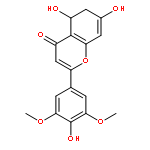
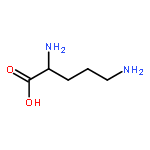
Co-reporter:Jingmiao Shi, Meng Lei, Wenkui Wu, Huayun Feng, Jia Wang, Shanshan Chen, Yongqiang Zhu, Shihe Hu, Zhaogang Liu, Cheng Jiang
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 8) pp:1958-1962
Publication Date(Web):15 April 2016
DOI:10.1016/j.bmcl.2016.03.007
A series of novel dipeptidyl boronic acid proteasome inhibitors constructed from αα- and αβ-amino acids were designed and synthesized. Their structures were elucidated by 1H NMR, 13C NMR, LC–MS and HRMS. These compounds were evaluated for their β5 subunit inhibitory activities of human proteasome. The results showed that dipeptidyl boronic acid inhibitors composed of αα-amino acids were as active as bortezomib. Interestingly, the activities of those derived from αβ-amino acids lost completely. Of all the inhibitors, compound 22 (IC50 = 4.82 nM) was the most potent for the inhibition of proteasome activity. Compound 22 was also the most active against three MM cell lines with IC50 values less than 5 nM in inhibiting cell growth assays. Molecular docking studies displayed that 22 fitted very well in the β5 subunit active pocket of proteasome.A series of novel dipeptidyl boronic acid proteasome inhibitors constructed from αα and αβ amino acids were designed and synthesized.
Co-reporter:Meng Liu, Jie Huang, Dong-Xing Chen, Cheng Jiang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 3) pp:431-434
Publication Date(Web):1 February 2015
DOI:10.1016/j.bmcl.2014.12.060
A series of indole-3-carboxylic acids were designed as novel small molecular non-ATP-competitive Plk1 inhibitors. The designed compounds were synthesized and evaluated. Most of the targeted compounds showed potent Plk1 inhibitory activities and anti-proliferative characters. Particularly, 4f and 4g showed Plk1 inhibitory activity with IC50 values of 0.41 and 0.13 μM, which were about 5 and 17 times more potent compared to thymoquinone, respectively. Compound 4g also showed inhibitory activity to HeLa and MCF-7 cell lines with IC50 values of 0.72 and 1.15 μM, which was almost 3 and 4 times more potent than thymoquinone. Study of mechanism of action suggested that 4g was an ATP-independent and substrate-dependent Plk1 inhibitor. Moreover, 4g showed excellent Plk1 inhibitory selectivity against Plk2 and Plk3. Fluorescein isothiocyanate Annexin V/propidium iodide (PI) double-staining assay and western-blot results indicate that induction of apoptosis by 4g is involved in its anti-tumor activity. This study may provide a support for further optimization of non-ATP-competitive Plk1 inhibitors.
Co-reporter:Dong-Xing Chen;Jie Huang;Meng Liu;Yun-Gen Xu
Archiv der Pharmazie 2015 Volume 348( Issue 1) pp:2-9
Publication Date(Web):
DOI:10.1002/ardp.201400294
A series of small-molecule Plk1 inhibitors targeting the substrate-binding pocket were designed through rational drug design for the first time. The designed compounds were synthesized and their activities were evaluated in vitro. Some of the targeted compounds showed potent Plk1 inhibitory activities and anti-proliferative characters. Particularly, 5i showed Plk1 inhibitory activity with an IC50 value of 0.68 µM. Compound 5i also showed cell growth inhibitory activity on HeLa cells with an IC50 value of 0.51 µM, which is about four times more potent compared to thymoquinone. The mechanism of action suggested that 5i was an ATP-independent and substrate-dependent Plk1 inhibitor. Compound 5i demonstrated excellent Plk1 inhibitory selectivity against Plk2, Plk3, and five serine/threonine and tyrosine kinases. Our discovery and structure–activity relationship study may provide useful lead compounds for further optimization of non-ATP-competitive Plk1 inhibitors.
Co-reporter:Jun-Peng Zhang, Jie Huang, Chao Liu, Xu-Fang Lu, Bao-Xiang Wu, Li Zhao, Na Lu, Qing-Long Guo, Zhi-Yu Li, Cheng Jiang
Chinese Chemical Letters 2014 Volume 25(Issue 7) pp:1025-1028
Publication Date(Web):July 2014
DOI:10.1016/j.cclet.2014.05.048
A series of new 3-benzoheterocyclic substituted pyridopyrimidines were designed and synthesized. Structures of the compounds were determined by IR, 1H NMR, and elemental analyses. The anti-proliferation activity of 13 novel compounds was evaluated in A549, HL-60, BGC-823 and SMMC-7721 cell lines. Compounds 3, 5, 7, 8, 9, 10 showed potent inhibitory activity against the four tested cancer cell lines. These six compounds were examined for Top I inhibition at 100 μmol/L by measuring the relaxation of supercoiled DNA in plasmid pBR322. Most of the tested compounds inhibited the enzyme at this concentration. The most potent compound 9 was as potent as camptothecin.Using a scaffold modification strategy, we identified a series of new 3-benzoheterocyclic substituted pyridopyrimidines as potential topoisomerase I inhibitors.

