Co-reporter:Yixin Han, Karla Mahender Reddy, and E. J. Corey
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 11, 2017
DOI:10.1021/acs.orglett.7b02498
A novel strategy for accessing cyclic α-amino ketones enantioselectively has opened a simple synthetic route to the antidepressant (2R,6R)-hydroxynorketamine and numerous analogues. Mechanistically guided catalyst selection was essential in an initial olefin epoxidation step. In a second crucial step, the epoxide was subjected to a novel O → N displacement that occurred with retention of configuration through the use of Al- or Ti-based azides, which promote epoxide activation and internal cis delivery of N3 to carbon.
Co-reporter:Rajender Vemula, Nathan C. Wilde, Rajendar Goreti, and E. J. Corey
Organic Letters July 21, 2017 Volume 19(Issue 14) pp:3883-3883
Publication Date(Web):July 11, 2017
DOI:10.1021/acs.orglett.7b01768
The previously unknown 5-spirocyclohexylisoimidazole has been made efficiently and simply by reaction of ammonia, glyoxal hydrate, and cyclohexanone. It is a very useful precursor for the diastereocontrolled synthesis of many C2-symmetric 1,2-diamines, a class which is important for the generation of a variety of C2-symmetric reagents and catalysts for enantioselective synthesis.
Co-reporter:Simon Breitler, Yixin Han, and E. J. Corey
Organic Letters December 15, 2017 Volume 19(Issue 24) pp:6686-6686
Publication Date(Web):December 1, 2017
DOI:10.1021/acs.orglett.7b03412
A novel method is demonstrated for the diastereoselective conversion of a monocyclic enone to a pair of bicyclic intermediates which enable the stereocontrolled enantioselective syntheses of retigeranic acids A and B.
Co-reporter:Eswar Bhimireddy and E. J. Corey
Journal of the American Chemical Society August 16, 2017 Volume 139(Issue 32) pp:11044-11044
Publication Date(Web):July 31, 2017
DOI:10.1021/jacs.7b07366
A method is described for the joining of two α-lithiated C(sp3) stereocenters efficiently and with retention of configuration. The key step involves the effective removal of two electrons from a chiral organocuprate R2CuLi, by i-propyl 2,4-dinitrobenzoate to form a Cu(III) complex that undergoes at −90 °C accelerated reductive elimination enantioselectively and exclusively without the formation of free radicals.
Co-reporter:Karla Mahender Reddy; Eswar Bhimireddy; Barla Thirupathi; Simon Breitler; Shunming Yu;E. J. Corey
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2443-2453
Publication Date(Web):January 26, 2016
DOI:10.1021/jacs.6b00100
The coordination of chiral ligands to Lewis acid metal derivatives, a useful strategy for enantioselective, electrophilic catalysis, generally leads to a lower level of catalytic activity than that of the original uncomplexed compound. Activation by further attachment of a proton or strong Lewis acid to the complex provides a way to overcome the deactivating effect of a chiral ligand. The research described herein has demonstrated that further enhancement of catalytic activity is possible by the judicious placement of fluorine substituents in the chiral ligand. This approach has led to a new, second-generation family of chiral oxazaborolidinium cationic species which can be used to effect many Diels–Alder reactions in >95% yield and >95% ee using catalyst loadings at the 1–2 mol % level. The easy recovery of the chiral ligand makes the application of these new catalysts especially attractive for large-scale synthesis.
Co-reporter:Barla Thirupathi, Simon Breitler, Karla Mahender Reddy, and E. J. Corey
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10842-10845
Publication Date(Web):August 16, 2016
DOI:10.1021/jacs.6b08018
The activation of second-generation fluorinated oxazaborolidines by the strong acid triflimide (Tf2NH) in CH2Cl2 solution leads to highly active chiral Lewis acids that are very effective catalysts for (4 + 2) cycloaddition. We report herein that this catalytic activity can be further enhanced by the use of Tf2NH in combination with the biscoordinating Lewis acid TiCl4 or SnCl4 as a coactivator. The effective increase in acidity of an exceedingly strong protic acid is greater for biscoordinating TiCl4 and SnCl4 than for monocoordinating salts, even the strong Lewis acids AlBr3 and BBr3 in CH2Cl2 or CH2Cl2/toluene. The increase in the effective acidity of Tf2NH can be understood in terms of a stabilized cyclic anionic complex of Tf2N– and TiCl4, which implies a broader utility than that described here. The utility of Tf2NH–TiCl4 activation of fluorinated oxazaborolidines is documented by examples including the first enantioselective (4 + 2) cycloaddition to α,β-unsaturated acid chlorides.
Co-reporter:Yixin Han, Simon Breitler, Shao-Liang Zheng, and E. J. Corey
Organic Letters 2016 Volume 18(Issue 23) pp:6172-6175
Publication Date(Web):November 18, 2016
DOI:10.1021/acs.orglett.6b03186
The enantioselective reduction of prochiral 4,4-disubstituted 2,5-cyclohexadienones to chiral 2-cyclohexenones has been accomplished by the use of a carefully selected chiral bisphosphine–CuI complex and diisobutylaluminum hydride–hexamethylphosphoric triamide complex. This reagent has provided access to a key bicyclic intermediate for the total synthesis of the natural enantiomer of the pentacyclic sesterterpene retigeranic acid that involves spatial discrimination between CH3 and CH2CH2R substituents, an operation that has been elusive previously. In addition, a second method for desymmetrization is described using catalytic enantioselective [4 + 2]-cycloaddition of cyclopentadiene to prochiral 4,4-disubstituted 2,5-cyclohexadienones.
Co-reporter:Goreti Rajendar;E. J. Corey
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5837-5844
Publication Date(Web):April 14, 2015
DOI:10.1021/jacs.5b03229
Three different methods have been developed that effectively utilize chiral oxiranes derived from Katsuki–Sharpless epoxidation of allylic alcohols as initiating groups for cationic cyclization of unsaturated substrates to form chiral polycycles. This type of transformation has previously been problematic. These employ either epoxy-methoximes, vinyl-substituted oxiranes, or hydroxymethyl oxiranes. All three approaches are described in detail. In addition, this research has led to possible explanations for previously encountered difficulties in this area and provided two new insights into the Lewis acid activation of oxiranes. The methodology described herein constitutes a valuable link between two powerful synthetic constructions, enantioselective Katsuki–Sharpless epoxidation and cationic polycyclization reactions.
Co-reporter:Karavadhi Surendra, Rajender Vemula, Yifeng Han, and E. J. Corey
Organic Letters 2015 Volume 17(Issue 7) pp:1621-1623
Publication Date(Web):March 11, 2015
DOI:10.1021/acs.orglett.5b00521
Lithiation of α–C-H groups in organic substrates by RLi or R2NLi followed by silylation with R′3SiCl generally provides analogous products regardless of the R′ group of R′3SiCl. A striking exception using 3,4-benzothiophane as substrate depending on whether R′ is methyl, phenyl, or isopropyl is demonstrated. With R′ = Me or Ph, the geminal α,α-bis-silylated products result whereas with i-Pr3SiCl the trans-α,α′-bis-silylated sulfide is formed. The latter pathway provides ready access to the C2-symmetric enantiomers of trans-2,5-bis(triisopropylsilyl)-3,4-benzothiophane.
Co-reporter:Karavadhi Surendra ;E. J. Corey
Journal of the American Chemical Society 2014 Volume 136(Issue 31) pp:10918-10920
Publication Date(Web):July 24, 2014
DOI:10.1021/ja506502p
The removal of the iodide ion from indium triiodide by means of reactive Ag(I) salts leads to the formation of the highly reactive ligandless cation InI2+, which is unusual in having two vacant low-lying p-orbitals. This bivalent Lewis acidity leads to an especially high affinity for the two orthogonal π-bonds of carbon–carbon triple bonds. Consequently, the double-coordinating InI2+ is an especially effective reagent for the selective activation of C≡C and the catalytic initiation of cationic cyclization processes. A number of such reactions are described to demonstrate synthetic utility.
Co-reporter:Yifeng Han, Yun Ma, Ivan Keresztes, David B. Collum, and E. J. Corey
Organic Letters 2014 Volume 16(Issue 17) pp:4678-4679
Publication Date(Web):August 26, 2014
DOI:10.1021/ol502348y
Benzylic C–H lithiation of 3,4-benzothiophane and subsequent treatment with triphenyl- or trimethylchlorosilane under a variety of conditions leads to α,α- rather than α,α′-bis-silylation products as a consequence of anion stabilization by R3Si and very fast deprotonation of the intermediate monosilylated product, even with a sterically bulky base such as lithium diisopropylamide.
Co-reporter:Karavadhi Surendra ; Goreti Rajendar ;E. J. Corey
Journal of the American Chemical Society 2013 Volume 136(Issue 2) pp:642-645
Publication Date(Web):December 20, 2013
DOI:10.1021/ja4125093
The 1:1 complex of o,o′-dichloro-R-BINOL and SbCl5 initiates the enantioselective cationic polycyclization of polyunsaturated substrates at a predictable π-bond which may be either terminal or, as shown herein, internal. The extension of this powerful construction to internal π-bonds expands the scope of this method and opens up very short pathways to numerous chiral polycyclic molecules, including natural products and their analogues. Especially simple synthetic routes are disclosed that provide access to dysideapalaunic acid, dehydroabietic acid, and epipodocarpic acid and illustrate the value of this enantioselective approach.
Co-reporter:Timothy R. Newhouse, Philip S. J. Kaib, Andrew W. Gross, and E. J. Corey
Organic Letters 2013 Volume 15(Issue 7) pp:1591-1593
Publication Date(Web):March 7, 2013
DOI:10.1021/ol400362t
A highly effective acid-catalyzed cyclopropyl ester to γ-lactone skeletal rearrangement has been demonstrated and applied to the synthesis of a variety of bi- and tricyclic functionalized lactones, rigid and highly compact structures for use as biological probes.
Co-reporter:Timothy R. Newhouse ; Xin Li ; Megan M. Blewett ; Clare M. C. Whitehead ;E. J. Corey
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17354-17357
Publication Date(Web):October 9, 2012
DOI:10.1021/ja305991k
A detailed stereomechanistic analysis has led to the design of a new tetradentate ligand for the enantioselective Ti(IV)-catalyzed oxidation of unsymmetrical sulfides to sulfoxides with high selectivity. The pathway of this oxidation and the closely related and long-known Kagan–Modena oxidation have been clarified to identify the likely origin of the enantioselectivity.
Co-reporter:Karavadhi Surendra ;E. J. Corey
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:11992-11994
Publication Date(Web):July 10, 2012
DOI:10.1021/ja305851h
This report describes the synthesis of a range of chiral polycyclic molecules (tricyclic to pentacyclic) from achiral polyene precursors by enantioselective proton-initiated polycyclization promoted by the 1:1 complex of o,o′-dichloro-BINOL and SbCl5. Excellent yields (ca. 90% per ring formed) and enantioselectivety (20:1 to 50:1) were obtained. The process is practical as well as efficient, because the chiral ligand is both readily prepared from R,R- or S,S-BINOL and easily recovered from the reaction mixture by extraction.
Co-reporter:Karavadhi Surendra ; Wenwei Qiu ;E. J. Corey
Journal of the American Chemical Society 2011 Volume 133(Issue 25) pp:9724-9726
Publication Date(Web):June 6, 2011
DOI:10.1021/ja204142n
