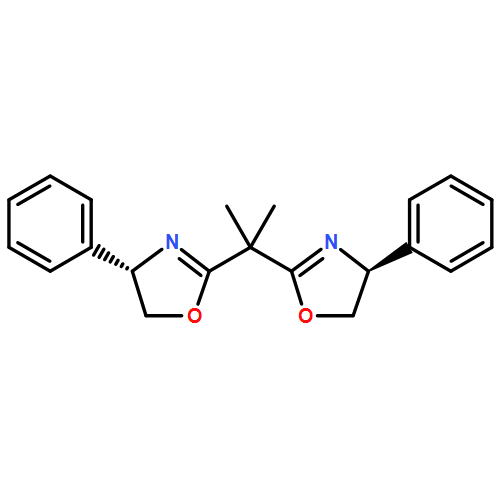
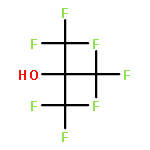
Co-reporter:Nadine Ritterskamp, Katherine Sharples, Emma Richards, Andrea Folli, Mario Chiesa, James A. Platts, and Damien M. Murphy
Inorganic Chemistry October 2, 2017 Volume 56(Issue 19) pp:11862-11862
Publication Date(Web):September 21, 2017
DOI:10.1021/acs.inorgchem.7b01874
The interaction of imidazole with a [Cu(acac)2] complex was studied using electron paramagnetic resonance (EPR), electron nuclear double resonance (ENDOR), hyperfine sublevel correlation spectroscopy (HYSCORE), and density functional theory (DFT). At low Im ratios (Cu:Im 1:10), a 5-coordinate [Cu(acac)2Imn=1] monoadduct is formed in frozen solution with the spin Hamiltonian parameters g1 = 2.063, g2 = 2.063, g3 = 2.307, A1 = 26, A2 = 15, and A3 = 472 MHz with Im coordinating along the axial direction. At higher Im concentrations (Cu:Im 1:50), a 6-coordinate [Cu(acac)2Imn=2] bis-adduct is formed with the spin Hamiltonian parameters g1 = 2.059, g2 = 2.059, g3 = 2.288, A1 = 30, A2 = 30, and A3 = 498 MHz with a poorly resolved 14N superhyperfine pattern. The isotropic EPR spectra revealed a distribution of species ([Cu(acac)2], [Cu(acac)2Imn=1], and [Cu(acac)2Imn=2]) at Cu:Im ratios of 1:0, 1:10, and 1:50. The superhyperfine pattern originates from two strongly coordinating N3 imino nitrogens of the Im ring. Angular selective 14N ENDOR analysis revealed the NA tensor of [34.8, 43.5, 34.0] MHz, with e2qQ/h = 2.2 MHz and η = 0.2 for N3. The hyperfine and quadrupole values for the remote N1 amine nitrogens (from HYSCORE) were found to be [1.5, 1.4, 2.5] MHz with e2qQ/h = 1.4 MHz and η = 0.9. 1H ENDOR also revealed three sets of HA tensors corresponding to the nearly equivalent H2/H4 protons in addition to the H5 and H1 protons of the Im ring. The spin Hamiltonian parameters for the geometry optimized structures of [Cu(acac)2Imn=2], including cis-mixed plane, trans-axial, and trans-equatorial, were calculated. The best agreement between theory and experiment indicated the preferred coordination is trans-equatorial [Cu(acac)2Imn=2]. A number of other Im derivatives were also investigated. 4(5)-methyl-imidazole forms a [Cu(acac)2(Im-3)n=2] trans-equatorial adduct, whereas the bulkier 2-methyl-imidazole (Im-2) and benzimidazole (Im-4) form the [Cu(acac)2(Im-2,4)n=1] monoadduct only. Our data therefore show that subtle changes in the substrate structure lead to controllable changes in coordination behavior, which could in turn lead to rational design of complexes for use in catalysis, imaging, and medicine.
