Co-reporter:S. Doherty;J. G. Knight;T. Backhouse;E. Abood;H. Alshaikh;I. J. S. Fairlamb;R. A. Bourne;T. W. Chamberlain;R. Stones
Green Chemistry (1999-Present) 2017 vol. 19(Issue 7) pp:1635-1641
Publication Date(Web):2017/04/03
DOI:10.1039/C6GC03528K
Phosphino-decorated polymer immobilised ionic liquid phase stabilised palladium nanoparticles (PdNP@PPh2-PIILP) and their PEGylated counterparts (PdNP@PPh2-PEGPIILP) are remarkably active and exceptionally selective catalysts for the aqueous phase hydrogenation of α,β-unsaturated aldehydes, ketones, esters and nitriles with PdNP@PPh2-PEGPIILP giving complete conversion and 100% selectivity for reduction of the CC bond, under mild conditions. This is the most selective PdNP-based system to be reported for the aqueous phase hydrogenation of this class of substrates.
Co-reporter:S. Doherty;J. G. Knight;T. Backhouse;E. Abood;H. Alshaikh;I. J. S. Fairlamb;R. A. Bourne;T. W. Chamberlain;R. Stones
Green Chemistry (1999-Present) 2017 vol. 19(Issue 7) pp:1635-1641
Publication Date(Web):2017/04/03
DOI:10.1039/C6GC03528K
Phosphino-decorated polymer immobilised ionic liquid phase stabilised palladium nanoparticles (PdNP@PPh2-PIILP) and their PEGylated counterparts (PdNP@PPh2-PEGPIILP) are remarkably active and exceptionally selective catalysts for the aqueous phase hydrogenation of α,β-unsaturated aldehydes, ketones, esters and nitriles with PdNP@PPh2-PEGPIILP giving complete conversion and 100% selectivity for reduction of the CC bond, under mild conditions. This is the most selective PdNP-based system to be reported for the aqueous phase hydrogenation of this class of substrates.
Co-reporter:Simon Doherty, Julian G. Knight, Daniel O. Perry, Nicholas A. B. Ward, Dror M. Bittner, William McFarlane, Corinne Wills, and Michael R. Probert
Organometallics 2016 Volume 35(Issue 9) pp:1265-1278
Publication Date(Web):April 27, 2016
DOI:10.1021/acs.organomet.6b00146
A homologous series of triaryl-like KITPHOS-type monophosphines containing one, two, or three bulky 12-phenyl-9,10-dihydro-9,10-ethenoanthracene (KITPHOS) units have been developed, and the influence of increasing steric bulk on their efficacy as ligands in gold(I)-catalyzed carbon–heteroatom bond-forming cyclizations has been investigated. Detailed solution NMR studies on Ph-TRISKITPHOS, its oxide, and the corresponding gold(I) chloride adduct identified a conformational exchange process involving a concerted librational motion of the individual anthracene-derived organic substituents about their P–C bonds. The cessation of this motion at reduced temperatures lowers the molecular symmetry such that the two C6H4 rings in each of the KITPHOS units become inequivalent; a lower energy process involving restricted rotation of the biaryl-like phenyl ring has also been identified. Electrophilic gold(I) complexes of these triaryl-like KITPHOS monophosphines catalyze the 5-exo-dig cycloisomerization of propargyl amides to afford the corresponding methylene oxazolines, which were used in a subsequent tandem carbonyl-ene reaction to afford functionalized 2-substituted oxazolines. A comparative survey revealed that catalyst efficiency for cycloisomerization decreases in the order MONOKITPHOS = BISKITPHOS > PPh3 > TRISKITPHOS. The optimum system also catalyzes the selective 6-endo-dig cyclization of 2-alkynylbenzyl alcohols, 2-alkynylbenzoic acid, and 2-phenylethynyl benzamides; gratifyingly, in several cases the yields obtained are markedly higher and/or reaction times significantly shorter than those previously reported for related gold catalysts. Moreover, these are the first examples of gold(I)-catalyzed 6-endo-dig cycloisomerizations involving 2-phenylethynyl benzamides and, reassuringly, the optimum gold(I)/MONOKITPHOS systems either rivaled or outperformed existing silver or palladium-based catalysts. The steric parameters of this homologous series of phosphines have been quantified and compared with selected triarylphosphines using a combination of Solid-G calculations, to determine the percentage of the metal coordination sphere shielded by the phosphine (the G parameter), and Salerno molecular buried volume calculations (SambVca) to determine the percent buried volume (%Vbur); the corresponding Tolman cone angles have also been determined from correlations.
Co-reporter:Simon Doherty, Julian G. Knight, Nicholas A. B. Ward, Daniel O. Perry, Dror M. Bittner, Michael R. Probert, and Stephen A. Westcott
Organometallics 2014 Volume 33(Issue 19) pp:5209-5219
Publication Date(Web):September 5, 2014
DOI:10.1021/om500520z
This paper reports the first examples of Suzuki–Miyaura cross-couplings involving aryl- and naphthylphosphonate-based boronate esters as the nucleophilic partner. A systematic comparison of the performance of biaryl-like KITPHOS- and XPHOS-based systems revealed that, between them, an electronically and sterically diverse range of substrates can be coupled with remarkable efficiency to afford high yields of the corresponding biaryl and heterobiaryl monophosphonates. The use of an aryl- and naphthylphosphonate-based boronate ester as the coupling partner presents an alternative and potentially complementary pathway to existing couplings in which the aryl- or naphthylphosphonate unit is typically introduced as the electrophile. The potential advantages associated with the use of this new class of coupling partner were clearly demonstrated by the palladium-catalyzed reaction between diethyl [2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]phosphonate and 1-bromo-2-methoxynaphthalene that gave the corresponding biaryl monophosphonate in 56% yield, a marked improvement on the 6% yield obtained from the reaction between 2-methoxy-1-naphthylboronic acid and diethyl (2-bromophenyl)phosphonate with the same catalyst under the same conditions. The potential utility of this new coupling combination was demonstrated by reducing one of the products, 2-methoxy-1-(2′-diethoxyphosphorylphenyl)naphthylene, to the corresponding primary phosphine, which was subsequently converted into a diastereoisomeric mixture of the R,R-hexane-2,5-diol-derived phospholane in reasonable yield.