InI3 and InBr3 have been found to be effective catalysts for the π activation of C≡C bonds to initiate the conversion of chiral propargylic alcohols or silyl ethers to polycyclic products in excellent yields and with high stereoselectivity. The method has been applied to the synthesis of chiral fused hexacyclic ring systems with the creation of multiple new stereocenters. The power and scope of the method are illustrated by a variety of examples.
Co-reporter:Wen-Wei Qiu, Karavadhi Surendra, Liang Yin, and E. J. Corey
Organic Letters 2011 Volume 13(Issue 21) pp:5893-5895
Publication Date(Web):October 12, 2011
DOI:10.1021/ol202621g
Fifteen examples are disclosed of efficient In(III)-catalyzed six-membered ring closure leading to bi-, tri-, and tetracyclic products.
Co-reporter:M. Kevin Brown ; Megan M. Blewett ; James R. Colombe ;E. J. Corey
Journal of the American Chemical Society 2010 Volume 132(Issue 32) pp:11165-11170
Publication Date(Web):July 28, 2010
DOI:10.1021/ja103103d
The experiments described here clarify the mechanism and origin of the enantioselectivity of the oxidation of racemic secondary alcohols catalyzed by chiral Mn(III)−salen complexes using HOBr, Br2/H2O/KOAc or PhI(OAc)2/H2O/KBr as a stoichiometric oxidant. Key points of the proposed pathway include (1) the formation of a Mn(V)−salen dibromide, (2) its subsequent reaction with the alcohol to give an alkoxy-Mn(V) species, and (3) carbonyl-forming elimination to produce the ketone via a highly organized transition state with intramolecular transfer of hydrogen from carbon to an oxygen of the salen ligand.
Co-reporter:Ryan A. Shenvi and E. J. Corey
Organic Letters 2010 Volume 12(Issue 15) pp:3548-3551
Publication Date(Web):July 12, 2010
DOI:10.1021/ol101410g
The presence of an ether oxygen within a chain undergoing cation−polyene cyclization has a profound influence on the stereochemistry of this important construction, apparently due to nucleophilic participation of oxygen in the cyclization process and formation of an oxonium intermediate, leading to bent fused ring systems.
Co-reporter:Oleg V. Larionov and E. J. Corey
Organic Letters 2010 Volume 12(Issue 2) pp:300-302
Publication Date(Web):December 15, 2009
DOI:10.1021/ol902643w
Alkynylaluminum reagents undergo enantioselective conjugate addition to cyclic α,β-enones in the presence of chiral bisphosphine complexes of Ni(II).
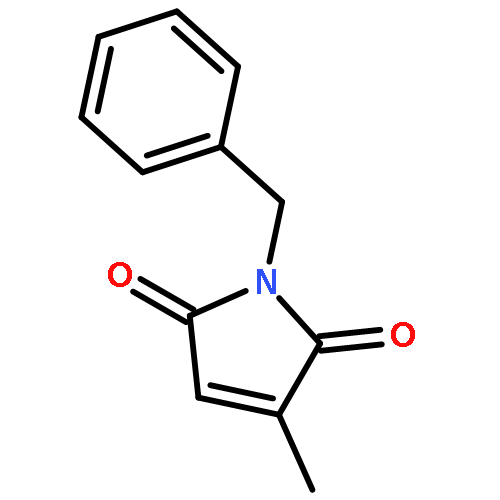
Co-reporter:Santanu Mukherjee and E. J. Corey
Organic Letters 2010 Volume 12(Issue 3) pp:632-635
Publication Date(Web):January 11, 2010
DOI:10.1021/ol902865d
Diels−Alder reactions of various combinations of maleimides and 1,3-dienes with cationic oxazaborolidines as catalysts have been shown to be highly efficient and enantioselective.
Co-reporter:Santanu Mukherjee and E. J. Corey
Organic Letters 2010 Volume 12(Issue 5) pp:1024-1027
Publication Date(Web):February 4, 2010
DOI:10.1021/ol100032u
The reaction rates and products in enantioselective Diels−Alder reactions with a range of dienophiles correlate with the expected degree of concertedness of bond formation in the transition state.
Co-reporter:Santanu Mukherjee, Alex P. Scopton and E. J. Corey
Organic Letters 2010 Volume 12(Issue 8) pp:1836-1838
Publication Date(Web):March 25, 2010
DOI:10.1021/ol1004802
Simple enantioselective routes to the two key intermediates shown above (at center) for the synthesis of laurenditerpenol have been developed using a Diels−Alder step and the same catalyst system for each.
Co-reporter:Xuri Gao, Ravi P. Singh and E. J. Corey
Organic Letters 2010 Volume 12(Issue 8) pp:1812-1814
Publication Date(Web):March 23, 2010
DOI:10.1021/ol100426m
An efficient enantioselective synthesis of the above C3-symmetric chiral quinuclidine starting with N-tert-butoxycarbonyl-4-pyridone has been developed.
Co-reporter:Rong-Jie Chein and E. J. Corey
Organic Letters 2010 Volume 12(Issue 1) pp:132-135
Publication Date(Web):December 3, 2009
DOI:10.1021/ol9025364
Electron-pair repulsion in 2-methoxyheteroarenes is important for N, but not for O or S heteroatoms.
Co-reporter:M. Kevin Brown and E. J. Corey
Organic Letters 2010 Volume 12(Issue 1) pp:172-175
Publication Date(Web):November 30, 2009
DOI:10.1021/ol9025793
This paper reports a method for highly enantioselective Diels−Alder reaction with an acetylene equivalent to produce chiral-bridged dienes. These dienes, by coordination to Rh(I), can serve as catalysts for the enantioselective addition of vinyl or aryl groups to α,β-unsaturated ketones.
Co-reporter:Ryan A. Shenvi ;E. J. Corey
Journal of the American Chemical Society 2009 Volume 131(Issue 16) pp:5746-5747
Publication Date(Web):April 1, 2009
DOI:10.1021/ja901400q
Short, practical, and scalable syntheses of (±)-7-methylomuralide and (−)-7-methylomuralide have been developed. Three consecutive tandem reaction pairs establish all of the carbons and the stereochemistry of the target molecule, vastly simplifying the synthetic scheme from N-trichloroethoxycarbonyl glycine. The chiral directing group controls the absolute stereochemistry of the key aldol reaction.
Co-reporter:Barbara Czakó ; László Kürti ; Akiko Mammoto ; Donald E. Ingber ;E. J. Corey
Journal of the American Chemical Society 2009 Volume 131(Issue 25) pp:9014-9019
Publication Date(Web):May 26, 2009
DOI:10.1021/ja902601e
The discovery that cortistatins A and J show noteworthy antiangiogenic activity prompted an investigation of the possibility that simpler and much more easily made compounds based on a steroid core might have useful bioactivity. These studies have led to the development of several potent, water-soluble compounds that may be suitable for local application to treat ocular wet macular degeneration, an important cause of blindness, as well as for treatment of various other angiogenesis-dependent diseases. One of these substances was tested in a mouse retinal angiogenesis model and found to inhibit angiogenesis at a locally administered dose of 500 pmol. Comparison of cell migration data for this and two other synthetic compounds with published data on cortistatin A indicate that they inhibit vascular endothelial growth factor-induced cell migration of human umbilical vein endothelial cells more strongly than cortistatin A.
Co-reporter:Karavadhi Surendra ;E. J. Corey
Journal of the American Chemical Society 2009 Volume 131(Issue 39) pp:13928-13929
Publication Date(Web):September 9, 2009
DOI:10.1021/ja906335u
The first enantioselective synthesis of lupeol has been developed by applying two carefully crafted cation-π cyclization stages to generate the pentacyclic structure with complete stereocontrol. The synthesis (Scheme 1) is noteworthy because of its brevity and also because it solves a longstanding problem in the field of natural product synthesis.
Co-reporter:Rong-Jie Chein, Ying-Yeung Yeung and E. J. Corey
Organic Letters 2009 Volume 11(Issue 7) pp:1611-1614
Publication Date(Web):March 11, 2009
DOI:10.1021/ol900258f
The oxazaborolidine-catalyzed reduction of 2,2-disubstituted cycloalkan-1,3-diones or hindered 2,2-disubstituted cyclic ketones using catecholborane as reductant proceeds with greater enantioselectivity when N,N-diethylaniline is added. It has now been shown that the effect of this additive is to catalyze the conversion of a harmful minor impurity in catalyst preparations to the active catalyst.
Co-reporter:László Kürti, Megan M. Blewett and E. J. Corey
Organic Letters 2009 Volume 11(Issue 20) pp:4592-4595
Publication Date(Web):September 15, 2009
DOI:10.1021/ol901859d
It is proposed that facial selectivity in the Jacobsen epoxidation is determined by electrostatic and steric factors with a two-step pathway involving a carbocationic intermediate.
Co-reporter:E.J. Corey
Angewandte Chemie International Edition 2009 Volume 48( Issue 12) pp:2100-2117
Publication Date(Web):
DOI:10.1002/anie.200805374
Co-reporter:E.J. Corey
Angewandte Chemie 2009 Volume 121( Issue 12) pp:2134-2151
Publication Date(Web):
DOI:10.1002/ange.200805374
Co-reporter:E. J. Corey and D. E. Koshland Jr.
ACS Chemical Biology 2007 Volume 2(Issue 7) pp:449
Publication Date(Web):July 20, 2007
DOI:10.1021/cb7001297
Co-reporter:E. J. Corey
Angewandte Chemie International Edition 2002 Volume 41(Issue 10) pp:
Publication Date(Web):15 MAY 2002
DOI:10.1002/1521-3773(20020517)41:10<1629::AID-ANIE1629>3.0.CO;2-0
The cover picture shows portraits of Otto Diels (top left) and Kurt Alder together with the original Diels–Alder reaction between cyclopentadiene and p-quinone. Also illustrated are the key Diels–Alder reactions that enabled the total synthesis of prostaglandins and taxol as they were carried out by the research groups of Corey and Nicolaou, respectively. This year marks the centennial anniversary of Alder's birth—and this date will be celebrated at Alder's University, the Universität Köln, by a symposium on the 27th May—while last year was the 125th anniversary of the birth of Diels and next year will signify the 75th anniversary of the first publication of Diels and Alder on the reaction of 1,3-dienes with olefins. The Diels–Alder reaction, as it has become known, is without doubt one of the most important reaction of organic syntheses, and thus it is a bonanza that Corey and Nicolaou et al. have written appropriate reviews, which can be found on pp. 1650–1667 and 1668–1698, respectively.
Co-reporter:E. J. Corey
Angewandte Chemie International Edition 2002 Volume 41(Issue 10) pp:
Publication Date(Web):15 MAY 2002
DOI:10.1002/1521-3773(20020517)41:10<1650::AID-ANIE1650>3.0.CO;2-B
One hundred years after the birth of Kurt Alder and seventy-five years after the discovery of his famous reaction, one of the most important and fascinating transformations in chemistry, research on that process continues to surprise, excite, delight, and inform the chemical community. This article is based on presentations given first at the University of Cologne, Germany (Kurt Alder lecture, 1992), then at the Roger Adams Award Symposium (1993), and later at the Bürgenstock Conference of 2001, and describes research by our group on the development and understanding of enantioselective versions of the Diels–Alder reactions. The elements of this review include 1) development of new chiral Lewis acid catalysts for highly enantioselective (>25:1) [4+2] cycloadditions; 2) the fine mechanistic details and pre-transition-state assemblies of these reactions; 3) the fundamental understanding of catalytic activity and enantioselectivity for highly enantioselective Diels–Alder processes; and 4) applications to the synthesis of complex molecules. The range and power of the Diels–Alder reaction have steadily increased over seven decades. The end of this remarkable development is not in sight, a high compliment to this field of Science and to its great inventor.
Co-reporter:E. J. Corey ;K. C. Nicolaou Dr.;Scott A. Snyder;Tamsyn Montagnon Dr.;Georgios E. Vassilikogiannakis Dr.
Angewandte Chemie 2002 Volume 114(Issue 10) pp:
Publication Date(Web):15 MAY 2002
DOI:10.1002/1521-3757(20020517)114:10<1703::AID-ANGE1703>3.0.CO;2-#
Das Titelbild zeigt Portraits von Otto Diels (links oben) und Kurt Alder zusammen mit der Original-Diels-Alder-Reaktion von Cyclopentadien mit p-Chinon. Zusätzlich sind die Diels-Alder-Reaktionen abgebildet, die Schlüsselschritte der Totalsynthesen der Prostaglandine und von Taxol waren, wie sie von Corey et al. bzw. Nicolaou et al. durchgeführt wurden. Entworfen wurde das Titelbild von Robbin Echon und Scott Snyder, beide am Scripps Research Institut in La Jolla, Kalifornien. Dieses Jahr jährt sich Alders Geburtstag zum hundertsten Mal – und dieses Datum wird an Alders Wirkungsstätte, der Universität Köln, mit einem Symposium am 27. Mai gefeiert –, letztes Jahr wäre Diels 125 Jahre alt geworden, und ins nächste Jahr fällt das 75. Jubiläum der ersten Publikation von Diels und Alder über die Reaktion von 1,3-Dienen mit Olefinen. Die Diels-Alder-Reaktion, wie sie bald genannt wurde, zählt sicherlich zu den allerwichtigsten Reaktionen der organischen Synthese, und es ist deshalb ein Glücksfall, dass E. J. Corey und K. C. Nicolaou et al. passende Übersichtsartikel schrieben, die sich auf den Seiten 1724–1741 bzw. 1742–1773 finden.