![(1R,2S,3R,4R,4aS,8aR)-3-Hydroxy-3,4,8,8a-tetramethyl-4-[(E)-2-(5- oxo-2,5-dihydro-3-furanyl)vinyl]-1,2,3,4,4a,5,6,8a-octahydronapht halene-1,2-diyl dinicotinate](http://img.cochemist.com/ccimg/176600/176520-13-1.png)
![(1R,2S,3R,4R,4aS,8aR)-3-Hydroxy-3,4,8,8a-tetramethyl-4-[(E)-2-(5- oxo-2,5-dihydro-3-furanyl)vinyl]-1,2,3,4,4a,5,6,8a-octahydronapht halene-1,2-diyl dinicotinate](http://img.cochemist.com/ccimg/176600/176520-13-1_b.png)




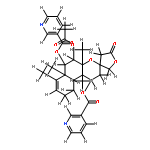











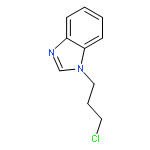
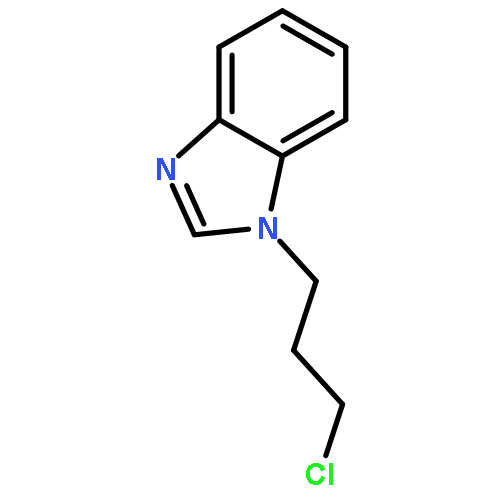
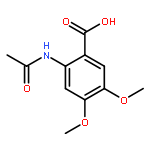
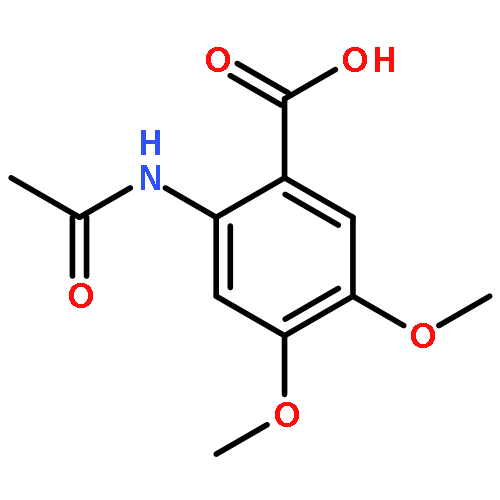


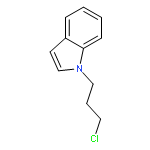





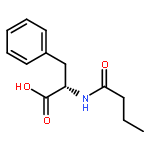
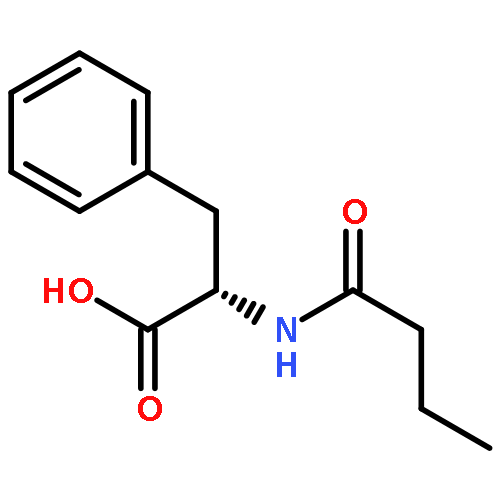


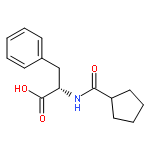
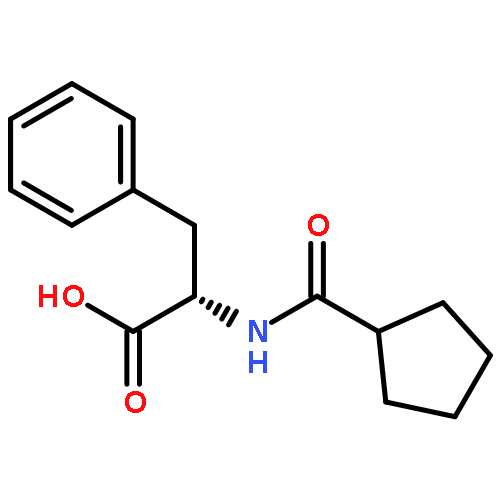


