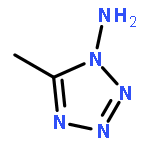
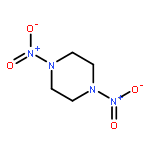
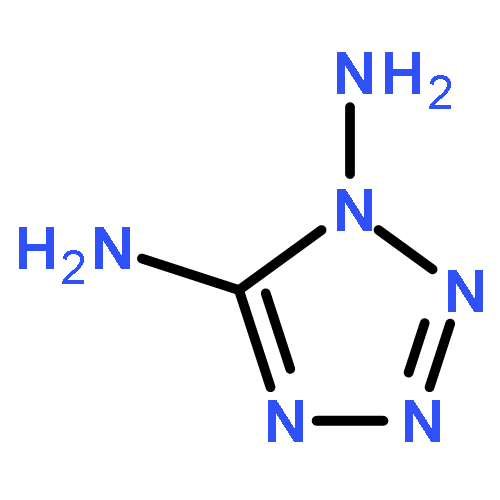
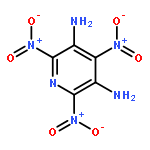
Co-reporter:Zheng Mei;Feng-Qi Zhao;Si-Yu Xu
RSC Advances (2011-Present) 2017 vol. 7(Issue 67) pp:42024-42029
Publication Date(Web):2017/08/29
DOI:10.1039/C7RA07693B
In order to solve a contradiction between early theoretical prediction and experiments concerning the γ → α phase transition of aluminum hydride, models of Li-doped AlH3 were constructed and investigated theoretically. Thermodynamic calculations show that the γ → α transition of pure AlH3 absorbs energy, and the changes in Gibbs free energy are in range of 1.74–1.99 kJ mol−1 at 298–380 K. These are opposite to the experimental fact that the γ- to α-phase transition takes place at 380 K. However, the changes in enthalpy and Gibbs free energy in the γ → α phase transition of Li-doped AlH3 are negative. The doping of Li decreases the activation energy of the γ → α transition and introduces more metastable states between them. As the doping content increases, both the changes in enthalpy and Gibbs free energy (ΔHγ→α and ΔGγ→α) decrease. The experimental ΔHγ→α value (−2.83 kJ mol−1) is between those of doped AlH3 with 1/23 and 1/11 Li-content (−0.87 and −5.62 kJ mol−1 for Al23LiH70 and Al11LiH34, respectively). Heat capacity CP(T) increases as the Li-doping content increases. The CP(T) of Al23LiH70 is consistent with the experiments. Considering the thermodynamic evidence and the experimental conditions for AlH3 preparation, the aluminum hydride synthesized by the reaction of LiAlH4 + AlCl3 is probably Li-doped with a Li content of 1/23. The changes in enthalpy and Gibbs free energy, as well as the activation energy for the γ → α phase transition can be increased if the Li-doped AlH3 is purified.
Co-reporter:Shan-Qisong Huang, Xiu-Lin Zeng, Si-Yu Xu, Xue-Hai Ju
Computational and Theoretical Chemistry 2016 Volume 1093() pp:91-97
Publication Date(Web):1 October 2016
DOI:10.1016/j.comptc.2016.08.015
First-principle calculations have been performed to study both O2 molecule and O atom adsorption on α-U(0 0 1) surface and subsurface. The results show that the long-bridge site is the most stable adsorption site for O2U(0 0 1) and OU(0 0 1) surface adsorption. The tetrahedral (Tet) 1 site is the most stable adsorption site for OU(0 0 1) subsurface adsorption. The partial electronic density of states (PDOSs) of O2U(0 0 1) and OU(0 0 1) shows the hybridization of U and O atoms orbitals causes the electrons of 5f/6d of U more non-local and leads to the formation of OU bond. The OU bond is partly ionic and partly covalent. The analysis of the diffusion path of O atom on U(0 0 1) shows that O atom can easily transfer from hollow (hol) 1 site to hollow 2 site with no energy barrier. O atom can hardly transfer from surface to the subsurface.First-principle calculations have been performed to study both O2 molecule and O atom adsorption in on-surface and subsurface for α-U(0 0 1) surface. The results show that the long-bridge site is the most stable adsorption site for O2U(0 0 1) and OU(0 0 1) surface adsorption. The tetrahedral (Tet) 1 site is the most stable adsorption site for OU(0 0 1) subsurface adsorption. The partial electronic density of states (PDOSs) of O2U(0 0 1) and OU(0 0 1) shows the hybridization of U and O atoms causes the electrons of 5f/6d of U more non-local and leads to the formation of OU bond. The OU bond is partly ionic and partly covalent. The analysis of the diffusion path of O atom on U(0 0 1) shows that O atom can easily transfer from hollow (hol) 1 site to hollow 2 site with no energy barrier. O atom can hardly transfer from surface to the subsurface.
Co-reporter:Jun Yin, Kadali Chaitanya, Xue-Hai Ju
Journal of Molecular Graphics and Modelling 2016 Volume 64() pp:40-50
Publication Date(Web):March 2016
DOI:10.1016/j.jmgm.2015.12.007
•The number and location of electron-withdrawing fluorine atoms at ATT affect the charge carrier transport significantly.•Introduction of moderate number of electron-withdrawing fluorine to π-conjugated planar molecule is favorable to electron transport.•The electron mobility of ATT3 reaches up to 0.48 cm2 V−1 s−1.The crystal structures of known anthra-tetrathiophene (ATT) and its three fluorinated derivatives (ATT1, ATT2 and ATT3) were predicted by the Monte Carlo-simulated annealing method with the embedded electrostatic potential (ESP) charges. The most stable crystal structures were further optimized by the density functional theory with the dispersion energy (DFT-D) method. In addition, the effect of the electron-withdrawing fluorine atoms on the molecular geometry, molecular stacking, electronic and transport properties of title compounds were investigated by the density functional theory and the incoherent charge-hopping model. The calculated results show that the introduction of fluorine atoms does not affect the molecular planarity but decreases the HOMO-LUMO gap, which is beneficial to electron injection and provides more charge carrier stabilization. The improved electron mobility from ATT to ATT3 is attributed to the favorable molecular packing with strong π–π interaction and the short stacking distance. ATT2 and ATT3 exhibit remarkable angular dependence of mobilities and anisotropic behaviors. The band structures reveal that all the paths with larger transfer integrals are along the directions of large dispersions in the valence band (VB) and conduction band (CB). ATT3 has the largest electron mobility (0.48 cm2 V−1 s−1) among the four compounds, indicating that fluorination is an effective approach to improve electron transport.
Co-reporter:Jun Yin, Kadali Chaitanya and Xue-Hai Ju
Journal of Materials Chemistry A 2015 vol. 3(Issue 14) pp:3472-3481
Publication Date(Web):13 Feb 2015
DOI:10.1039/C4TC02655A