Co-reporter:E. J. Corey
Angewandte Chemie 2002 Volume 114(Issue 10) pp:
Publication Date(Web):15 MAY 2002
DOI:10.1002/1521-3757(20020517)114:10<1724::AID-ANGE1724>3.0.CO;2-Q
Einhundert Jahre nach der Geburt von Kurt Alder und fünfundsiebzig Jahre nach der Entdeckung seiner berühmten Reaktion, einer der bedeutendsten und faszinierendsten Umwandlungen in der Chemie, ist die Forschung auf diesem Gebiet für Chemiker in der ganzen Welt noch immer überraschend, spannend, begeisternd und informativ. Dieser Aufsatz beruht auf Vorträgen, die an der Universität zu Köln (Kurt-Alder-Vortrag, 1992), beim Roger-Adams-Award-Symposium 1993 und anlässlich der Bürgenstock-Konferenz 2001 gehalten wurden. Er beschreibt die Forschung unserer Arbeitsgruppe in Harvard über die Entwicklung und das Verständnis enantioselektiver Varianten von Diels-Alder-Reaktionen und umfasst 1) die Entwicklung neuer chiraler Lewis-Säure-Katalysatoren für hoch enantioselektive (>25:1 Enantioselektivität) [4+2]-Cycloadditionen, 2) mechanistische Einzelheiten und Anordnungen in frühen Übergangszuständen dieser Reaktionen, 3) den fundamentalen Einblick in die katalytische Aktivität und Enantioselektion bei hoch enantioselektiven Diels-Alder-Reaktionen und 4) Anwendungen für die Synthese komplexer Moleküle. Der Anwendungsbereich und die Leistungsfähigkeit der Diels-Alder-Reaktion haben über sieben Jahrzehnte stetig zugenommen, und ein Ende dieser bemerkenswerten Entwicklung ist nicht in Sicht – eine hohe Anerkennung für dieses Gebiet der Wissenschaft und ihren großen Erfinder.
Co-reporter:E. J. Corey and Thomas W. Lee
Chemical Communications 2001 (Issue 15) pp:1321-1329
Publication Date(Web):11 Jul 2001
DOI:10.1039/B104800G
X-Ray crystallographic studies have provided experimental
evidence for the existence of intramolecular formyl C–H hydrogen
bonds to oxygen or fluorine ligands in complexes of aldehydes and boron
Lewis acids. This type of hydrogen bond can be regarded as
‘induced’ or ‘cooperative’ in the sense that its
strength can be expected to increase as the bonding between the formyl
oxygen and the Lewis acid becomes stronger. Coplanarity of the formyl group
and the metal–X subunit to which it is bound in a five-membered ring
effectively restricts rotation about the donor–acceptor bond between
the formyl oxygen and the metal center of the Lewis acid, thus creating an
additional organizing element in these complexes. This organizing element
provides a simple and logical basis for understanding the mechanistic basis
for enantioselectivity in many reactions of achiral aldehydes which are
catalyzed by chiral Lewis acids. These reactions include aldol, allylation
and ene addition to the formyl CO group and Diels–Alder
reactions of α,β-unsaturated aldehydes with 1,3-dienes. The idea
of the induced formyl C–H hydrogen bond can serve as a guide in the
design of new enantioselective catalysts as well as a mechanistic principle
for understanding preferred transition state assemblies.
Co-reporter:Henning Steinhagen;E. J. Corey
Angewandte Chemie 1999 Volume 111(Issue 13‐14) pp:
Publication Date(Web):12 JUL 1999
DOI:10.1002/(SICI)1521-3757(19990712)111:13/14<2054::AID-ANGE2054>3.0.CO;2-4
DieeinfachstedenkbareMethodezurHerstellungvono-Azaxylylenen, die baseninduzierte Eliminierung von Chlorwasserstoff aus Amid- oder Sulfonamidderivaten von o-(Chlormethyl)anilin, war bisher nicht beschrieben worden. Diese vielseitige Methode eröffnet einen einfachen und stereoselektiven Zugang zu polycyclischen Hydrochinolinderivaten, wie in Gleichung (1) an einem Beispiel gezeigt ist.
Co-reporter:E. J. Corey;Fu-Yao Zhang
Angewandte Chemie 1999 Volume 111(Issue 13‐14) pp:
Publication Date(Web):12 JUL 1999
DOI:10.1002/(SICI)1521-3757(19990712)111:13/14<2057::AID-ANGE2057>3.0.CO;2-N
Wahlweise Amprenavir 1 oder sein C(2)-Diastereomer lassen sich sehr einfach herstellen, wenn die Nitroaldolreaktion in Gegenwart eines der beiden Ammonium-Ionen 2 durchgeführt wird. Das Strukturelement 1,3-Diamino-2-hydroxypropyl von 1 liegt in vielen Peptidmimetika und HIV-Protease-Inhibitoren vor. Die hier vorgestellte neue Strategie sollte daher auch für die direkte und stereokontrollierte Synthese solcher Verbindungen nützlich sein.
Co-reporter:Henning Steinhagen;E. J. Corey
Angewandte Chemie International Edition 1999 Volume 38(Issue 13‐14) pp:
Publication Date(Web):12 JUL 1999
DOI:10.1002/(SICI)1521-3773(19990712)38:13/14<1928::AID-ANIE1928>3.0.CO;2-1
Surprisingly, thesimplestmethodpossibleforo-azaxylyleneproduction, base-induced elimination of hydrogen chloride from amide or sulfonamide derivatives of o-(chloromethyl)aniline, has never been reported. This powerful approach provides easy and stereoselective access to polycyclic hydroquinolines, as shown for an example in Equation (1).
Co-reporter:E. J. Corey;Fu-Yao Zhang
Angewandte Chemie International Edition 1999 Volume 38(Issue 13‐14) pp:
Publication Date(Web):12 JUL 1999
DOI:10.1002/(SICI)1521-3773(19990712)38:13/14<1931::AID-ANIE1931>3.0.CO;2-4
Either amprenavir (1) or its C(2) diastereomer can be synthesized in a simple way by the use of a nitroaldol reaction carried out in the presence of one or the other of the two ammonium ions 2. The 1,3-diamino-2-hydroxypropyl structural element of 1 is also found in many other peptidomimetics and HIV protease inhibitors. Described here is a new strategy for possible application to direct and stereocontrolled synthesis of such compounds.
Co-reporter:Elias J. Corey;Christopher J. Helal
Angewandte Chemie International Edition 1998 Volume 37(Issue 15) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z
High enantioselectivity can be achieved when chiral oxazaborolidines are used as catalysts in the reduction of ketones by borane. In the transition state on the way to the complex chiral compounds, the two reactants are activated and held in close proximity by the catalyst, as shown below.
Co-reporter:Elias J. Corey;Guanglin Luo;Linus Shouzhong Lin
Angewandte Chemie International Edition 1998 Volume 37(Issue 8) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980504)37:8<1126::AID-ANIE1126>3.0.CO;2-0
Coupling between a sulfone and an acylsilane fragment (see below) constitutes the first step in the exceptionally short total syntheses of two chiral hexacyclic hydrocarbons isolated from Eocene Messel shale (Germany). Like the first step, subsequent steps also employ a powerful synthetic tactic: polycyclization of a specially constructed chiral, multiply unsaturated oxirane. TBS=tBuMe2Si.
Co-reporter:Elias J. Corey;Angel Guzman-Perez
Angewandte Chemie International Edition 1998 Volume 37(Issue 4) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V
A long-standing challenge to synthesis can now be met through the use of new and powerful catalytic asymmetric reactions for the assembly of complex chiral molecules with quaternary stereocenters from achiral building blocks. The reaction sequence shown below is just one example discussed in this review.
Co-reporter:E. J. Corey;Weidong Li;Tohru Nagamitsu
Angewandte Chemie International Edition 1998 Volume 37(Issue 12) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980703)37:12<1676::AID-ANIE1676>3.0.CO;2-T
A selective, irreversible inhibitor of proteasome function, lactacystin (1) is an important experimental tool in cell biology. An efficient and direct enantioselective synthesis of lactacystin proceeds via the intermediates shown below. This process allows for the first time easy access to analogues of lactacystin in which the isopropyl substituent is replaced by other lipophilic groups. PMB=p-methoxybenzyl.
Co-reporter:Elias J. Corey;Christopher J. Helal
Angewandte Chemie 1998 Volume 110(Issue 15) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980803)110:15<2092::AID-ANGE2092>3.0.CO;2-M
Eine hohe Enantioselektivität kann in der durch komplexe chirale Oxazaborolidine katalysierten Reduktion von Ketonen erreicht werden. Der Katalysator aktiviert im Übergangszustand die beiden Reaktanten und hält sie in enger Nachbarschaft zueinander (siehe Schema unten).
Co-reporter:Elias James Corey;Guanglin Luo;Linus Shouzhong Lin
Angewandte Chemie 1998 Volume 110(Issue 8) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980420)110:8<1147::AID-ANGE1147>3.0.CO;2-X
Die Kupplung zwischen einem Sulfon- und einem Acylsilanfragment (siehe unten) bildet den ersten Schritt der außergewöhnlich kurzen, ersten Totalsynthesen zweier chiraler hexacyclischer Kohlenwasserstoffe, die aus dem aus dem Eozän stammenden Messel-Schiefer (Deutschland) isoliert wurden. Ebenso wie bei diesem Schritt kommt auch beim folgenden eine leistungsfähige Synthesetaktik zum Einsatz: die Polycyclisierung des zunächst gebildeten chiralen, mehrfach ungesättigten Oxirans. TBS = tBuMe2Si.
Co-reporter:E. J. Corey;Angel Guzman-Perez
Angewandte Chemie 1998 Volume 110(Issue 4) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980216)110:4<402::AID-ANGE402>3.0.CO;2-6
Scheinbar wie durch Magie können komplexe chirale Moleküle mit quartären Stereozentren aus achiralen Bausteinen zusammengesetzt werden, indem eine Vielzahl neuer und effektiver katalytischer asymmetrischer Reaktionen angewendet wird. Die unten gezeigte Reaktionsfolge gibt ein Beispiel.
Co-reporter:E. J. Corey;Weidong Li;Tohru Nagamitsu
Angewandte Chemie 1998 Volume 110(Issue 12) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980619)110:12<1784::AID-ANGE1784>3.0.CO;2-#
Ein selektiver, irreversibler Proteasom-Inhibitor ist Lactacystin 1, das daher ein wichtiges Werkzeug in der Zellbiologie geworden ist. Eine effiziente und direkte enantioselektive Synthese von Lactacystin verläuft über die unten dargestellten Zwischenprodukte. Dieser Ansatz eröffnet erstmals auch einen einfachen Zugang zu Analoga von Lactacystin, bei denen der Isopropylsubstituent durch andere lipophile Gruppen ersetzt ist. PMB=p-Methoxybenzyl.
Co-reporter:E.J. Corey, David Barnes-Seeman, Thomas W. Lee
Tetrahedron: Asymmetry 1997 Volume 8(Issue 22) pp:3711-3713
Publication Date(Web):27 November 1997
DOI:10.1016/S0957-4166(97)00528-4
The diastereoselective rearrangement of α,α-dichloromethylboronate derivatives of chiral 1,2-diols (Matteson rearrangement) can be understood readily in terms of a bidentate interaction between the catalytic Lewis acid (ZnCl2) and the substrate, leading to favored transition states such as 4 and 5.Graphic
Co-reporter:Renad I. Zhdanov, Elias J. Corey
Steroids (15 September 2009) Volume 74(Issue 9) pp:723-724
Publication Date(Web):15 September 2009
DOI:10.1016/j.steroids.2009.03.004
The “Torgov reaction” opened an original pathway for the total chemical synthesis of steroid hormones, which is still used for the large-scale industrial production of steroid hormones at factories of Schering AG in Berlin, Germany.
Co-reporter:Renad I. Zhdanov, Elias J. Corey
Steroids (15 September 2009) Volume 74(Issue 9) pp:723-724
Publication Date(Web):15 September 2009
DOI:10.1016/j.steroids.2009.03.004
The “Torgov reaction” opened an original pathway for the total chemical synthesis of steroid hormones, which is still used for the large-scale industrial production of steroid hormones at factories of Schering AG in Berlin, Germany.