Co-reporter:S. Doherty;P. Goodrich;C. Hardacre;V. Pârvulescu;C. Paun
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 2) pp:295-302
Publication Date(Web):
DOI:10.1002/adsc.200700425
Abstract
Lewis acid complexes based on copper(II) and an imidazolium-tagged bis(oxazoline) have been used to catalyse the asymmetric Mukaiyama aldol reaction between methyl pyruvate and 1-methoxy-1-trimethylsilyloxypropene under homogeneous and heterogeneous conditions. Although the ees obtained in ionic liquid were similar to those found in dichloromethane, there was a significant rate enhancement in the ionic liquid with reactions typically reaching completion within 2 min compared with only 55 % conversion after 60 min in dichloromethane. However, this rate enhancement was offset by lower chemoselectivity in ionic liquids due to the formation of 3-hydroxy-1,3-diphenylbutan-1-one as a by-product. Supporting the catalyst on silica or an imidazolium-modified silica using the ionic liquid or in an ionic liquid-diethyl ether system completely suppressed the formation of this by-product without reducing the enantioselectivity. Although the heterogeneous systems were characterised by a drop in catalytic activity the system could be recycled up to five times without any loss in conversion or ee.
Co-reporter:Simon Doherty;JulianG. Knight;CatherineH. Smyth ;GraemeA. Jorgenson
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 11-12) pp:1801-1806
Publication Date(Web):
DOI:10.1002/adsc.200800307
Abstract
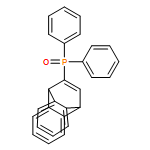
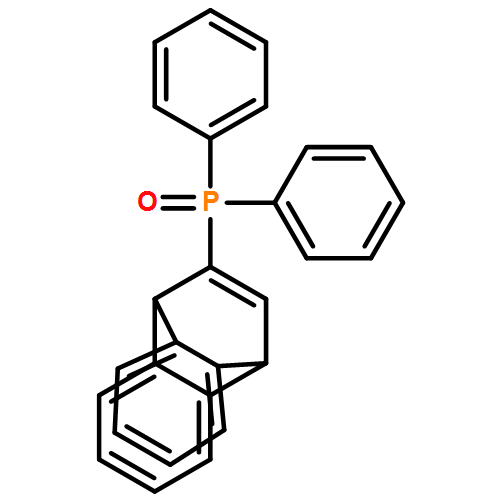
Electron-rich, bicyclic biaryl-like KITPHOS monophosphines have been prepared via Diels–Alder cycloaddition between 1-alkynylphosphine oxides and anthracene in an operationally straightforward and highly modular synthetic protocol that will allow access to an architecturally and electronically diverse family of ligands. Palladium complexes of these ligands are highly efficient catalysts for the Buchwald–Hartwig amination and Suzuki–Miyaura coupling of a wide range of aryl chlorides, which for the vast majority of substrate combinations outperform their o-(dicyclohexylphosphino)biphenyl-based counterparts.
Co-reporter:Simon Doherty;Julian G. Knight;Amy McRae;Ross W. Harrington ;William Clegg
European Journal of Organic Chemistry 2008 Volume 2008( Issue 10) pp:1759-1766
Publication Date(Web):
DOI:10.1002/ejoc.200700922
Abstract
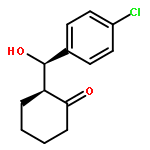
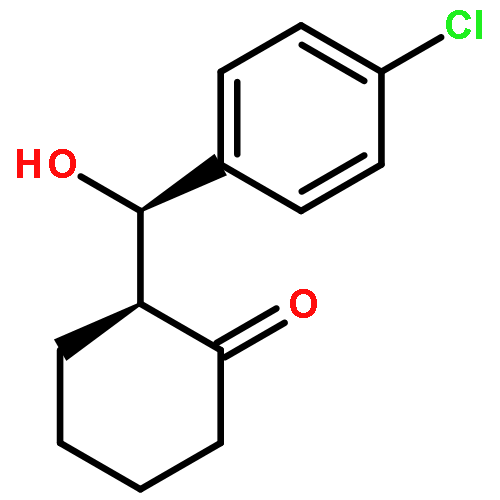
Oxazoline-substituted prolinamides catalyse the direct asymmetric aldol reaction between cyclohexanone and a range of aldehydes to give excellent conversions and enantioselectivities up to 84 % under optimum conditions. Reactions were highly substrate-specific with electron-deficient aldehydes giving the highest yields and ee values. The absolute configuration of the 4-chlorobenzaldehyde-derived product was unequivocally established as (2S,1′R) by single-crystal X-ray analysis, and the stereochemistry of the product was shown to be determined principally by the stereochemistry of the proline fragment.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Simon Doherty;Peter Goodrich;Christopher Hardacre;Julian G. Knight;Mimi T. Nguyen;Vasile I. Pârvulescu;Cristina Paun
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 6) pp:
Publication Date(Web):17 APR 2007
DOI:10.1002/adsc.200600531
Imidazolium-tagged bis(oxazolines) have been prepared and used as chiral ligands in the copper(II)-catalysed Diels–Alder reaction of N-acryloyl- and N-crotonoyloxazolidinones with cyclopentadiene and 1,3-cyclohexadiene in the ionic liquid 1-ethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide, [emim][NTf2]. A significant and substantial enhancement in the rate and enantioselectivity was achieved in [emim][NTf2] compared with dichloromethane. For example, complete conversion and enantioselectivities up to 95 % were obtained for the reaction between N-acryloyloxazolidinone and cyclopentadiene within 2 min in [emim][NTf2] whereas the corresponding reaction in dichloromethane required 60 min to reach completion and gave an ee of only 16 %. The enhanced rates obtained in the ionic liquid enabled a catalyst loading as low as 0.5 mol % to give complete conversion within 2 min while retaining the same level of enantioselectivity. The imidazolium-tagged catalysts can be recycled ten times without any loss in activity or enantioselectivity and showed much higher affinity for the ionic liquid phase during the recycle procedure than the analogous uncharged ligand.
Co-reporter:Simon Doherty, Julian G. Knight and Michael Betham
Chemical Communications 2006 (Issue 1) pp:88-90
Publication Date(Web):14 Nov 2005
DOI:10.1039/B512556A
Palladium complexes of 2-pyridyldiphenylphosphine anchored on polystyrene, polymethylmethacrylate and styrene–methylmethacrylate copolymer form highly active heterogeneous catalysts for the alkoxycarbonylation of terminal alkynes with activities approaching those obtained under homogeneous conditions.