A novel crystal structure of octaseleno[8]circulene (C16Se8, we named it “selflower”) was predicted on the basis of a sym-tetraselenatetrathio[8]circulene crystal (C16S4Se4, selenosulflower). The charge transport properties of selenosulflower and its selenium analogue of selflower as potential ambipolar materials were investigated by the density functional theory (DFT) coupled with the incoherent charge-hopping model. Insights into their geometric and electronic structures, frontier molecular orbitals, reorganization energies and transfer integrals, anisotropic mobilities as well as band structures of the two novel materials are provided in detail. The gap of the frontier molecular orbitals decreases when all sulfur atoms of C16S4Se4 are substituted by selenium, which improves the charge transfer efficiency. The predicted hole and electron mobilities of C16Se8 are 1.03 and 1.26 cm2 V−1 s−1, respectively. C16S4Se4 has a hole mobility of 0.49 cm2 V−1 s−1 and an electron mobility of 0.74 cm2 V−1 s−1. Both circulenes exhibit electron-dominated ambipolar performance. The small reorganization energy and larger transfer integral originating from the face to face π–π stacking lead to large charge mobility for the novel compound C16Se8. From the viewpoint of transfer integral, the electron coupling among the dominant hopping pathways indicates that the charge transport processes take place in the parallel dimers with π–π interaction. The two materials exhibit a remarkable angular dependence of mobilities and anisotropic behaviors. The newly designed “selflower” C16Se8 is a novel organic semiconductor and worth synthesizing.
Co-reporter:Kadali Chaitanya, Xue-Hai Ju and B. Mark Heron
RSC Advances 2015 vol. 5(Issue 6) pp:3978-3998
Publication Date(Web):21 Nov 2014
DOI:10.1039/C4RA09914A
The structural and electronic properties of five known triarylamine derived sensitizers (A1, A1-F, C218, D2 and Y123) and their associated hypothetical dyes (C218-F, D2-F, Y123-F, Y1234 and Y1234-F) have been studied using density functional theory and time-dependent density functional theory. The sensitizers primarily comprise of a triphenylamine, a 4,4′-dihexylcyclopenta[2,1-b:3,4-b]dithiophene and a cyanoacrylic acid as the electron donating, π-spacer and accepting units, respectively. The π-system is extended by incorporation of either a benzo[c][1,2,5]thiadiazol-4,7-diyl unit or an ortho-fluorophenyl unit or both. To gain insight into the effect of elongation of the π-system on the electronic properties of dye sensitized TiO2 interfaces, first-principles calculations have been carried out on sensitizer molecules co-adsorbed on the (101) surface of the anatase TiO2. The theoretical results revealed that elongating the π-system of the sensitizers with both the benzothiadiazole and ortho-fluorophenyl units increases the molecular extinction coefficient, the excited state lifetime and the light harvesting efficiency but decreases the band gap and the reorganization energy relative to the structurally comparable reference dye Y123. The calculated short circuit current density and level alignment quality showed that the π-system in the triarylamine sensitizers elongated with both benzothiadiazole and ortho-fluorophenyl units broadens their potential use in DSSCs due to the enhanced values as compared to the reference dye. The results obtained in this study will provide a valuable reference for the strategy of inserting various π-spacers in triarylamine sensitizers for dye sensitized solar cell applications.
Co-reporter:Jun Yin, Kadali Chaitanya and Xue-Hai Ju
RSC Advances 2015 vol. 5(Issue 80) pp:65192-65202
Publication Date(Web):22 Jul 2015
DOI:10.1039/C5RA06418J
To gain a better understanding of the fluorination effect on charge transport properties, the charge transport properties of the six fused thiophene derivatives 2,6-diphenylbisthieno[3,2-b:2′,3′-d]thiophene (DP-DTT), 6,6′-diphenyl-2,2′-bibisthieno[3,2-b:2′,3′-d]thiophene (DP-BDTT), 2-(pentafluorophenyl)-6-phenylbisthieno[3,2-b:2′,3′-d]thiophene (FPP-DTT), 6,6′-bis(pentafluorophenyl)-2,2′-bibisthieno[3,2-b:2′,3′-d]thiophene (FPP-BDTT), 2,6-dipentafluorophenyl-bisthieno[3,2-b:2′,3′-d]thiophene (DFP-DTT) and 6,6′-dipentafluorophenyl-2,2′-bibisthieno[3,2-b:2′,3′-d]thiophene (DFP-BDTT) were explored by density functional theory (DFT) coupled with the incoherent charge-hopping model at the molecular and crystal levels. The crystal structures of the title compounds are either predicted by the dispersion-corrected density functional method (DFD-D) or retrieved from the Cambridge Crystallographic Database. Introducing electron-withdrawing fluorine atoms to the end phenyl of the DTT and BDTT molecules can decrease the HOMO–LUMO gap, which is beneficial to the conductivity. FPP-BDTT has the largest electron mobility among the six compounds because it has a small electron reorganization energy and large transfer integral. The efficient overlaps of π-orbitals and smaller π–π stacking distance are proved to be the main reasons for the good hole transport property of DFP-DTT. Additionally, FPP-BDTT and DFP-BDTT have shown remarkable anisotropic behaviors and the maximal charge mobilities are along a specific crystal axis direction with strong π–π interactions, which further confirms our finding that the fluorination effect may be an effective way to improve charge mobilities.
Co-reporter:Dr. Yang-Yang Wu; Feng-Qi Zhao; Si-Yu Xu; Xue-Hai Ju
Chemistry – An Asian Journal 2015 Volume 10( Issue 2) pp:362-369
Publication Date(Web):
DOI:10.1002/asia.201403033
Abstract
We designed a cyclic borane (B6H12) molecule with a benzene-like structure, in which the six B atoms are located in the same plane. Three methods of B3LYP, MP2, and CCSD with the 6-311++G** basis were used to investigate its structure, electronic property, and stability. Next, we calculated the stability and electronic property of three hydroboron derivatives with fused rings of B10H18, B14H24, and B16H26. Finally, we investigated three types of novel two-dimensional infinite hydroboron sheets with diborane as a building block. The results of the phonon spectra ensure the dynamic stability of these predicted structures. Furthermore, the three types of hydroboron sheets are shown to have different band gap energies of less than 3.0 eV. Some investigations on the optical properties have also been performed. The predicted sheets are candidates for semiconductors, whose band gap energy can be tuned by the positions of the bridge hydrogen atoms in the sheets.