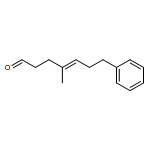
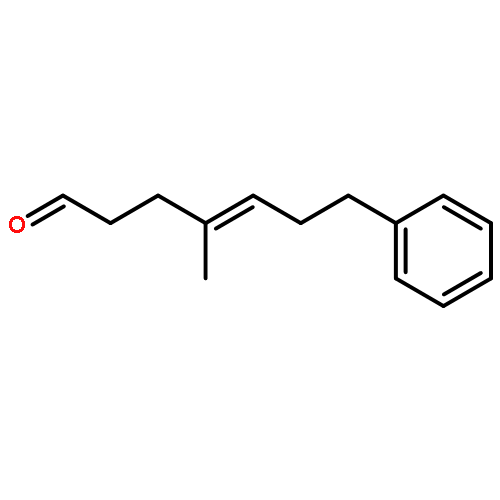
![(2r,5r)-1-[2-[(2r,5r)-2,5-diphenylphospholan-1-yl]ethyl]-2,5-diphenylphospholane](http://img.cochemist.com/ccimg/528600/528565-79-9.png)
![(2r,5r)-1-[2-[(2r,5r)-2,5-diphenylphospholan-1-yl]ethyl]-2,5-diphenylphospholane](http://img.cochemist.com/ccimg/528600/528565-79-9_b.png)