![Phosphine oxide, [3-[2-(diphenylphosphinyl)ethynyl]-5,6,7,8-tetrahydro-1,4-dimethyl-2-naphthalenyl]diphenyl-](/data/chemimg/3726400/959864-35-8.png)
![Phosphine oxide, [3-[2-(diphenylphosphinyl)ethynyl]-5,6,7,8-tetrahydro-1,4-dimethyl-2-naphthalenyl]diphenyl-](/data/chemimg/3726400/959864-35-8_b.png)
![5,5'-Bi-1H-isoindole, 6,6'-bis(diphenylphosphino)-2,2',3,3'-tetrahydro-2,2'-bis[(4-methylphenyl)sulfonyl]-](/data/chemimg/3720700/959864-40-5.png)
![5,5'-Bi-1H-isoindole, 6,6'-bis(diphenylphosphino)-2,2',3,3'-tetrahydro-2,2'-bis[(4-methylphenyl)sulfonyl]-](/data/chemimg/3720700/959864-40-5_b.png)


![Phosphine, (6-methoxy[1,1'-biphenyl]-2,2'-diyl)bis[diphenyl-](/data/chemimg/3506400/871707-43-6.png)
![Phosphine, (6-methoxy[1,1'-biphenyl]-2,2'-diyl)bis[diphenyl-](/data/chemimg/3506400/871707-43-6_b.png)






![Benzenamine, 2-[(3aS,8aR)-3a,8a-dihydro-8H-indeno[1,2-d]oxazol-2-yl]-](/data/chemimg/3721500/602313-61-1.png)
![Benzenamine, 2-[(3aS,8aR)-3a,8a-dihydro-8H-indeno[1,2-d]oxazol-2-yl]-](/data/chemimg/3721500/602313-61-1_b.png)




![5,5'-Bi-1H-isoindole, 6,6'-bis(diphenylphosphinyl)-2,2',3,3'-tetrahydro-2,2'-bis[(4-methylphenyl)sulfonyl]-](/data/chemimg/3720700/959864-34-7.png)
![5,5'-Bi-1H-isoindole, 6,6'-bis(diphenylphosphinyl)-2,2',3,3'-tetrahydro-2,2'-bis[(4-methylphenyl)sulfonyl]-](/data/chemimg/3720700/959864-34-7_b.png)
![[5,5'-Bi-2H-indene]-2,2,2',2'-tetracarboxylic acid, 6,6'-bis(diphenylphosphinyl)-1,1',3,3'-tetrahydro-, 2,2,2',2'-tetramethyl ester](/data/chemimg/3720700/959864-33-6.png)
![[5,5'-Bi-2H-indene]-2,2,2',2'-tetracarboxylic acid, 6,6'-bis(diphenylphosphinyl)-1,1',3,3'-tetrahydro-, 2,2,2',2'-tetramethyl ester](/data/chemimg/3720700/959864-33-6_b.png)