Co-reporter:Stefan Pelties, Emma Carter, Andrea Folli, Mary F. Mahon, Damien M. Murphy, Michael K. Whittlesey, and Robert Wolf
Inorganic Chemistry 2016 Volume 55(Issue 21) pp:11006
Publication Date(Web):October 12, 2016
DOI:10.1021/acs.inorgchem.6b01540
Potassium graphite reduction of the half-sandwich Ni(II) ring-expanded diamino/diamidocarbene complexes CpNi(RE-NHC)Br gave the Ni(I) derivatives CpNi(RE-NHC) (where RE-NHC = 6-Mes (1), 7-Mes (2), 6-MesDAC (3)) in yields of 40%–50%. The electronic structures of paramagnetic 1–3 were investigated by CW X-/Q-band electron paramagnetic resonance (EPR) and Q-band 1H electron nuclear double resonance (ENDOR) spectroscopy. While small variations in the g-values were observed between the diaminocarbene complexes 1 and 2, pronounced changes in the g-values were detected between the almost isostructural species (1) and diamidocarbene species (3). These results highlight the sensitivity of the EPR g-tensor to changes in the electronic structure of the Ni(I) centers generated by incorporation of heteroatom substituents onto the backbone ring positions. Variable-temperature EPR analysis also revealed the presence of a second Ni(I) site in 3. The experimental g-values for these two Ni(I) sites detected by EPR in frozen solutions of 3 are consistent with resolution on the EPR time scale of the disordered components evident in the X-ray crystallographically determined structure and the corresponding density functional theory (DFT)-calculated g-tensor. Q-band 1H ENDOR measurements revealed a small amount of unpaired electron spin density on the Cp rings, consistent with the calculated SOMO of complexes 1–3. The magnitude of the 1H A values for 3 were also notably larger, compared to 1 and 2, again highlighting the influence of the diamidocarbene on the electronic properties of 3.
Co-reporter:Mary R. Healy; Emma Carter; Ian A. Fallis; Ross S. Forgan; Ross J. Gordon; Eduardo Kamenetzky; Jason B. Love; Carole A. Morrison; Damien M. Murphy;Peter A. Tasker
Inorganic Chemistry 2015 Volume 54(Issue 17) pp:8465-8473
Publication Date(Web):August 19, 2015
DOI:10.1021/acs.inorgchem.5b01180
Copper complexes of the phenolic oxime family of ligands (3-X-salicylaldoximes) are used extensively as metal solvent extractants. Incorporation of electronegative substituents in the 3-position, ortho to the phenol group, can be used to buttress the interligand H-bonding, leading to an enhancement in extractant strength. However, investigation of the relevant H-bonding in these complexes can be exceedingly difficult. Here, we have combined EPR, ENDOR, DFT, and X-ray crystallography to study this effect. Analysis of the 1H ENDOR data revealed a variation in the Cu···H16 (oxime proton) distance from 2.92 Å for the unsubstituted complex [Cu(L2)2] to 3.65 Å for the X = CH2N(C6H13)2 substituted complex [Cu(L3)2]. DFT calculations showed that this variation is caused by changes to the length and strength of the H-bond between the oximic hydrogen and the phenolate oxygen. Noticeable changes to the Cu···H15 (azomethine proton) distances and the Cu···N bonding parameters were also observed in the two complexes, as revealed through the NA and NQ ENDOR data. Distortions in the structure of the complex and variations in the oximic proton to phenolate oxygen H-bond strength caused by the substituent (X) were confirmed by DFT and X-ray crystallography. DFT directly evidenced the importance of the interaction between H16 and the amine nitrogen of CH2N(C6H13)2 in the buttressed complex and indicated that the high strength of this interaction may not necessarily lead to an enhancement of copper extraction, as it can impose an unfavorable geometry in the inner coordination sphere of the complex. Therefore, ENDOR, DFT, and X-ray structural data all indicate that the aminomethyl substituent (X) ortho to the phenolic oxygen atom provides a particularly strong buttressing of interligand H-bonding in these copper complexes and that these outer sphere interactions can significantly influence structure and stability.