![BICYCLO[2.2.2]OCT-5-ENE-2,3-DIMETHANOL, (1R,2S,3S,4S)-](http://img.cochemist.com/ccimg/167900/167819-35-4.png)
![BICYCLO[2.2.2]OCT-5-ENE-2,3-DIMETHANOL, (1R,2S,3S,4S)-](http://img.cochemist.com/ccimg/167900/167819-35-4_b.png)


![(1R,2S,4S)-3,3-Dimethylbicyclo[2.2.1]heptane-2-methanol](http://img.cochemist.com/ccimg/81700/81601-68-5.png)
![(1R,2S,4S)-3,3-Dimethylbicyclo[2.2.1]heptane-2-methanol](http://img.cochemist.com/ccimg/81700/81601-68-5_b.png)

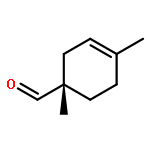
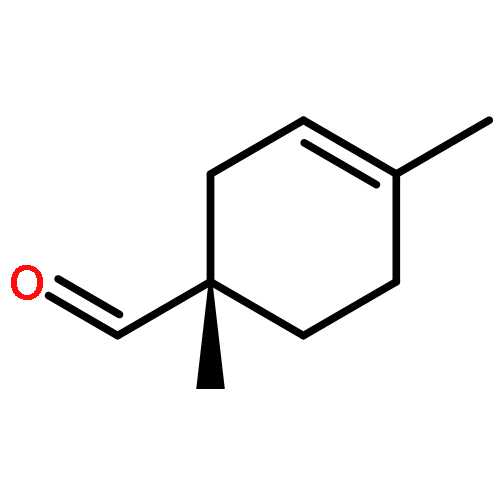
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2S,4R)-](http://img.cochemist.com/ccimg/72300/72204-08-1.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2S,4R)-](http://img.cochemist.com/ccimg/72300/72204-08-1_b.png)
![(7aS)-tetrahydro-1H,3H-Pyrrolo[1,2-c]oxazole-1,3-dione](http://img.cochemist.com/ccimg/45800/45736-33-2.png)
![(7aS)-tetrahydro-1H,3H-Pyrrolo[1,2-c]oxazole-1,3-dione](http://img.cochemist.com/ccimg/45800/45736-33-2_b.png)