Co-reporter:Kadali Chaitanya, Xue-Hai Ju and B. Mark Heron
RSC Advances 2014 vol. 4(Issue 51) pp:26621-26634
Publication Date(Web):04 Jun 2014
DOI:10.1039/C4RA02473G
The density functional theory and time-dependent density functional theory calculations of the electronic structures and electronic absorption spectra of a series of zinc porphyrin based sensitizers were reported. The sensitizers comprise of either 10H-phenothiazin-3-yl or bis(4-(hexyloxy)phenyl)amino and acene bridged carboxylic acid as electron donating and accepting units, respectively. The dye–(TiO2)36 anatase nanoparticle systems were also simulated to show the electronic structure on the interface. The calculated results show that a strong electron-donating capacity of the donor group attached at the meso-position opposite to the anchoring group of the dye will increase the molecular extinction coefficient, excited state lifetime, light harvesting efficiency and decrease the reorganization energy as compared to the structurally similar reference dye YD2-o-C8. The calculated short circuit current density and level alignment quality clearly indicate that the zinc-porphyrin dyes substituted with 10H-phenothiazin-3-yl donor and either 4-ethynylbenzoic acid or 4-ethynyl-1-naphthoic acid offer potential for use in DSSCs due to their large values when compared to the reference dye. The results obtained in this study will certainly provide a useful reference to the future design of tetra-substituted zinc porphyrins for dye sensitized solar cell applications.
Co-reporter:Yang-Yang Wu, Feng-Qi Zhao, Xue-Hai Ju
Computational and Theoretical Chemistry 2014 Volume 1027() pp:151-159
Publication Date(Web):1 January 2014
DOI:10.1016/j.comptc.2013.11.001
•Structures and stabilities of charged MgBn clusters were predicted.•The geometry of the stablest neutral clusters change from 2D to 3D as size increases.•The geometries of some neutral clusters change dramatically with different charges.•Anionic and cationic clusters have larger average bonding energy than the neutral.•Odd–even alteration occurs in some properties of the neutral and cationic clusters.The structures and stabilities of the neutral and the positively/negatively charged Mg-doped boron clusters, i.e., MgBn±m (n = 1–7 and m = 0, 1), have been studied at the UB3LYP/6-311+G* level. Several possible multiplicities of each cluster were tested to determine the most stable structure among the isomers. For the neutral clusters, when n ⩽ 5, the lowest-energy geometry is in favor of a planar structure. From n = 6 on, the most stable geometry tends to be quasi-planar or three-dimensional. When the clusters are positively or negatively charged, the lowest-energy structures of the clusters with some sizes change dramatically from their neutral analogs, such as MgB7-, MgB2- and MgB3+. The average binding energy per atom, fragmentation energy and second order energy difference were evaluated. The results indicated that the neutral and cationic clusters possess higher stability when n = 3 and 5, and the anionic is more stable only at n = 3. The adiabatic ionization potential (AIP) and adiabatic electron affinity (AEA) were also calculated for the neutral clusters to investigate their electronic properties. MgB6 has both the highest AIP and adiabatic AEA. Odd–even alteration was found in the HOMO–LUMO gap energy for the neutral and cationic clusters. NBO analysis was done to study the natural population.Graphical abstractThe structures and stabilities of the neutral, positive/negative charged Mg-doped boron clusters, i.e., MgBn±m (n = 1–7 and m = 0, 1), have been studied at the UB3LYP/6-311+G* level. Several possible multiplicities of each cluster were tried to determine the most stable structure among the isomers. The average binding energy, fragmentation energy, second order energy difference, adiabatic ionization potential (AIP) and adiabatic electron affinity (AEA) were determined.
Co-reporter:Jian-Ying Zhao, Feng-Qi Zhao, Si-Yu Xu, Xue-Hai Ju
Journal of Molecular Graphics and Modelling 2014 Volume 48() pp:9-17
Publication Date(Web):March 2014
DOI:10.1016/j.jmgm.2013.11.002
•Adsorption and dissociation of CO2 on doping Al12X were investigated by DFT methods.•Adsorption energies, binding energies and energy barriers for CO2 were determined.•Doping atoms and spin states influence geometries, electronic properties and energies.•Energy barrier for Al12Fe is the lowest among all the clusters.The adsorption and decomposition of CO2 molecule on X-centered icosahedronal Al12X clusters (doping atom X = Al, Be, Zn, Fe, Ni, Cu, B, C, Si, P) were investigated by the DFT methods of PW91 and PWC. Adsorption energies, chemisorption energies and energy barriers of physic- and chemisorptions for CO2 were determined. It was found that the doping atoms and spin states have important influences on the Al12X geometries, electronic properties and energies of the adsorption processes. CO2 chemisorption on the Al12C cluster is energetically and kinetically unfavorable. CO2 decomposition on the metallic doping Al12X (X = Fe, Ni, Cu) clusters has relatively low energy barriers. On contrary, the barriers are large when X = B, C, Si and P. The energy barriers for CO2 chemisorption and decomposition on the Al12Fe cluster are 5.23 kJ/mol and 38.53 kJ/mol, respectively. These values are the lowest among all the clusters being discussed. The adsorption and decomposition of CO2 on the Al12X cluster can be tuned by X doping.The adsorption and dissociation of CO2 molecule on neutral X-centered icosahedronal Al12X clusters (X = Al, Be, Zn, Fe, Ni, Cu, B, C, Si, P) were investigated by the DFT methods of functional PW91 and PWC. Adsorption energies, binding energies and barriers between the physic- and chemi-sorption states for CO2 were determined. The adsorption and dissociation of CO2 on the Al13X cluster can be tuned by controllable X doping.