Co-reporter:Evi Vinck ; Emma Carter ; Damien M. Murphy ;Sabine Van Doorslaer
Inorganic Chemistry 2012 Volume 51(Issue 15) pp:8014-8024
Publication Date(Web):July 25, 2012
DOI:10.1021/ic300058p
The interactions of a weak organic acid (acetic acid, HOAc) with a toluene solution of the CoII–Schiff base type complex, (R,R′)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexane-diamino CoII (labeled [Co(1)]), was investigated using EPR, HYSCORE, and DFT computations. This activated [CoII(1)] system is extremely important within the context of asymmetric catalysts (notably the hydrolytic kinetic resolution of epoxides) despite the lack of detailed structural information about the nature of the paramagnetic species present. Under anaerobic conditions, the LS [CoII(1)] complex with a |yz, 2A2⟩ ground state is converted into a low-spin (LS) and a high-spin (HS) complex in the presence of the acid. The newly formed LS state is assigned to the coordinated [CoII(1)]-(HOAc) complex, possessing a |z2, 2A1⟩ ground state (species A; gx = 2.42, gy = 2.28, gz = 2.02, Ax = 100, Ay = 120, Az = 310 MHz). The newly formed HS state is assigned to an acetate coordinated [CoII(1)]–(OAc–) complex, possessing an S = 3/2 spin ground state (species B, responsible for a broad EPR signal with g ≈ 4.6). These spin ground states were confirmed with DFT calculations using the hybrid BP86 and B3LYP functionals. Under aerobic conditions, the LS and HS complexes (species A and B) are not observed; instead, a new HS complex (species C) is formed. This complex is tentatively assigned to a paramagnetic superoxo bridged dimer (AcO–)[CoII(1)···O2–CoIII(1)](HOAc), as distinct from the more common diamagnetic peroxo bridged dimers. Species C is characterized by a very broad HS EPR signal (gx = 5.1, gy = 3.9, gz = 2.1) and is reversibly formed by oxygenation of the LS [CoII(1)]–(HOAc) complex to the superoxo complex [CoIII(1)O2–](HOAc), which subsequently forms the association complex C by interaction with the HS [CoII(1)](OAc–) species. The LS and HS complexes were also identified using other organic acids (benzoic and propanoic acid). Thermal annealing–quenching experiments revealed the additional presence of [CoIII(1)O2–](HOAc) adducts, corroborating the presence of species C and the presence of diamagnetic dimer complexes in the solution, such as the EPR silent (HOAc)[CoIII(1)(O22-)CoIII(1)](HOAc). Overall, it appears that a facile interconversion of the [Co(1)] complex, possessing a LS ground state, occurs in the presence of acetic acid, producing both HS and LS CoII states, prior to formation of the oxidized active form of the catalyst, [CoIII(1)](OAc–).
Co-reporter:Mari Elena Owen, Emma Carter, Graham J. Hutchings, Benjamin D. Ward and Damien M. Murphy
Dalton Transactions 2012 vol. 41(Issue 36) pp:11085-11092
Publication Date(Web):06 Jul 2012
DOI:10.1039/C2DT31273E
X- and Q-band EPR and ENDOR spectroscopy was used to study the structure of a series of heteroleptic and homoleptic copper bis(oxazoline) complexes, based on the (−)-2,2′-isopropylidenebis[(4S)-4-phenyl-2-oxazoline] ligand and bearing different counterions (chloride versus triflate); labelled [CuII(1a–c)]. The geometry of the two heteroleptic complexes, [CuII(1a)] and [CuII(1c)], depended on the choice of counterion. Formation of the homoleptic complex was only evident when the CuII(OTf)2 salt was used (CuII(Cl)2 inhibited the transformation from heteroleptic to homoleptic complexes). The hyperfine and quadrupole parameters for the surrounding ligand nuclei were determined by ENDOR. Well resolved 19F and 1H couplings confirmed the presence of both coordinated water and TfO− counterions in [Cu(1a)].
Co-reporter:William D. Woodul ; Emma Carter ; Robert Müller ; Anne F. Richards ; Andreas Stasch ; Martin Kaupp ; Damien M. Murphy ; Matthias Driess ;Cameron Jones
Journal of the American Chemical Society 2011 Volume 133(Issue 26) pp:10074-10077
Publication Date(Web):June 11, 2011
DOI:10.1021/ja204344e
Stoichiometric reduction of the bulky β-diketiminato germanium(II) chloride complex [(ButNacnac)GeCl] (ButNacnac = [{N(Dip)C(But)}2CH]−, Dip = C6H3Pri2-2,6) with either sodium naphthalenide or the magnesium(I) dimer [{(MesNacnac)Mg}2] (MesNacnac = [(MesNCMe)2CH]−, Mes = mesityl) afforded the radical complex [(ButNacnac)Ge:]• in moderate yields. X-ray crystallographic, EPR/ENDOR spectroscopic, computational, and reactivity studies revealed this to be the first authenticated monomeric, neutral germanium(I) radical.