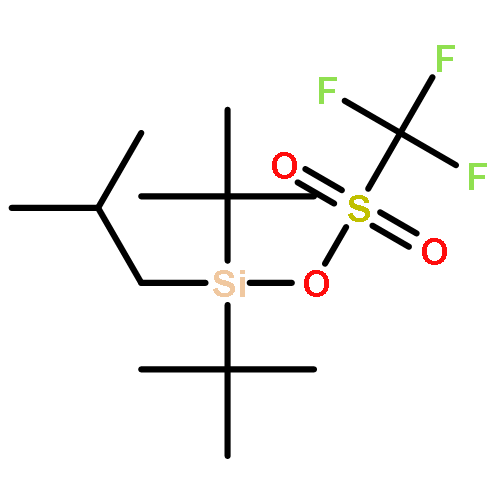
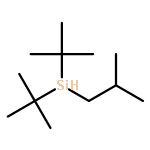
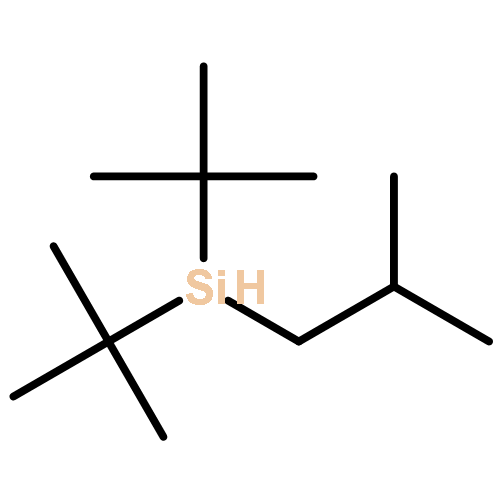
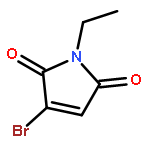
![2,6-DIOXABICYCLO[2.2.1]HEPTANE, 1-METHYL-4-(PHENYLMETHYL)-](http://img.cochemist.com/ccimg/862500/862457-29-2.png)
![2,6-DIOXABICYCLO[2.2.1]HEPTANE, 1-METHYL-4-(PHENYLMETHYL)-](http://img.cochemist.com/ccimg/862500/862457-29-2_b.png)
![1H-TETRAZOLE, 5-[(2-METHYLPROPYL)SULFONYL]-1-PHENYL-](http://img.cochemist.com/ccimg/852200/852178-76-8.png)
![1H-TETRAZOLE, 5-[(2-METHYLPROPYL)SULFONYL]-1-PHENYL-](http://img.cochemist.com/ccimg/852200/852178-76-8_b.png)