Co-reporter:Jian-Ying Zhao, Yu Zhang, Feng-Qi Zhao, and Xue-Hai Ju
The Journal of Physical Chemistry A 2013 Volume 117(Issue 47) pp:12519-12528
Publication Date(Web):November 7, 2013
DOI:10.1021/jp405934w
The adsorption of a CO2 molecule on neutral and charged X-centered icosahedron Al12X±z clusters (X = Al, Be, Zn, Ni, Cu, B, P; z = 0, 1) was investigated by the density functional PW91 and PWC methods. Optimized configurations corresponding to physisorption and chemisorption of CO2 were identified. The adsorption energies, activation barriers, and binding energies involving both the physisorption (Al12X±z·CO2–I) and chemisorption (Al12X±z·CO2–II) for CO2 were determined. The chemisorption of a CO2 molecule on the Al12X clusters (X is a metallic doping element) requires relatively low activation barriers. The lowest barrier was found to be with the Al12Be cluster. For the Al12X– clusters, the barriers are all higher than those of the neutral analogues. For the Al12X+ clusters, two corresponding configurations are linked by a low-energy barrier, and CO2 molecule chemisorption on the Al12Be+ cluster has the lowest barrier. The adsorption energies are larger than the energy barriers, which facilitates the chemisorption. The results show that carbon dioxide adsorbed on the Al12X±z clusters can be tuned by controllable X doping and the total number of valence electrons and suggest the potential application of Al12X±z nanostructures for carbon dioxide capture and activation.
Co-reporter:Kadali Chaitanya, B. Mark Heron, Xue-Hai Ju
Dyes and Pigments (June 2017) Volume 141() pp:
Publication Date(Web):June 2017
DOI:10.1016/j.dyepig.2017.03.012
•The first-principles investigation of the influence of a local electric field on the dye – semiconductor interface.•Due to the Stark effect, the WS-51 dye exhibited a blue shifted absorption spectrum with rising electric field strength.•Doping a nitrogen atom in TiO2 moderates the electric field intensity at the dye – semiconductor interface.The primary objective of this paper is to study the influence of the local electric field at the dye/semiconductor (pure and nitrogen doped TiO2) interface on the performance of dye sensitized solar cells (DSSCs). In this regard, as a first step, we explored the influence of electric field on the performance of the WS-51 dye by computing its properties in an electric field up to 15 × 10−4 a.u. The electronic and optical properties of an established, efficient, donor-acceptor-π-acceptor dye in different electric field strengths were probed with density functional theory (DFT) and time-dependent DFT. The calculated results indicate that, under an electric field, the dye shows significant changes in absorption spectrum due to considerable changes in molecular structure. The TD-DFT results indicate that the absorption spectrum of the dye in acetonitrile solution have shown a blue shift with decreasing molecular extinction coefficient by rising electric field strength. Secondly, we investigated the dye adsorbed on Ti48O96 and Ti48O95N clusters. In the applied field off state, the absorption spectrum of the dye/Ti48O95N system is red shifted with an improved molecular extinction coefficient as compared to the dye/Ti48O96 system, indicating that the nitrogen doped TiO2 surface is more favourable for enhancing the efficiency of the DSSCs. Finally, the calculated results suggested that the light harvesting efficiency (LHE) of the dye/Ti48O95N system is higher than that of the dye/Ti48O96 system. But, under an electric field the LHE of the dye on Ti48O96 and Ti48O95N clusters is likely to decrease with increasing electric field strength. The photon to current response of the DSSCs is limited by the local electric field generated at the dye – semiconductor interface. Finally, results indicate that the doping of a nitrogen atom in TiO2 moderates the electric field intensity at the dye – semiconductor interface. Therefore, the results obtained in this study will provide a valuable reference for understanding the role of local electric field for the further optimization of DSSCs.
Co-reporter:Jun Yin, Kadali Chaitanya and Xue-Hai Ju
Journal of Materials Chemistry A 2015 - vol. 3(Issue 14) pp:NaN3481-3481
Publication Date(Web):2015/02/13
DOI:10.1039/C4TC02655A
A novel crystal structure of octaseleno[8]circulene (C16Se8, we named it “selflower”) was predicted on the basis of a sym-tetraselenatetrathio[8]circulene crystal (C16S4Se4, selenosulflower). The charge transport properties of selenosulflower and its selenium analogue of selflower as potential ambipolar materials were investigated by the density functional theory (DFT) coupled with the incoherent charge-hopping model. Insights into their geometric and electronic structures, frontier molecular orbitals, reorganization energies and transfer integrals, anisotropic mobilities as well as band structures of the two novel materials are provided in detail. The gap of the frontier molecular orbitals decreases when all sulfur atoms of C16S4Se4 are substituted by selenium, which improves the charge transfer efficiency. The predicted hole and electron mobilities of C16Se8 are 1.03 and 1.26 cm2 V−1 s−1, respectively. C16S4Se4 has a hole mobility of 0.49 cm2 V−1 s−1 and an electron mobility of 0.74 cm2 V−1 s−1. Both circulenes exhibit electron-dominated ambipolar performance. The small reorganization energy and larger transfer integral originating from the face to face π–π stacking lead to large charge mobility for the novel compound C16Se8. From the viewpoint of transfer integral, the electron coupling among the dominant hopping pathways indicates that the charge transport processes take place in the parallel dimers with π–π interaction. The two materials exhibit a remarkable angular dependence of mobilities and anisotropic behaviors. The newly designed “selflower” C16Se8 is a novel organic semiconductor and worth synthesizing.