Co-reporter:Damien M. Murphy, Ignacio Caretti, Emma Carter, Ian A. Fallis, Marcus C. Göbel, James Landon, Sabine Van Doorslaer, and David J. Willock
Inorganic Chemistry 2011 Volume 50(Issue 15) pp:6944-6955
Publication Date(Web):June 27, 2011
DOI:10.1021/ic200113u
Single enantiomers of R/S-methylbenzylamine (MBA) were found to selectively form adducts with two chiral Cu–salen complexes, [CuII(1)] (H21 = N,N′-bis(3,5-ditert-butylsalicylidene)-1,2-diaminocyclohexane) and [CuII(2)] (H22 = N,N′-bis-salicylidene-1,2-cyclohexanediamino). The axial g/A spin Hamiltonian parameters of the Cu–MBA adducts were typical of 5-coordinate species. Enantiomer discrimination in the MBA binding was directly evidenced by W-band CW EPR, revealing an 86 ± 5% preference for formation of the R,R-[Cu(1)] + S-MBA adducts compared to R,R-[Cu(1)] + R-MBA; this was reduced to a 57 ± 5% preference for R,R-[Cu(2)] + S-MBA following removal of the tert-butyl groups. The structure of these diastereomeric adducts was further probed by different hyperfine techniques (ENDOR and HYSCORE), although no structural differences were detected between these adducts using these techniques. The diastereomeric adducts were found to possess lower symmetry, as evidenced by rhombic g tensors and inequivalent Himine couplings. This was caused by the selective binding mode of MBA onto one side of the chiral CuII complex. DFT calculations were performed on the R,R-[Cu(1)] + S-MBA and R,R-[Cu(1)] + R-MBA adducts. A distinct difference in orientation and binding mode of the MBA was identified in both adducts, confirming the experimental results. The preferred heterochiral R,R-[Cu(1)] + S-MBA adduct was found to be 5 kJ mol–1 lower in energy compared to the homochiral adduct. A delicate balance of steric repulsion between the α-proton (attached to the asymmetric carbon atom) of MBA and the methine proton (attached to the asymmetric carbon atom) of [Cu(1)] was crucial in the stereoselective binding.
Co-reporter:Damien M. Murphy;Lucia E. McDyre;Emma Carter;Andreas Stasch;Cameron Jones
Magnetic Resonance in Chemistry 2011 Volume 49( Issue 4) pp:159-163
Publication Date(Web):
DOI:10.1002/mrc.2721
Abstract
In this paper, the paramagnetic properties of a novel magnesium ketyl radical (compound 1), formed by reduction of benzophenone with a dimeric Mg(I) complex in the presence of dimethylaminopyridine, are described. Using CW EPR, ENDOR and special TRIPLE resonance, the spin distribution in the radical has been explored at variable temperatures (200–298 K). At 298 K, most of the unpaired spin is found to be confined to the (OCPh2•) fragment based on the hyperfine couplings (hfc's) of o-H = 8.30, m-H = 3.00 and p-H = 9.95 MHz. Smaller hfc's to 25 Mg (5.54 MHz) and 14NDMAP (0.90 MHz) were also evidenced in the 298 K EPR spectrum, indicating some spin delocalisation onto the Mg(Nacnac)(DMAP) fragment. At lower temperatures, restricted rotations of the diphenyl rings create an inequivalent spin distribution in the two rings, with o1-H = 8.80, o2-H = 7.85, m-H = 3.00 and p-H = 10.00 MHz. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:E. Carter, I.A. Fallis, D.M. Murphy, D.J. Willock, S. Van Doorslaer, E. Vinck
Chemical Physics Letters 2010 Volume 486(1–3) pp:74-79
Publication Date(Web):5 February 2010
DOI:10.1016/j.cplett.2009.12.066
Abstract
The role of H-bonds in controlling the binding mode of epoxides, namely propylene oxide (3) and cis-2,3-epoxybutane (4), to the vanadyl salen-type complexes, N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexane-diamino-vanadium (IV) oxide, [VO(1)], and VO(3,5-tBu2-salophen), [VO(2)], have been examined using cw-EPR, cw-ENDOR and HYSCORE spectroscopy. One of the methine protons from the cyclohexyl backbone in [VO(1)] has previously been shown to form a weak H-bond with the epoxide oxygen atom. The absence of this methine proton in the salophen derivative [VO(2)] removes this H-bonding ability of the complex and in turn weakens the epoxide interaction.
Co-reporter:Jason Green, Emma Carter, Damien M. Murphy
Chemical Physics Letters 2009 Volume 477(4–6) pp:340-344
Publication Date(Web):6 August 2009
DOI:10.1016/j.cplett.2009.07.002
Co-reporter:DamienM. Murphy Dr.;IanA. Fallis Dr.;DavidJ. Willock Dr.;James Lon;Emma Carter Dr.;Evi Vinck
Angewandte Chemie International Edition 2008 Volume 47( Issue 8) pp:1414-1416
Publication Date(Web):
DOI:10.1002/anie.200703537
Co-reporter:DamienM. Murphy Dr.;IanA. Fallis Dr.;DavidJ. Willock Dr.;James Lon;Emma Carter Dr.;Evi Vinck
Angewandte Chemie 2008 Volume 120( Issue 8) pp:1436-1438
Publication Date(Web):
DOI:10.1002/ange.200703537
Co-reporter:Robert J. Baker Dr.;Robert D. Farley Dr.;Cameron Jones ;David P. Mills;Marc Kloth Dr. Dr.