![Magnesium, bromo[(3,5-dimethoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/844600/844503-91-9.png)
![Magnesium, bromo[(3,5-dimethoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/844600/844503-91-9_b.png)
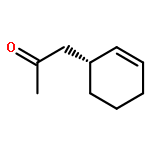
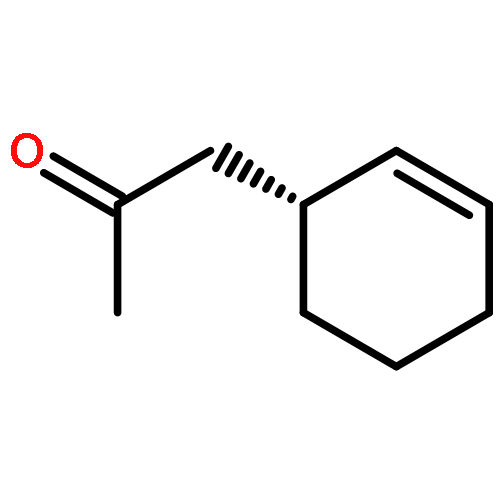
![Cyclohexanone, 3-[(trimethylsilyl)ethynyl]-, (3S)-](http://img.cochemist.com/ccimg/767400/767332-16-1.png)
![Cyclohexanone, 3-[(trimethylsilyl)ethynyl]-, (3S)-](http://img.cochemist.com/ccimg/767400/767332-16-1_b.png)
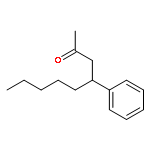
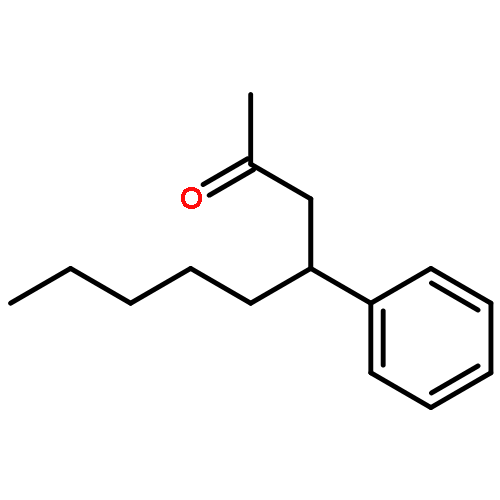
![BENZENE, [(3E,7E)-4,8,12-TRIMETHYL-3,7,11-TRIDECATRIENYL]-](http://img.cochemist.com/ccimg/405600/405506-90-3.png)
![BENZENE, [(3E,7E)-4,8,12-TRIMETHYL-3,7,11-TRIDECATRIENYL]-](http://img.cochemist.com/ccimg/405600/405506-90-3_b.png)
![Phenol, 4-[[tris(1-methylethyl)silyl]oxy]-](/data/chemimg/3735700/223689-82-5.png)
![Phenol, 4-[[tris(1-methylethyl)silyl]oxy]-](/data/chemimg/3735700/223689-82-5_b.png)


![Silane, [(1-methylene-2-propenyl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/139300/139278-54-9.png)
![Silane, [(1-methylene-2-propenyl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/139300/139278-54-9_b.png)
![Aluminum, bis(2-methylpropyl)[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/135500/135491-34-8.png)
![Aluminum, bis(2-methylpropyl)[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/135500/135491-34-8_b.png)

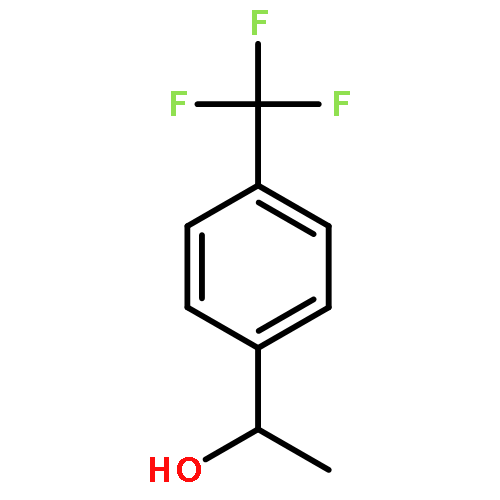
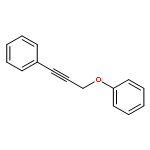
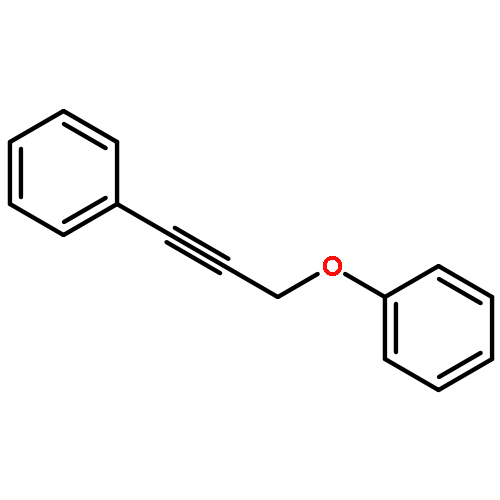


![1H-Cyclopenta[c]furan-1,6(3H)-dione, tetrahydro-3,3-dimethyl-, cis-](http://img.cochemist.com/ccimg/91500/91492-10-3.png)
![1H-Cyclopenta[c]furan-1,6(3H)-dione, tetrahydro-3,3-dimethyl-, cis-](http://img.cochemist.com/ccimg/91500/91492-10-3_b.png)
![Pyrrolidine, 2-[(phenylmethoxy)methyl]-, (2S)-](http://img.cochemist.com/ccimg/89600/89597-97-7.png)
![Pyrrolidine, 2-[(phenylmethoxy)methyl]-, (2S)-](http://img.cochemist.com/ccimg/89600/89597-97-7_b.png)
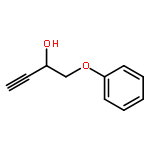


![Bicyclo[2.2.1]hept-5-ene-2,3-dimethanol, (1R,2S,3S,4S)-](http://img.cochemist.com/ccimg/78100/78037-77-1.png)
![Bicyclo[2.2.1]hept-5-ene-2,3-dimethanol, (1R,2S,3S,4S)-](http://img.cochemist.com/ccimg/78100/78037-77-1_b.png)
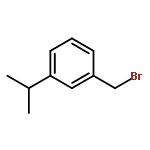
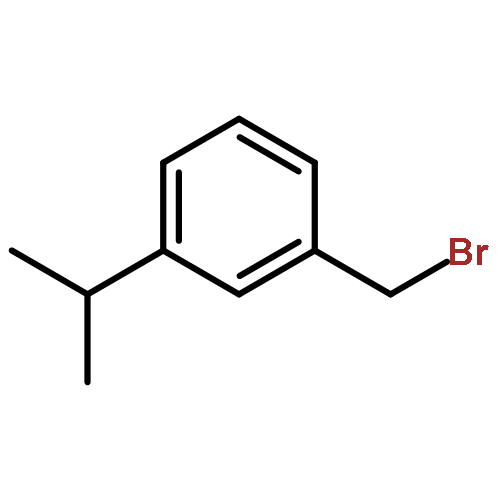
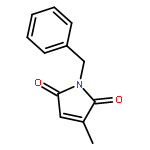






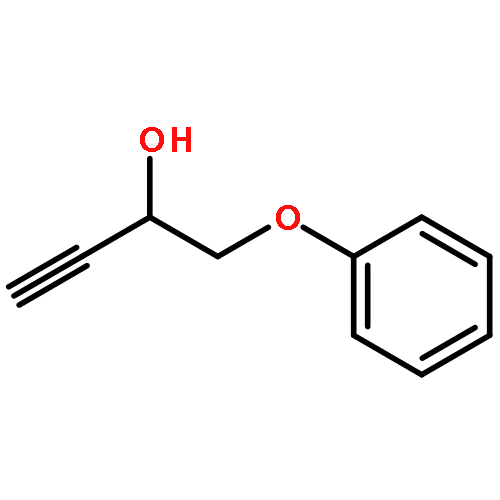
![Benzene, 1-methoxy-4-[(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/55600/55539-39-4.png)
![Benzene, 1-methoxy-4-[(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/55600/55539-39-4_b.png)



