, and the kinetics of acid-catalysed ligand substitution.
Co-reporter:Ahmed Alwaaly and Richard A. Henderson
Chemical Communications 2014 vol. 50(Issue 68) pp:9669-9671
Publication Date(Web):04 Jul 2014
DOI:10.1039/C4CC04197F
Rates of proton transfers between lutH+ (lut = 2,6-dimethylpyridine) and [Ni(XPh)(PhP{CH2CH2PPh2}2)]+ (X = O, S or Se) are slow and show little variation (kO:kS:kSe = 1:12:9). This unusual behaviour is a consequence of sterics affecting the optimal interaction between the reactants prior to proton transfer.
Co-reporter:Dilek Nartop, William Clegg, Ross W. Harrington, Richard A. Henderson and Corinne Y. Wills
Dalton Transactions 2014 vol. 43(Issue 8) pp:3372-3382
Publication Date(Web):18 Dec 2013
DOI:10.1039/C3DT53359J
The kinetics of the reactions between [Ni(MeOH)6]2+ (hereafter Ni2+) and a variety of neutral Schiff base multidentate ligands have been measured in methanol at 25.0 °C using stopped-flow spectrophotometry. The ligands contain a variety of different potential donor sites (phenolic OH, imine N, pyridyl N and NH groups), different structural components and substituents. The kinetic studies explore how systematic changes to the composition of the ligands affect the rates of binding. The results are consistent with the Eigen–Wilkins mechanism in which the ligand initially forms an outer-sphere association with Ni2+ prior to dissociation of a coordinated solvent molecule and binding to the metal ion. The general features that emerge from these studies are as follows. (i) For ligands with the same donor set, the rates of binding are all similar irrespective of changes to the ligand framework (bridge and substituents). (ii) Comparison of structurally analogous ligands shows that the presence of pyridyl or NH groups in the multidentate results in significantly faster reactions. (iii) With ligands containing multiple NH groups, the rate of ligand binding increases as the number of NH groups increases. The extent to which these kinetic features can be attributed to preferential binding of particular donor groups is discussed.
Co-reporter:Ahmed Alwaaly and Richard A. Henderson
Chemical Communications 2014 - vol. 50(Issue 68) pp:NaN9671-9671
Publication Date(Web):2014/07/04
DOI:10.1039/C4CC04197F
Rates of proton transfers between lutH+ (lut = 2,6-dimethylpyridine) and [Ni(XPh)(PhP{CH2CH2PPh2}2)]+ (X = O, S or Se) are slow and show little variation (kO:kS:kSe = 1:12:9). This unusual behaviour is a consequence of sterics affecting the optimal interaction between the reactants prior to proton transfer.
Co-reporter:Thaer M. M. Al-Rammahi and Richard A. Henderson
Dalton Transactions 2016 - vol. 45(Issue 1) pp:NaN314-314
Publication Date(Web):2015/11/20
DOI:10.1039/C5DT04008F
Kinetic studies on the acid-catalyzed substitution reactions of the teminal chloro-ligands in [Fe4S4Cl4]2− by PhS− in the presence of the acids NHR3+ (R = Me, Prn or Bun) are reported. Although these acids have very similar pKas (17.6–18.4) the reactions show a variety of different kinetics, some of which are inconsistent with a mechanism involving simple protonation of the cluster followed by substitution of a terminal ligand. The observed behaviour is more consistent with the recently proposed mechanism in which Fe–(μ3-SH) bond elongation/cleavage occurs upon protonation of a μ3-S, and suggests that both the acidity and bulk of the acid is important in the protonation step. Other studies have determined the activation parameters (ΔH‡ and ΔS‡) for both the protonation and substitution steps of the acid-catalyzed substitution reactions of [Fe4S4X4]2− (X = Cl or SEt). A significantly negative ΔS‡ is observed for the substitution steps of both clusters indicating associative pathways. This is inconsistent with earlier interpretation of the kinetics of these reactions (based exclusively on the dependence of the rate on the concentration of nucleophile) and indicates that there is no dissociative substitution mechanism and the pathway associated with a zero order dependence on the concentration of PhS− involves associative substitution with the solvent (MeCN) being the nucleophile.