Chemistry - A European Journal 2005 Volume 11(Issue 10) pp:
Publication Date(Web):10 MAR 2005
DOI:10.1002/chem.200401103
Paramagnetic diazabutadienegallium(II or III) complexes, [(Ar-DAB)2Ga] and [{(Ar-DAB.)GaX}2] (X=Br or I; Ar-DAB={N(Ar)C(H)}2, Ar=2,6-diisopropylphenyl), have been prepared by reactions of an anionic gallium N-heterocyclic carbene analogue, [K(tmeda)][:Ga(Ar-DAB)], with either “GaI” or [MoBr2(CO)2(PPh3)2]. A related InIII complex, [(Ar-DAB.)InCl2(thf)], has also been prepared. These compounds were characterised by X-ray crystallography and EPR/ENDOR spectroscopy. The EPR spectra of all metal(III) complexes incorporating the Ar-DAB ligand, [(Ar-DAB.)MX2(thf)n] (M=Al, Ga or In; X=Cl or I; n=0 or 1) and [(Ar-DAB)2Ga], confirmed that the unpaired spin density is primarily ligand centred, with weak hyperfine couplings to Al (a=2.85 G), Ga (a=17—25 G) or In (a=26.1 G) nuclei. Changing the N substituents of the diazabutadiene ligand to tert-butyl groups in the gallium complex, [(tBu-DAB.)GaI2] (tBu-DAB={N(tBu)C(H)}2), changes the unpaired electron spin distribution producing 1H and 14N couplings of 1.4 G and 8.62 G, while the aryl-substituted complex, [(Ar-DAB.)GaI2], produces couplings of about 5.0 G. These variations were also manifested in the gallium couplings, namely aGa ∼1.4 G for [(tBu-DAB.)GaI2] and aGa ∼25 G for [(Ar-DAB.)GaI2]. The EPR spectra of the gallium(II) and indium(II) diradical complexes, [{(Ar-DAB.)GaBr}2], [{(Ar-DAB.)GaI}2], [{(tBu-DAB.)GaI}2] and [{(Ar-DAB.)InCl}2], revealed doublet ground states, indicating that the GaGa and InIn bonds prevent dipole–dipole coupling of the two unpaired electrons. The EPR spectrum of the previously reported complex, [(Ar-BIAN.)GaI2] (Ar-BIAN=bis(2,6-diisopropylphenylimino)acenaphthene) is also described. The hyperfine tensors for the imine protons, and the aryl and tert-butyl protons were obtained by ENDOR spectroscopy. In [(Ar-DAB.)GaI2], gallium hyperfine and quadrupolar couplings were detected for the first time.
Co-reporter:Sarah L Baum, Ian G.M Anderson, Richard R Baker, Damien M Murphy, Christopher C Rowlands
Analytica Chimica Acta 2003 Volume 481(Issue 1) pp:1-13
Publication Date(Web):28 March 2003
DOI:10.1016/S0003-2670(03)00078-3
It has been shown in previous work that free radicals are generated in the mainstream smoke of cigarettes. The most direct method for the detection and quantification of these radicals is electron spin resonance (ESR) spectroscopy in conjunction with the spin trapping method. However, the nature of the spin adduct spectrum and the concentration of the radicals trapped in solution, will vary markedly depending on the experimental conditions employed. In order to apply ESR–spin trapping for analytical experiments in the quantification of free radicals in cigarette smoke, a rigorous set of experimental protocols must be developed. In the current paper, experiments were conducted in order to determine the optimal conditions for maximum signal intensities and reproducibility of results. A set of experimental protocols is therefore described for free radical quantification. These tests were optimised using the University of Kentucky IR4F reference cigarette and also applied to a set of commercial cigarette samples. The results show that radical concentrations in smoke vary amongst cigarettes in both the gas phase and particulate phases. Using the series of commercial cigarettes, where many parameters change from cigarette to cigarette, no statistically significant correlations were found between radical levels and total particulate matter in smoke. However, a weak correlation was found between the gas phase radical levels and total particulate matter levels in smoke. There may also be a complex effect of tobacco type on radical levels in smoke.