![2H-Indene-2,2-dicarboxylic acid, 5-(diphenylphosphinyl)-6-[2-(diphenylphosphinyl)ethynyl]-1,3-dihydro-4,7-dimethyl-, 2,2-dimethyl ester](/data/chemimg/3720700/1067274-35-4.png)
![2H-Indene-2,2-dicarboxylic acid, 5-(diphenylphosphinyl)-6-[2-(diphenylphosphinyl)ethynyl]-1,3-dihydro-4,7-dimethyl-, 2,2-dimethyl ester](/data/chemimg/3720700/1067274-35-4_b.png)



![[5,5'-Bi-2H-indene]-2,2,2',2'-tetracarboxylic acid, 6,6'-bis(diphenylphosphino)-1,1',3,3'-tetrahydro-, 2,2,2',2'-tetramethyl ester](/data/chemimg/3720700/959864-39-2.png)
![[5,5'-Bi-2H-indene]-2,2,2',2'-tetracarboxylic acid, 6,6'-bis(diphenylphosphino)-1,1',3,3'-tetrahydro-, 2,2,2',2'-tetramethyl ester](/data/chemimg/3720700/959864-39-2_b.png)










![9-Oxabicyclo[6.1.0]nonane, (1R,8S)-rel-](http://img.cochemist.com/ccimg/5000/4925-71-7.png)
![9-Oxabicyclo[6.1.0]nonane, (1R,8S)-rel-](http://img.cochemist.com/ccimg/5000/4925-71-7_b.png)


![ETHANONE, 1-(2'-METHYL[1,1'-BIPHENYL]-2-YL)-](http://img.cochemist.com/ccimg/217500/217468-51-4.png)
![ETHANONE, 1-(2'-METHYL[1,1'-BIPHENYL]-2-YL)-](http://img.cochemist.com/ccimg/217500/217468-51-4_b.png)


![2-Pyridinamine, N-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/722500/722498-54-6.png)
![2-Pyridinamine, N-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/722500/722498-54-6_b.png)









![CYCLOHEXANONE, 2-[(R)-2-FURANYLHYDROXYMETHYL]-, (2S)-](http://img.cochemist.com/ccimg/882500/882497-82-7.png)
![CYCLOHEXANONE, 2-[(R)-2-FURANYLHYDROXYMETHYL]-, (2S)-](http://img.cochemist.com/ccimg/882500/882497-82-7_b.png)






![2-Oxazolidinone, 3-[(1R,2R,4R)-bicyclo[2.2.1]hept-5-en-2-ylcarbonyl]-](http://img.cochemist.com/ccimg/131600/131564-05-1.png)
![2-Oxazolidinone, 3-[(1R,2R,4R)-bicyclo[2.2.1]hept-5-en-2-ylcarbonyl]-](http://img.cochemist.com/ccimg/131600/131564-05-1_b.png)
![PHOSPHONIC ACID, [1-(2-METHYLPHENYL)-2-NAPHTHALENYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/571300/571206-39-8.png)
![PHOSPHONIC ACID, [1-(2-METHYLPHENYL)-2-NAPHTHALENYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/571300/571206-39-8_b.png)


![PYRIDINE, 2-[(4-ETHENYLPHENYL)PHENYLPHOSPHINO]-](http://img.cochemist.com/ccimg/875800/875778-90-8.png)
![PYRIDINE, 2-[(4-ETHENYLPHENYL)PHENYLPHOSPHINO]-](http://img.cochemist.com/ccimg/875800/875778-90-8_b.png)



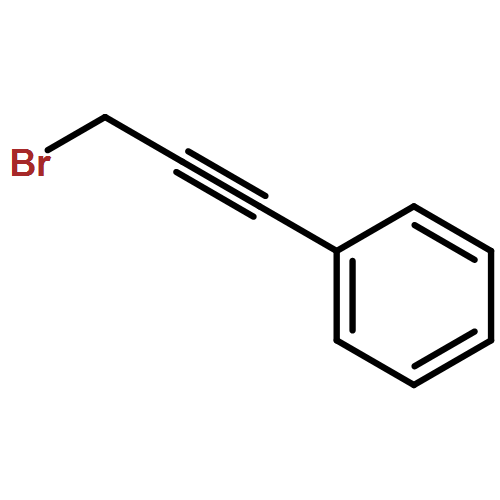




![1H-PYRAZOLE, 1-(4'-METHYL[1,1'-BIPHENYL]-2-YL)-](http://img.cochemist.com/ccimg/864100/864074-48-6.png)
![1H-PYRAZOLE, 1-(4'-METHYL[1,1'-BIPHENYL]-2-YL)-](http://img.cochemist.com/ccimg/864100/864074-48-6_b.png)