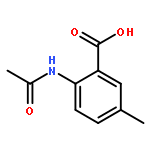
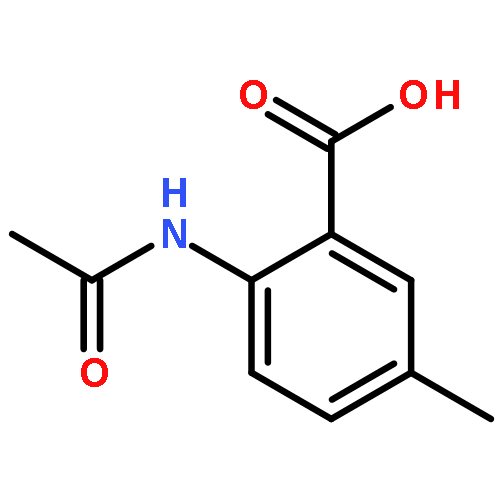
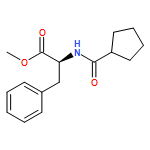
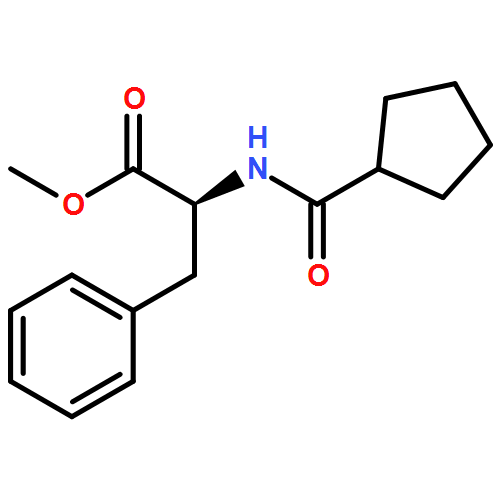






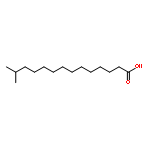
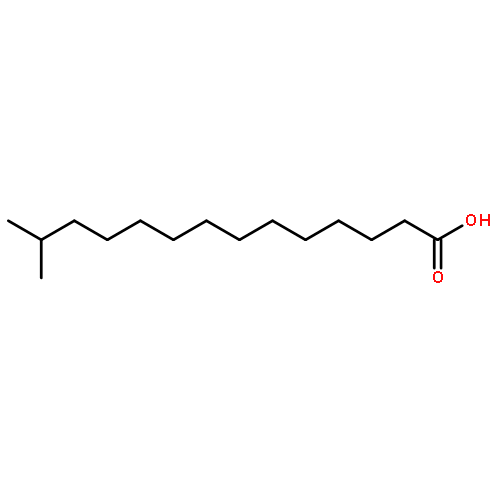


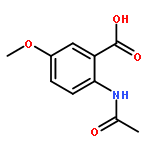
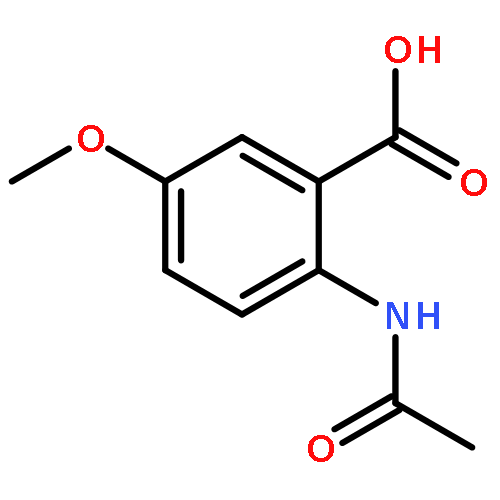



![[(2r,3r,4s,5r,6s)-3,4,5,6-tetrakis[(3,4,5-trihydroxybenzoyl)oxy]oxan-2-yl]methyl 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/15000/14937-32-7.png)
![[(2r,3r,4s,5r,6s)-3,4,5,6-tetrakis[(3,4,5-trihydroxybenzoyl)oxy]oxan-2-yl]methyl 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/15000/14937-32-7_b.png)




![1H-Imidazo[1,2-b]pyrazole,2,3-dihydro-](http://img.cochemist.com/ccimg/6800/6714-29-0.png)
![1H-Imidazo[1,2-b]pyrazole,2,3-dihydro-](http://img.cochemist.com/ccimg/6800/6714-29-0_b.png)

















![2-[(2E,6E,10E,14E,18E,22E,26Z,30E,34E)-3,7,11,15,19,23,27,31,35,39-decamethyltetraconta-2,6,10,14,18,22,26,30,34,38-decaen-1-yl]-3-methylnaphthalene-1,4-dione](http://img.cochemist.com/ccimg/600/523-40-0.png)
![2-[(2E,6E,10E,14E,18E,22E,26Z,30E,34E)-3,7,11,15,19,23,27,31,35,39-decamethyltetraconta-2,6,10,14,18,22,26,30,34,38-decaen-1-yl]-3-methylnaphthalene-1,4-dione](http://img.cochemist.com/ccimg/600/523-40-0_b.png)