![1,3,5,7-TETRAAZASPIRO[3.4]OCTANE, 1,3,5,7-TETRANITRO-](http://img.cochemist.com/ccimg/883300/883244-94-8.png)
![1,3,5,7-TETRAAZASPIRO[3.4]OCTANE, 1,3,5,7-TETRANITRO-](http://img.cochemist.com/ccimg/883300/883244-94-8_b.png)
![1,2,4,6-Tetraazaspiro[2.3]hexane, 1,2,4,6-tetranitro-](http://img.cochemist.com/ccimg/883300/883244-93-7.png)
![1,2,4,6-Tetraazaspiro[2.3]hexane, 1,2,4,6-tetranitro-](http://img.cochemist.com/ccimg/883300/883244-93-7_b.png)
![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7.png)
![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7_b.png)








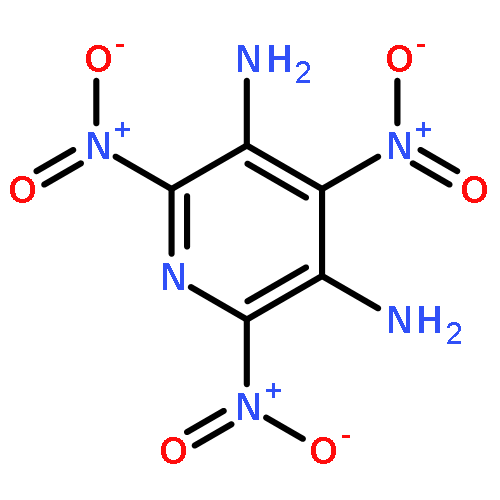
![2H-Imidazo[4,5-b]pyrazin-2-one, octahydro-1,3,4,7-tetranitro-](http://img.cochemist.com/ccimg/160100/160013-48-9.png)
![2H-Imidazo[4,5-b]pyrazin-2-one, octahydro-1,3,4,7-tetranitro-](http://img.cochemist.com/ccimg/160100/160013-48-9_b.png)
![2,4,6,8,9,10-Hexaazatricyclo[3.3.1.13,7]decane, 2,4,6,8,9,10-hexanitro-](http://img.cochemist.com/ccimg/157200/157110-14-0.png)
![2,4,6,8,9,10-Hexaazatricyclo[3.3.1.13,7]decane, 2,4,6,8,9,10-hexanitro-](http://img.cochemist.com/ccimg/157200/157110-14-0_b.png)
![1,3,5,7-Tetraazaspiro[3.3]heptane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/155600/155582-25-5.png)
![1,3,5,7-Tetraazaspiro[3.3]heptane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/155600/155582-25-5_b.png)