![PHOSPHONOUS DICHLORIDE, [BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76505-20-9.png)
![PHOSPHONOUS DICHLORIDE, [BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76505-20-9_b.png)
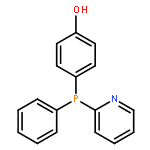

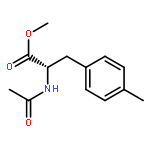







![[1,1'-biphenyl]-2,2'-diylbis(diphenylphosphane oxide)](/data/chemimg/2253300/174467-54-0.png)
![[1,1'-biphenyl]-2,2'-diylbis(diphenylphosphane oxide)](/data/chemimg/2253300/174467-54-0_b.png)


![Morpholine, 4-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/84800/84736-47-0.png)
![Morpholine, 4-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/84800/84736-47-0_b.png)










![Benzenamine, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/1787400/209850-73-7.png)
![Benzenamine, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/1787400/209850-73-7_b.png)




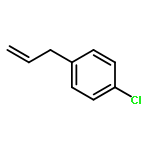
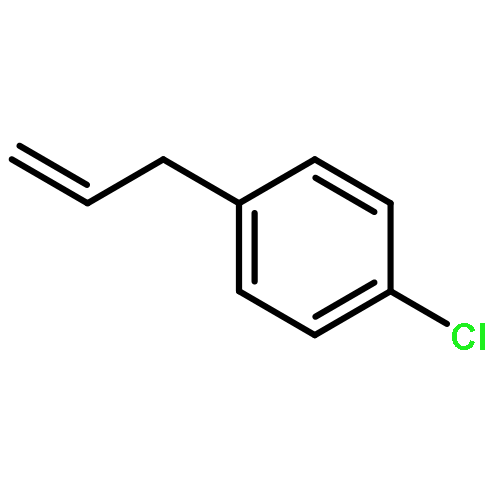










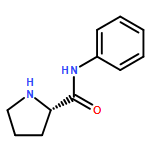
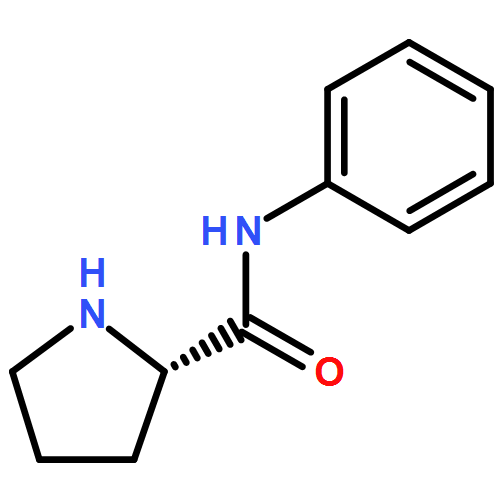




![4-Cyclohexene-1,2-dicarboxylic acid,bis[(1R,2S,5R)-5-methyl-2-(1-methylethyl)cyclohexyl] ester, (1S,2S)-](http://img.cochemist.com/ccimg/109100/109062-95-5.png)
![4-Cyclohexene-1,2-dicarboxylic acid,bis[(1R,2S,5R)-5-methyl-2-(1-methylethyl)cyclohexyl] ester, (1S,2S)-](http://img.cochemist.com/ccimg/109100/109062-95-5_b.png)






![Isobenzofuran, 1,3-dihydro-1-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/64500/64421-13-2.png)
![Isobenzofuran, 1,3-dihydro-1-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/64500/64421-13-2_b.png)
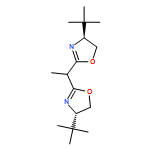
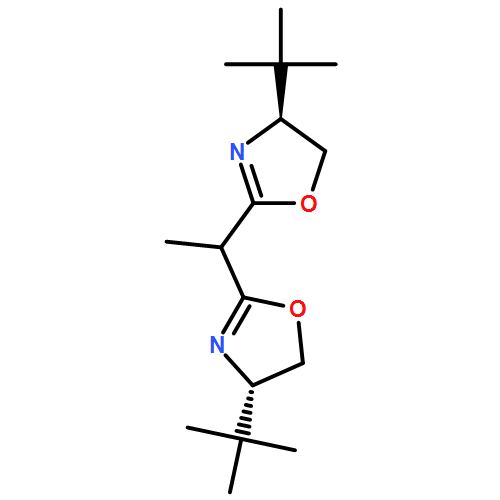
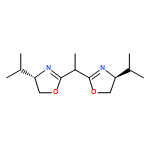
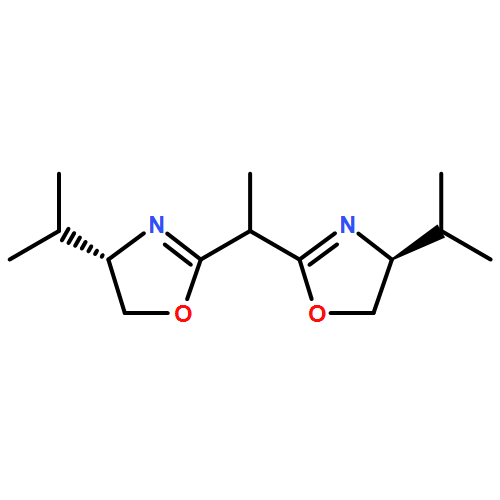
![Dichloro[(R)-(+)-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl]palladium(II), min.](http://img.cochemist.com/ccimg/115900/115826-95-4.png)
![Dichloro[(R)-(+)-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl]palladium(II), min.](http://img.cochemist.com/ccimg/115900/115826-95-4_b.png)
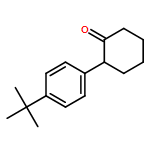