Co-reporter:Robert D. Farley;Elio Giamello;Christopher C. Rowls;Ian J. Purnell;Mario Chiesa;Maria Cristina Paganini
Magnetic Resonance in Chemistry 2002 Volume 40(Issue 6) pp:381-386
Publication Date(Web):5 APR 2002
DOI:10.1002/mrc.1028
A variety of surface anion vacancies, or point defects, are created by high-temperature activation of a series of polycrystalline alkaline earth metal oxides (MgO, CaO and SrO). Subsequent UV irradiation of the activated oxide under a hydrogen atmosphere results in the generation of surface colour centres [FS+(H)], by electron trapping at these anion vacancies. The paramagnetic properties of these colour centres were studied by EPR and ENDOR spectroscopy. 1H ENDOR spectroscopy revealed that a well defined heterogeneity of trapped electron species exists on each oxide surface, as characterized by the different superhyperfine couplings between the trapped electron and the nearby proton of the FS+ (H) centre. On MgO and CaO two dominant FS+ (H) centres were identified (labelled sites I and II) whereas on SrO three FS+ (H) species were found (sites I, II and III). The possible surface sites responsible for electron stabilization are discussed, and include a 3C corner mono-vacancy, a 4C mono-vacancy and an anion–cation di-vacancy. The results indicate that regardless of the oxide used, a common degree of morphological similarities exists on each oxide. Copyright © 2002 John Wiley & Sons, Ltd.
Co-reporter:Yvonne Traa, Damien M. Murphy, Robert D. Farley and Graham J. Hutchings
Physical Chemistry Chemical Physics 2001 vol. 3(Issue 6) pp:1073-1080
Publication Date(Web):20 Feb 2001
DOI:10.1039/B010083H
A CuNaY catalyst was prepared and used to study the enantioselective
aziridination of styrene, with PhINTs as the nitrogen source, in
the presence of a bis(oxazoline) chiral modifier. The chiral modifier used
was a diimine ligand, (S)-(−)-2,2′-isopropylidenebis(4-phenyl-2-oxazoline).
EPR spectroscopy provides the first direct experimental evidence for the
formation of a copper(II)-bis(oxazoline) complex inside
the Y zeolite pores after stirring the calcined catalysts with the chiral
ligand using acetonitrile as solvent. The copper complexes possess square
pyramidal and square planar symmetries, with spin Hamiltonian parameters analogous
to those of the equivalent homogeneous complex dissolved in solution. These
copper(II) complexes accounted for at least 40% of all
available copper within the ion exchanged CuNaY catalyst and represent one
Cu(II)-bis(oxazoline) complex per supercage. The remaining
uncomplexed Cu(II) ions remain solvated to the acetonitrile molecules.
After the aziridination reaction was carried out in the presence of styrene
and PhINTs, EPR evidenced the selective loss of the signal due to
the copper(II)-bis(oxazoline) complex with square pyramidal
and square planar symmetries but practically no loss in overall Cu(II)
content. This was explained on the grounds of a changing co-ordination
environment of the encapsulated complex. However when PhINTs was added
separately to the catalyst a dramatic loss in Cu(II) signal intensity
was observed. These results are discussed in terms of the reaction mechanism
in operation.
Co-reporter:Damien M Murphy, Christopher C Rowlands
Current Opinion in Solid State and Materials Science 2001 Volume 5(Issue 1) pp:97-104
Publication Date(Web):January 2001
DOI:10.1016/S1359-0286(00)00035-8
EMR techniques have been extensively used in the past year to explore problems relevant to heterogeneous catalysis, including surface defects and radicals, redox processes with supported transition metal ions and in situ studies at elevated temperatures. The combination of these techniques with computational methods, can now be used to provide detailed structural information of active sites even in polycrystalline materials. Developments in high field EMR and the utilisation of pulsed EMR methods are providing marked improvements in sensitivity and spatial resolution of paramagnetic surface states.
Co-reporter:Emma Carter, David Collison, Ruth Edge, Emma C. Fitzgerald, Hannah N. Lancashire, Damien M. Murphy, Joseph J. W. McDouall, Joseph Sharples and Mark W. Whiteley
Dalton Transactions 2010 - vol. 39(Issue 47) pp:NaN11431-11431
Publication Date(Web):2010/10/27
DOI:10.1039/C0DT00642D
The paramagnetic aryl-alkynyl complexes [Mo(CCAr)(dppe)(η-C7H7)]+ (dppe = Ph2PCH2CH2PPh2; Ar = C6H5, [1]+; C6D5, [2]+; C6H4-4-F, [3]+; C6H4-4-Me, [5]+) and [Mo(CCBut)(dppe)(η-C7H7)]+ [4]+, have been investigated in a combined EPR and ENDOR study. Direct experimental evidence for the delocalisation of unpaired spin density over the framework of an aryl-alkynyl ligand has been obtained. The X-band solution EPR spectrum of the 4-fluoro derivative, [3]+, exhibits resolved hyperfine coupling to the remote para position of the aryl group [aiso(19F) = 4.5 MHz, (1.6 G)] in addition to couplings attributable to 95/97Mo, 31P and 1H of the C7H7 ring. A full analysis of the 1H ENDOR spectra is restricted by the low g anisotropy of the system which prevents the use of orientation selection. However, inter-comparison of the 1H cw-ENDOR frozen solution spectra of [1]+, [2]+, [4]+ and [5]+, combined with spectral simulation informed by calculated values derived from DFT investigations, has facilitated estimation of the experimental aiso(1H) hyperfine couplings of [1]+ including the ortho, ±3.7 MHz (±1.3 G) and para, ±3.9 MHz (±1.4 G) positions of the C6H5 substituent of the aryl-alkynyl ligand.