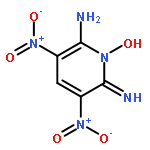
![Imidazo[4,5-d]imidazol-2(1H)-one, hexahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/130300/130256-72-3.png)
![Imidazo[4,5-d]imidazol-2(1H)-one, hexahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/130300/130256-72-3_b.png)
![2,4,6,8-Tetraazabicyclo[3.3.1]nonane-3,7-dione, 2,4,6,8-tetranitro-](http://img.cochemist.com/ccimg/115100/115029-36-2.png)
![2,4,6,8-Tetraazabicyclo[3.3.1]nonane-3,7-dione, 2,4,6,8-tetranitro-](http://img.cochemist.com/ccimg/115100/115029-36-2_b.png)

![1,3,4,5,7,8-hexanitrooctahydrodiimidazo[4,5-b:4',5'-e]pyrazine-2,6(1H,3H)-dione](http://img.cochemist.com/ccimg/115100/115029-33-9.png)
![1,3,4,5,7,8-hexanitrooctahydrodiimidazo[4,5-b:4',5'-e]pyrazine-2,6(1H,3H)-dione](http://img.cochemist.com/ccimg/115100/115029-33-9_b.png)


![4,7-dinitro-4,5,6,7-tetrahydro[1,2,5]oxadiazolo[3,4-b]pyrazine](http://img.cochemist.com/ccimg/98800/98778-15-5.png)
![4,7-dinitro-4,5,6,7-tetrahydro[1,2,5]oxadiazolo[3,4-b]pyrazine](http://img.cochemist.com/ccimg/98800/98778-15-5_b.png)