![2'-Methyl-[1,1'-biphenyl]-4-carbonitrile](http://img.cochemist.com/ccimg/189900/189828-30-6.png)
![2'-Methyl-[1,1'-biphenyl]-4-carbonitrile](http://img.cochemist.com/ccimg/189900/189828-30-6_b.png)




![Cyclohexanone, 2-[(R)-(4-bromophenyl)hydroxymethyl]-, (2S)-](http://img.cochemist.com/ccimg/281700/281676-87-7.png)
![Cyclohexanone, 2-[(R)-(4-bromophenyl)hydroxymethyl]-, (2S)-](http://img.cochemist.com/ccimg/281700/281676-87-7_b.png)




![Titanium,trichloro[(1,2,3,4,5-h)-1-methyl-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/1300/1282-31-1.png)
![Titanium,trichloro[(1,2,3,4,5-h)-1-methyl-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/1300/1282-31-1_b.png)






![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7_b.png)
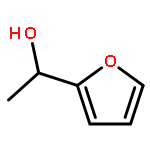
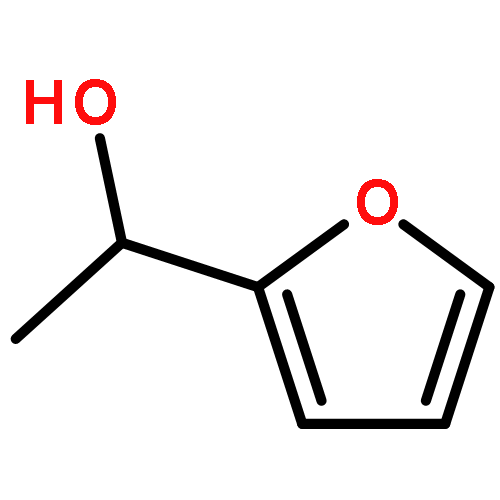








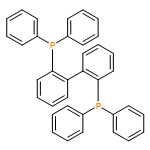
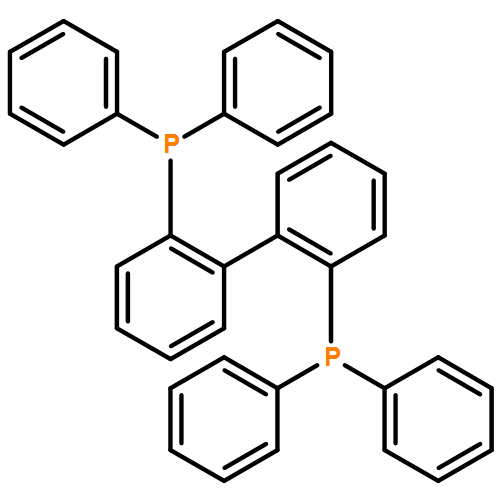










![1-[(4-ETHENYLPHENYL)METHYL]PYRROLIDINE](http://img.cochemist.com/ccimg/60500/60472-54-0.png)
![1-[(4-ETHENYLPHENYL)METHYL]PYRROLIDINE](http://img.cochemist.com/ccimg/60500/60472-54-0_b.png)


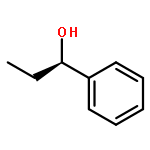
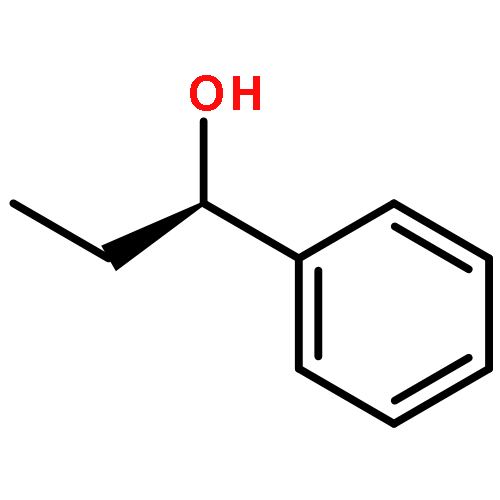
![Cyclohexanone, 2-[(R)-hydroxy(4-nitrophenyl)methyl]-, (2S)-rel-](/data/chemimg/664300/71444-30-9.png)
![Cyclohexanone, 2-[(R)-hydroxy(4-nitrophenyl)methyl]-, (2S)-rel-](/data/chemimg/664300/71444-30-9_b.png)
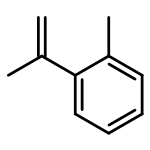
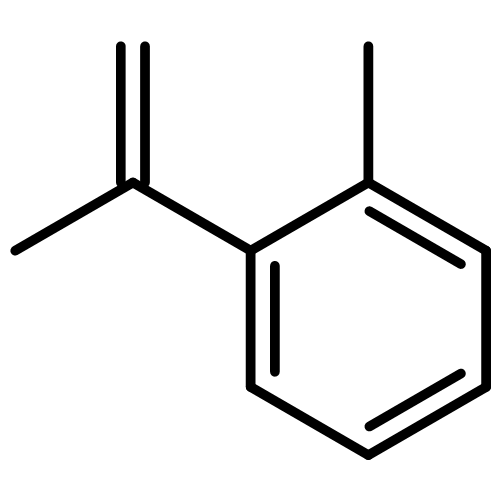