Co-reporter:Lucia E. McDyre, Tracy Hamilton, Damien M. Murphy, Kingsley J. Cavell, William F. Gabrielli, Martin J. Hanton and David M. Smith
Dalton Transactions 2010 - vol. 39(Issue 33) pp:NaN7799-7799
Publication Date(Web):2010/07/21
DOI:10.1039/C0DT00127A
The preparation and characterisation of the Cr(I) complexes [Cr(CO)4L]+ (L = Ph2PN(R)PPh2, Ph2P(R)PPh2), which are used as pre-catalysts for the selective oligomerization of ethylene, are reported. The electronic properties and structural features of these complexes in frozen solution have been established via continuous wave X-band Electron Paramagnetic Resonance (cw-EPR) and continuous wave 1H, 14N and 31P Electron Nuclear Double Resonance (cw-ENDOR) spectroscopy. The EPR spectra are dominated by the g anisotropy, with notably large PA couplings from the two equivalent 31P nuclei. The spin Hamiltonian parameters (g⊥ (gxx = gyy) > ge > g∥ (gzz)) are consistent with a low-spin d5 system possessing C2v symmetry, with a SOMO where the metal contribution is primarily dxy for all complexes. The isotropic Fermi contact term (Paiso, determined by EPR and ENDOR) was found to be largest for complexes containing ligands e, d, f and g, indicating that the 31P 3 s character in the SOMO is higher for the PNP type ligands than the PCP type. Subtle structural differences in the complexes were also identified through variations in the Δg shifts (identified by EPR), and through differences in the phenyl ring conformations (identified by 1H ENDOR). Attempts to correlate trends in EPR-derived parameters with data measured for catalysis using these pre-catalysts are also made, but no clear connections were found.
Co-reporter:Mari Elena Owen, Emma Carter, Graham J. Hutchings, Benjamin D. Ward and Damien M. Murphy
Dalton Transactions 2012 - vol. 41(Issue 36) pp:NaN11092-11092
Publication Date(Web):2012/07/06
DOI:10.1039/C2DT31273E
X- and Q-band EPR and ENDOR spectroscopy was used to study the structure of a series of heteroleptic and homoleptic copper bis(oxazoline) complexes, based on the (−)-2,2′-isopropylidenebis[(4S)-4-phenyl-2-oxazoline] ligand and bearing different counterions (chloride versus triflate); labelled [CuII(1a–c)]. The geometry of the two heteroleptic complexes, [CuII(1a)] and [CuII(1c)], depended on the choice of counterion. Formation of the homoleptic complex was only evident when the CuII(OTf)2 salt was used (CuII(Cl)2 inhibited the transformation from heteroleptic to homoleptic complexes). The hyperfine and quadrupole parameters for the surrounding ligand nuclei were determined by ENDOR. Well resolved 19F and 1H couplings confirmed the presence of both coordinated water and TfO− counterions in [Cu(1a)].