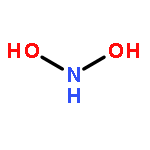
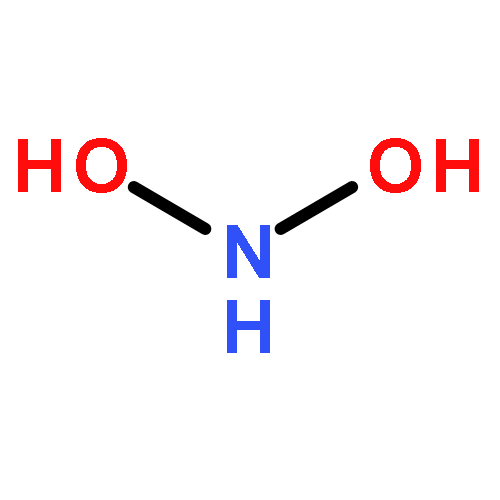


![2,4,8,10-Tetraazaspiro[5.5]undecane, 2,4,8,10-tetranitro-](http://img.cochemist.com/ccimg/84700/84606-37-1.png)
![2,4,8,10-Tetraazaspiro[5.5]undecane, 2,4,8,10-tetranitro-](http://img.cochemist.com/ccimg/84700/84606-37-1_b.png)


![PYRAZINO[2,3-B]PYRAZINE, DECAHYDRO-1,4,5,8-TETRANITRO-, TRANS-](http://img.cochemist.com/ccimg/83700/83673-31-8.png)
![PYRAZINO[2,3-B]PYRAZINE, DECAHYDRO-1,4,5,8-TETRANITRO-, TRANS-](http://img.cochemist.com/ccimg/83700/83673-31-8_b.png)


![3,7-Diazabicyclo[3.3.1]nonane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/81400/81340-15-0.png)
![3,7-Diazabicyclo[3.3.1]nonane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/81400/81340-15-0_b.png)
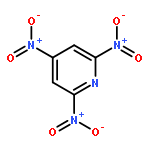
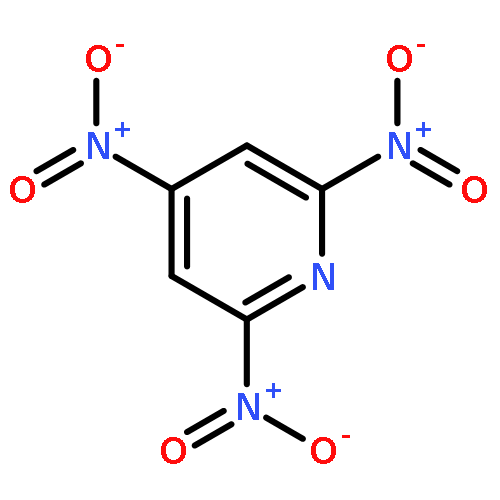
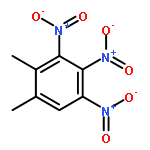
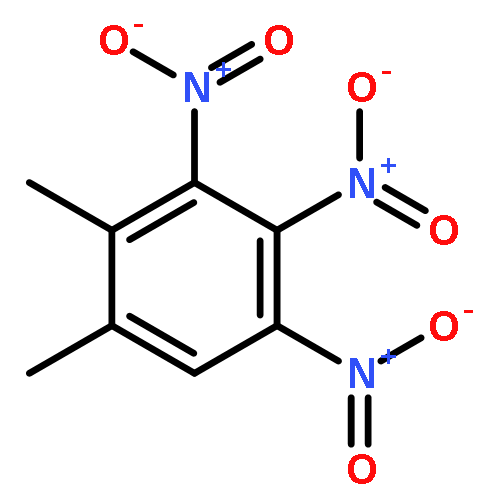
![1,3,7,9-Tetraazaspiro[4.5]decane, 1,3,7,9-tetranitro-](http://img.cochemist.com/ccimg/65500/65479-81-4.png)
![1,3,7,9-Tetraazaspiro[4.5]decane, 1,3,7,9-tetranitro-](http://img.cochemist.com/ccimg/65500/65479-81-4_b.png)
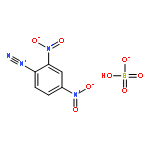