![1-[4-(2-methylphenyl)phenyl]ethanone](http://img.cochemist.com/ccimg/57000/56917-39-6.png)
![1-[4-(2-methylphenyl)phenyl]ethanone](http://img.cochemist.com/ccimg/57000/56917-39-6_b.png)










![1,2-Bis[(Z)-1-(diphenylphosphino)prop-1-ylidene]cyclohexane, min. 98% Et2-CYCLO-NUPHOS](http://img.cochemist.com/ccimg/420200/420120-78-1.png)
![1,2-Bis[(Z)-1-(diphenylphosphino)prop-1-ylidene]cyclohexane, min. 98% Et2-CYCLO-NUPHOS](http://img.cochemist.com/ccimg/420200/420120-78-1_b.png)




![2-Pentanone, 4-[(2-hydroxypropyl)imino]-](http://img.cochemist.com/ccimg/27400/27372-93-6.png)
![2-Pentanone, 4-[(2-hydroxypropyl)imino]-](http://img.cochemist.com/ccimg/27400/27372-93-6_b.png)







![Pyridine, 2-[1,1':3',1''-terphenyl]-2'-yl-](/data/chemimg/129800/219843-49-9.png)
![Pyridine, 2-[1,1':3',1''-terphenyl]-2'-yl-](/data/chemimg/129800/219843-49-9_b.png)






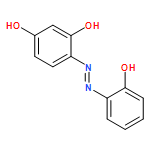
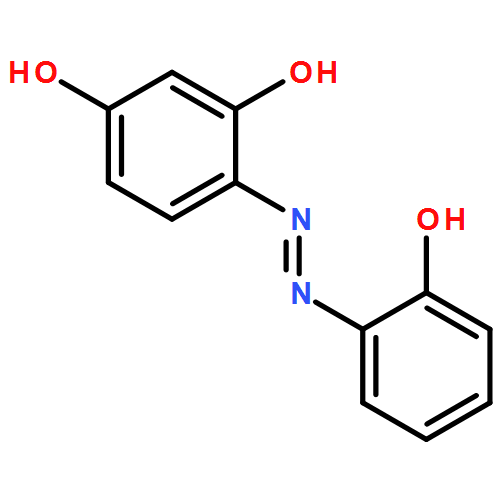





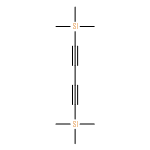
-](http://img.cochemist.com/ccimg/34800/34767-55-0.png)
-](http://img.cochemist.com/ccimg/34800/34767-55-0_b.png)


















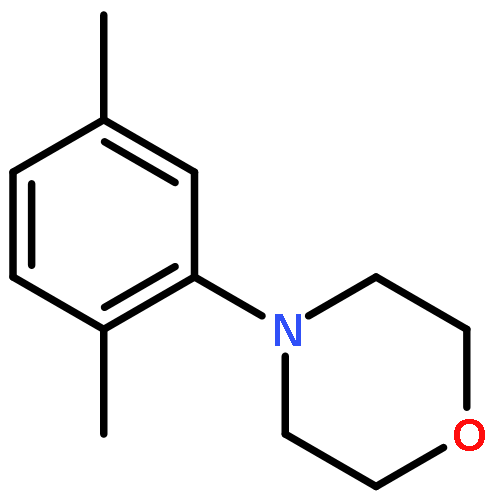
![Pyridine, 2-[1,1'-biphenyl]-2-yl-](/data/chemimg/129800/219843-48-8.png)
![Pyridine, 2-[1,1'-biphenyl]-2-yl-](/data/chemimg/129800/219843-48-8_b.png)








![Cyclohexanone, 2-[(R)-(2-chlorophenyl)hydroxymethyl]-, (2S)-](http://img.cochemist.com/ccimg/921000/920956-95-2.png)
![Cyclohexanone, 2-[(R)-(2-chlorophenyl)hydroxymethyl]-, (2S)-](http://img.cochemist.com/ccimg/921000/920956-95-2_b.png)


![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5.png)
![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5_b.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2_b.png)
![Cyclohexanone, 2-[(R)-hydroxyphenylmethyl]-, (2S)-](http://img.cochemist.com/ccimg/156100/156039-16-6.png)
![Cyclohexanone, 2-[(R)-hydroxyphenylmethyl]-, (2S)-](http://img.cochemist.com/ccimg/156100/156039-16-6_b.png)