![Naphth[1,2-b]oxirene, 1a,2,3,7b-tetrahydro-, (1aS,7bR)-](http://img.cochemist.com/ccimg/58900/58800-12-7.png)
![Naphth[1,2-b]oxirene, 1a,2,3,7b-tetrahydro-, (1aS,7bR)-](http://img.cochemist.com/ccimg/58900/58800-12-7_b.png)









![POLY[(METHYL-1,4-PIPERAZINEDIYL)(3-OXO-1,3-PROPANEDIYL)IMINO(CARBOXYMETHYLENE)IMINO(1-OXO-1,3-PROPANEDIYL)], HYDROCHLORIDE](http://img.cochemist.com/ccimg/691900/691850-66-5.png)
![POLY[(METHYL-1,4-PIPERAZINEDIYL)(3-OXO-1,3-PROPANEDIYL)IMINO(CARBOXYMETHYLENE)IMINO(1-OXO-1,3-PROPANEDIYL)], HYDROCHLORIDE](http://img.cochemist.com/ccimg/691900/691850-66-5_b.png)









![2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(E)-nitrilomethylidyne]]bis-Phenol](/data/chemimg/215057-24-2.png)
![2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(E)-nitrilomethylidyne]]bis-Phenol](/data/chemimg/215057-24-2_b.png)


















![Phenol, 2,2'-[1,2-ethanediylbis(nitrilomethylidyne)]bis-, (E,E)-](http://img.cochemist.com/ccimg/129500/129409-01-4.png)
![Phenol, 2,2'-[1,2-ethanediylbis(nitrilomethylidyne)]bis-, (E,E)-](http://img.cochemist.com/ccimg/129500/129409-01-4_b.png)



![2-Propanamine,2-methyl-N-[(1-oxido-4-pyridinyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/66900/66893-81-0.png)
![2-Propanamine,2-methyl-N-[(1-oxido-4-pyridinyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/66900/66893-81-0_b.png)






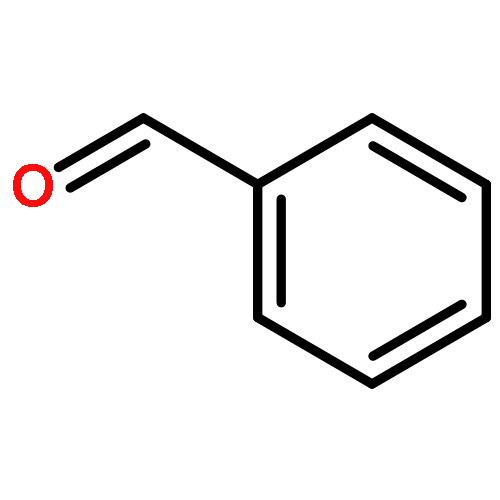


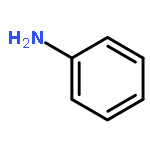
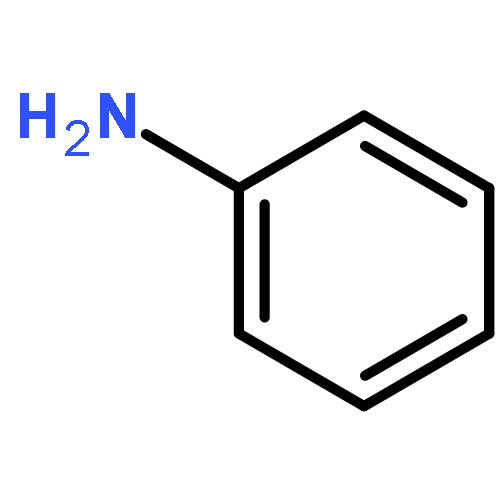
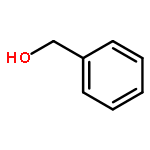













![Chromium,tetracarbonyl[1,1'-(1,2-ethanediyl)bis[1,1-diphenylphosphine-kP]]-, (OC-6-22)-](http://img.cochemist.com/ccimg/29900/29890-04-8.png)
![Chromium,tetracarbonyl[1,1'-(1,2-ethanediyl)bis[1,1-diphenylphosphine-kP]]-, (OC-6-22)-](http://img.cochemist.com/ccimg/29900/29890-04-8_b.png)
![Aluminate(1-),tetrakis[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)-2-propanolato-kO]-, lithium (1:1), (T-4)-](http://img.cochemist.com/ccimg/275000/274933-96-9.png)
![Aluminate(1-),tetrakis[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)-2-propanolato-kO]-, lithium (1:1), (T-4)-](http://img.cochemist.com/ccimg/275000/274933-96-9_b.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/149800/149769-84-6.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/149800/149769-84-6_b.png)
![Pyridine, 2-[(4S)-4,5-dihydro-4-(phenylmethyl)-2-oxazolyl]-](/data/chemimg/354800/108915-08-8.png)
![Pyridine, 2-[(4S)-4,5-dihydro-4-(phenylmethyl)-2-oxazolyl]-](/data/chemimg/354800/108915-08-8_b.png)




![Sodium;(2s)-2-azaniumyl-3-[[(2r)-2,3-di(hexadecanoyloxy)propoxy]-oxidophosphoryl]oxypropanoate](http://img.cochemist.com/ccimg/145900/145849-32-7.png)
![Sodium;(2s)-2-azaniumyl-3-[[(2r)-2,3-di(hexadecanoyloxy)propoxy]-oxidophosphoryl]oxypropanoate](http://img.cochemist.com/ccimg/145900/145849-32-7_b.png)
![Pyridine, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/108000/108915-04-4.png)
![Pyridine, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/108000/108915-04-4_b.png)