![N-(azidomethyl)-N'-{[(azidomethyl)(nitro)amino]methyl}-N,N'-dinitromethanediamine](http://img.cochemist.com/ccimg/62300/62209-57-8.png)
![N-(azidomethyl)-N'-{[(azidomethyl)(nitro)amino]methyl}-N,N'-dinitromethanediamine](http://img.cochemist.com/ccimg/62300/62209-57-8_b.png)
![3,6-DINITRO-1,3A,4,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE-2,5-DIONE](http://img.cochemist.com/ccimg/55600/55510-04-8.png)
![3,6-DINITRO-1,3A,4,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE-2,5-DIONE](http://img.cochemist.com/ccimg/55600/55510-04-8_b.png)
![Imidazo[4,5-d]imidazole-2,5(1H,3H)-dione,tetrahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/55600/55510-03-7.png)
![Imidazo[4,5-d]imidazole-2,5(1H,3H)-dione,tetrahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/55600/55510-03-7_b.png)
![1,4-Benzenediamine, N,N-dimethyl-N'-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/54100/54050-72-5.png)
![1,4-Benzenediamine, N,N-dimethyl-N'-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/54100/54050-72-5_b.png)

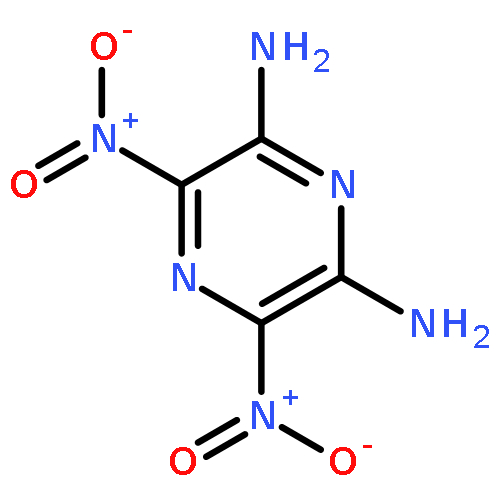
















































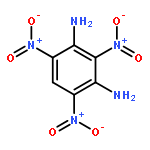

![1,3-Propanediol,2,2-bis[(nitrooxy)methyl]-, 1-nitrate](http://img.cochemist.com/ccimg/1700/1607-17-6.png)
![1,3-Propanediol,2,2-bis[(nitrooxy)methyl]-, 1-nitrate](http://img.cochemist.com/ccimg/1700/1607-17-6_b.png)
















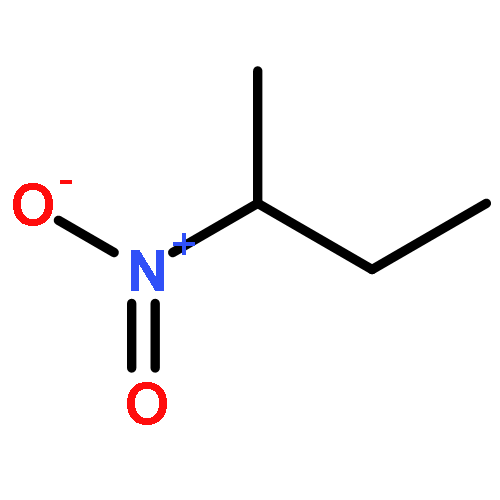



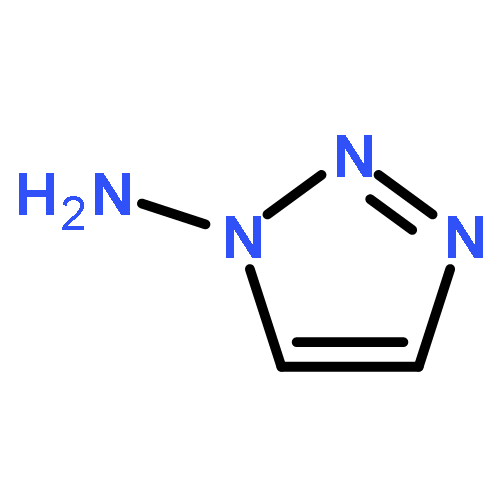
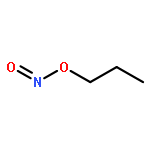

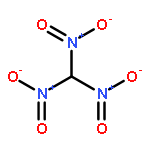










![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4_b.png)