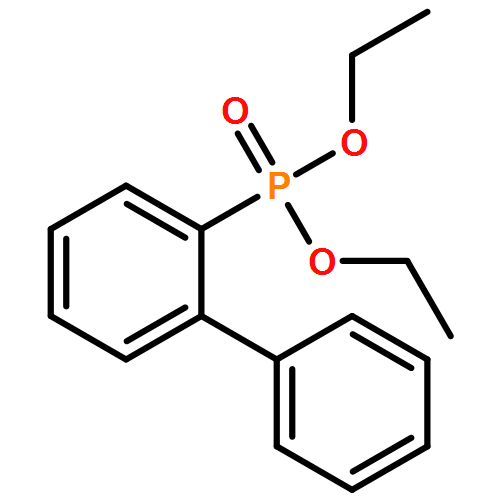




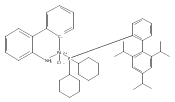
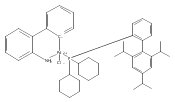




![Benzenamine, 2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-](/data/chemimg/108800/194350-71-5.png)
![Benzenamine, 2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-](/data/chemimg/108800/194350-71-5_b.png)


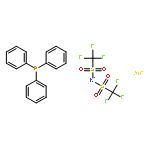
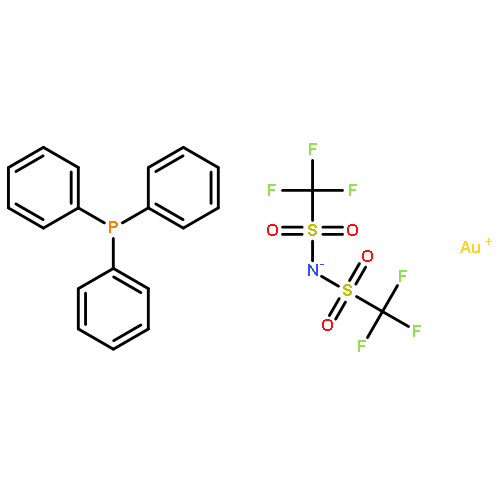

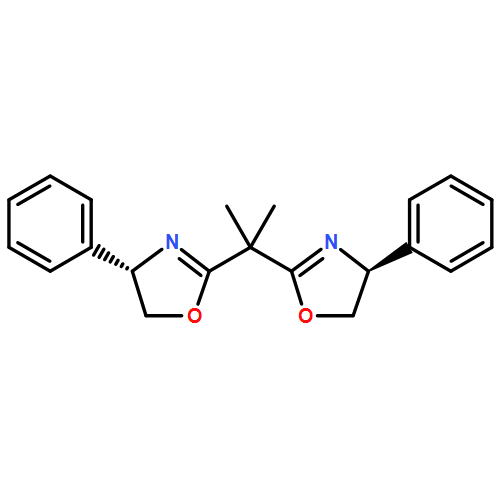
![Cyclohexanone, 2-[(R)-hydroxy(4-nitrophenyl)methyl]-, (2S)-](/data/chemimg/104400/351533-35-2.png)
![Cyclohexanone, 2-[(R)-hydroxy(4-nitrophenyl)methyl]-, (2S)-](/data/chemimg/104400/351533-35-2_b.png)
![Ethanone,1-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2142-66-7.png)
![Ethanone,1-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2142-66-7_b.png)
![[(2s,3r)-3-(methylsulfonyloxymethyl)-2-bicyclo[2.2.1]heptanyl]methyl Methanesulfonate](http://img.cochemist.com/ccimg/2500/2434-88-0.png)
![[(2s,3r)-3-(methylsulfonyloxymethyl)-2-bicyclo[2.2.1]heptanyl]methyl Methanesulfonate](http://img.cochemist.com/ccimg/2500/2434-88-0_b.png)




![Bicyclo[2.2.1]heptane-2,3-dimethanol,2,3-dimethanesulfonate, (1R,2R,3R,4S)-rel-](http://img.cochemist.com/ccimg/2500/2434-90-4.png)
![Bicyclo[2.2.1]heptane-2,3-dimethanol,2,3-dimethanesulfonate, (1R,2R,3R,4S)-rel-](http://img.cochemist.com/ccimg/2500/2434-90-4_b.png)
![Ruthenium,[1,1'-(1R)-[1,1'-binaphthalene]-2,2'-diylbis[1,1-diphenylphosphine-kP]]dichloro[(1R,2R)-1,2-diphenyl-1,2-ethanediamine-kN1,kN2]-, (OC-6-13)-](http://img.cochemist.com/ccimg/212200/212143-23-2.png)
![Ruthenium,[1,1'-(1R)-[1,1'-binaphthalene]-2,2'-diylbis[1,1-diphenylphosphine-kP]]dichloro[(1R,2R)-1,2-diphenyl-1,2-ethanediamine-kN1,kN2]-, (OC-6-13)-](http://img.cochemist.com/ccimg/212200/212143-23-2_b.png)