Co-reporter:Ahmed Alwaaly, William Clegg, Richard A. Henderson, Michael R. Probert and Paul G. Waddell
Dalton Transactions 2015 - vol. 44(Issue 7) pp:NaN3317-3317
Publication Date(Web):2015/01/06
DOI:10.1039/C4DT03543G
The complexes [Ni(S2CR)(triphos)]BPh4 (R = Me, Et, Bun or Ph; triphos = PhP{CH2CH2PPh2}2) have been prepared and characterised. X-ray crystallography (for R = Et, Ph, C6H4Me-4, C6H4OMe-4 and C6H4Cl-4) shows that the geometry of the five-coordinate nickel in the cation is best described as distorted trigonal bipyramidal, containing a bidentate carboxydithioate ligand with the two sulfur atoms spanning axial and equatorial sites, the other axial site being occupied by the central phosphorus of triphos. The reactions of [Ni(S2CR)(triphos)]+ with mixtures of HCl and Cl− in MeCN to form equilibrium solutions containing [Ni(SH(S)CR)(triphos)]2+ have been studied using stopped-flow spectrophotometry. The kinetics show that proton transfer is slower than the diffusion-controlled limit and involves at least two coupled equilibria. The first step involves the rapid association between [Ni(S2CR)(triphos)]+ and HCl to form the hydrogen-bonded precursor, {[Ni(S2CR)(triphos)]+⋯HCl} (KR1) and this is followed by the intramolecular proton transfer (kR2) to produce [Ni(SH(S)CR)(triphos)]2+. In the reaction of [Ni(S2CMe)(triphos)]+ the rate law is consistent with the carboxydithioate ligand undergoing chelate ring-opening after protonation. It seems likely that chelate ring-opening occurs for all [Ni(S2CR)(triphos)]+, but only with [Ni(S2CMe)(triphos)]+ is the protonation step sufficiently fast that chelate ring-opening is rate-limiting. With all other systems, proton transfer is rate-limiting. DFT calculations indicate that protonation can occur at either sulfur atom, but only protonation at the equatorial sulfur results in chelate ring-opening. The ways in which protonation of either sulfur atom complicates the analyses and interpretation of the kinetics are discussed.
Co-reporter:Dilek Nartop, William Clegg, Ross W. Harrington, Richard A. Henderson and Corinne Y. Wills
Dalton Transactions 2014 - vol. 43(Issue 8) pp:NaN3382-3382
Publication Date(Web):2013/12/18
DOI:10.1039/C3DT53359J
The kinetics of the reactions between [Ni(MeOH)6]2+ (hereafter Ni2+) and a variety of neutral Schiff base multidentate ligands have been measured in methanol at 25.0 °C using stopped-flow spectrophotometry. The ligands contain a variety of different potential donor sites (phenolic OH, imine N, pyridyl N and NH groups), different structural components and substituents. The kinetic studies explore how systematic changes to the composition of the ligands affect the rates of binding. The results are consistent with the Eigen–Wilkins mechanism in which the ligand initially forms an outer-sphere association with Ni2+ prior to dissociation of a coordinated solvent molecule and binding to the metal ion. The general features that emerge from these studies are as follows. (i) For ligands with the same donor set, the rates of binding are all similar irrespective of changes to the ligand framework (bridge and substituents). (ii) Comparison of structurally analogous ligands shows that the presence of pyridyl or NH groups in the multidentate results in significantly faster reactions. (iii) With ligands containing multiple NH groups, the rate of ligand binding increases as the number of NH groups increases. The extent to which these kinetic features can be attributed to preferential binding of particular donor groups is discussed.
Co-reporter:Ahmed Alwaaly, Ian Dance and Richard A. Henderson
Chemical Communications 2014 - vol. 50(Issue 37) pp:NaN4802-4802
Publication Date(Web):2014/03/10
DOI:10.1039/C4CC00922C
Density functional calculations show that Fe–S clusters undergo unexpected large structural changes when protonated at S. Protonation of prototypical cubanoid [Fe4S4X4]2− to [Fe4S3(SH)X4]− (X = Cl, SR, OR) results in formation of doubly-bridging SH, severance of one Fe–S bond, and creation of a three-coordinate Fe. These findings explain previously enigmatic results concerning the reactivity of these clusters, including the rates of protonation, pKa data, and the kinetics of acid-catalysed ligand substitution.
Co-reporter:Ahmed Alwaaly, William Clegg, Ross W. Harrington, Athinoula L. Petrou and Richard A. Henderson
Dalton Transactions 2015 - vol. 44(Issue 26) pp:NaN11983-11983
Publication Date(Web):2015/06/08
DOI:10.1039/C5DT01716E
Earlier kinetic studies on the protonation of the coordinated thiolate in the square-planar [Ni(SC6H4R′-4)(triphos)]+ (R′ = NO2, Cl, H, Me or MeO) by lutH+ (lut = 2,6-dimethylpyridine) indicate a two-step mechanism involving initial formation of a (kinetically detectable) precursor intermediate, {[Ni(SC6H4R′-4)(triphos)]⋯Hlut}2+ (KR1), followed by an intramolecular proton transfer step (kR2). The analogous [Ni(SR)(triphos)]BPh4 {R = Et, But or Cy; triphos = PhP(CH2CH2PPh2)2} have been prepared and characterized by spectroscopy and X-ray crystallography. Similar to the aryl thiolate complexes, [Ni(SR)(triphos)]+ are protonated by lutH+ in an equilibrium reaction but the observed rate law is simpler. Analysis of the kinetic data for both [Ni(SR)(triphos)]+ and [Ni(SC6H4R′-4)(triphos)]+ shows that both react by the same mechanism, but that KR1 is largest when the thiolate is poorly basic, or the 4-R′ substituent in the aryl thiolates is electron-withdrawing. These results indicate that it is both NH⋯S hydrogen bonding and encapsulation of the bound lutH+ (by the phenyl groups on triphos) which stabilize the precursor intermediate.
Co-reporter:Thaer M. M. Al-Rammahi and Richard A. Henderson
Dalton Transactions 2016 - vol. 45(Issue 4) pp:NaN1381-1381
Publication Date(Web):2015/12/08
DOI:10.1039/C5DT04523A
The mechanism of the acid-catalyzed substitution reaction of the terminal chloro-ligands in [Fe4S4Cl4]2− by PhS− in the presence of NHBun3+ involves rate-limiting proton transfer from NHBun3+ to the cluster (k0 = 490 ± 20 dm3 mol−1 s−1). A variety of small molecules and ions (L = substrate = Cl−, Br−, I−, RNHNH2 (R = Me or Ph), Me2NNH2, HCN, NCS−, N3−, ButNC or pyridine) bind to [Fe4S4Cl4]2− and this affects the rate of subsequent protonation of [Fe4S4Cl4(L)]n−. Where the kinetics allow, the equilibrium constants for the substrates binding to [Fe4S4Cl4]2− (KL) and the rates of proton transfer from NHBun3+ to [Fe4S4Cl4(L)]n− (kL0) have been determined. The results indicate the following general features. (i) Bound substrates increase the rate of protonation of the cluster, but the rate increase is modest (kL0/k0 = 1.6 to ≥72). (ii) When KL is small, so is kL0/k0. (iii) Binding substrates which are good σ-donors or good π-acceptors lead to the largest kL0/k0. This behaviour is discussed in terms of the recent proposal that protonation of [Fe4S4Cl4]2− at a μ3-S, is coupled to concomitant Fe–(μ3-SH) bond elongation/cleavage.









![1-Methyl-7-oxabicyclo[4.1.0]hepta-2,4-diene](http://img.cochemist.com/ccimg/17100/17091-93-9.png)
![1-Methyl-7-oxabicyclo[4.1.0]hepta-2,4-diene](http://img.cochemist.com/ccimg/17100/17091-93-9_b.png)





![7-Oxabicyclo[4.1.0]hepta-2,4-diene](http://img.cochemist.com/ccimg/1500/1488-25-1.png)
![7-Oxabicyclo[4.1.0]hepta-2,4-diene](http://img.cochemist.com/ccimg/1500/1488-25-1_b.png)





![1,6-DIMETHYL-7-OXABICYCLO[4.1.0]HEPTA-2,4-DIENE](http://img.cochemist.com/ccimg/73700/73654-32-7.png)
![1,6-DIMETHYL-7-OXABICYCLO[4.1.0]HEPTA-2,4-DIENE](http://img.cochemist.com/ccimg/73700/73654-32-7_b.png)