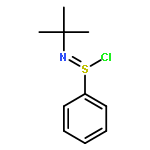

![BENZO[B]THIOPHENE, 2,3-DIHYDRO-, 1-OXIDE, (1R)-](http://img.cochemist.com/ccimg/41500/41469-54-9.png)
![BENZO[B]THIOPHENE, 2,3-DIHYDRO-, 1-OXIDE, (1R)-](http://img.cochemist.com/ccimg/41500/41469-54-9_b.png)
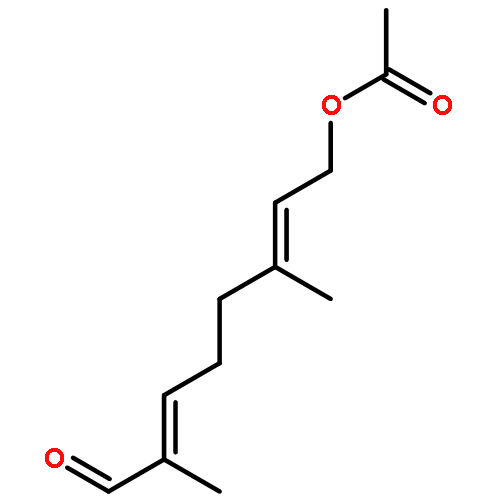
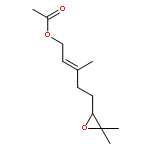
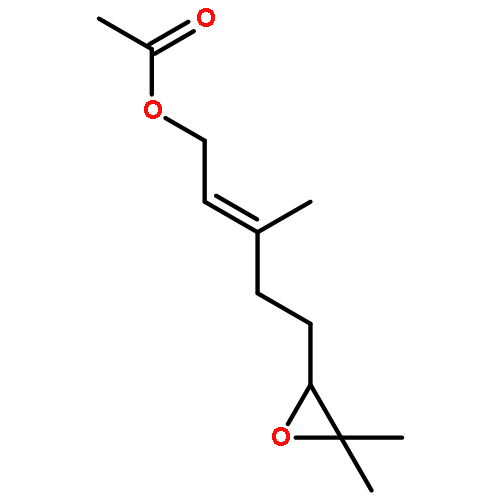




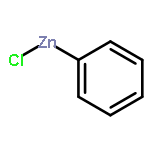










![Bicyclo[2.2.1]hepta-2,5-diene-2-carboxylic acid, ethyl ester](http://img.cochemist.com/ccimg/24500/24449-16-9.png)
![Bicyclo[2.2.1]hepta-2,5-diene-2-carboxylic acid, ethyl ester](http://img.cochemist.com/ccimg/24500/24449-16-9_b.png)

![Benzene, [(3E)-4,8-dimethyl-3,7-nonadienyl]-](http://img.cochemist.com/ccimg/22600/22555-66-4.png)
![Benzene, [(3E)-4,8-dimethyl-3,7-nonadienyl]-](http://img.cochemist.com/ccimg/22600/22555-66-4_b.png)




![Bicyclo[2.2.2]oct-5-en-2-one, (1R)-](http://img.cochemist.com/ccimg/16200/16196-15-9.png)
![Bicyclo[2.2.2]oct-5-en-2-one, (1R)-](http://img.cochemist.com/ccimg/16200/16196-15-9_b.png)
![Benzo[b]thiophene,2,3-dihydro-, 1,1-dioxide](http://img.cochemist.com/ccimg/14400/14315-13-0.png)
![Benzo[b]thiophene,2,3-dihydro-, 1,1-dioxide](http://img.cochemist.com/ccimg/14400/14315-13-0_b.png)
























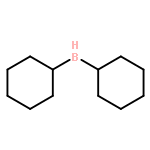
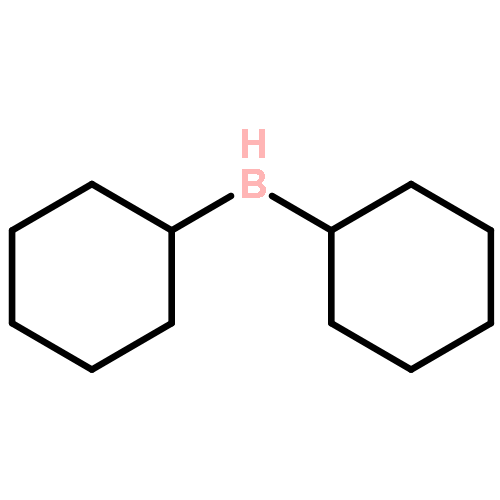
![Benzene, 1-[(R)-ethylsulfinyl]-4-methyl-](http://img.cochemist.com/ccimg/1600/1519-40-0.png)
![Benzene, 1-[(R)-ethylsulfinyl]-4-methyl-](http://img.cochemist.com/ccimg/1600/1519-40-0_b.png)






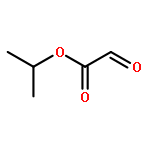
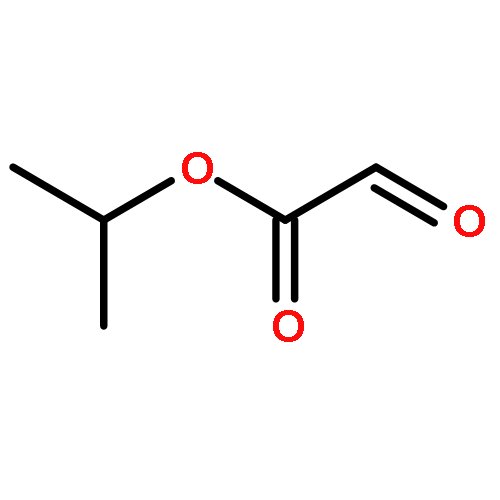


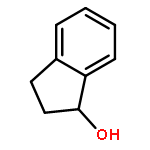
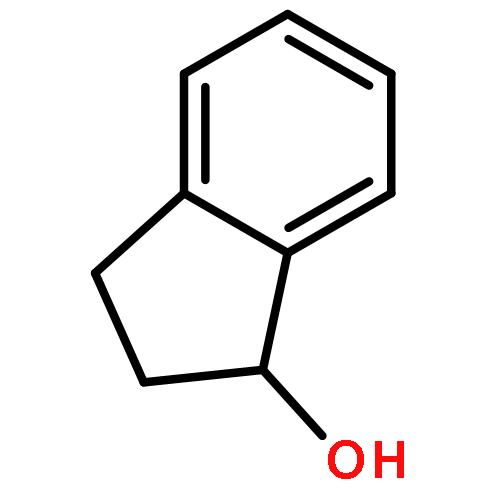
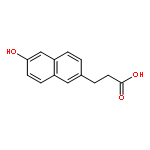
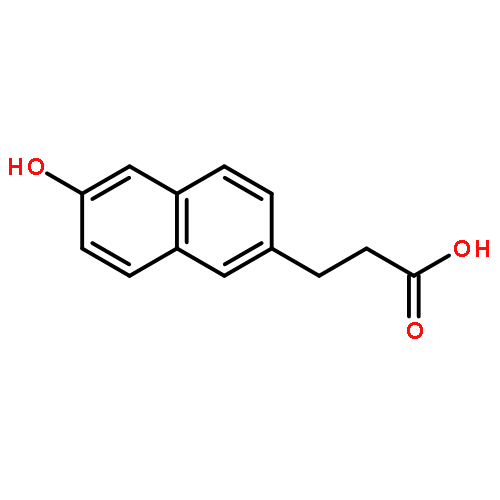






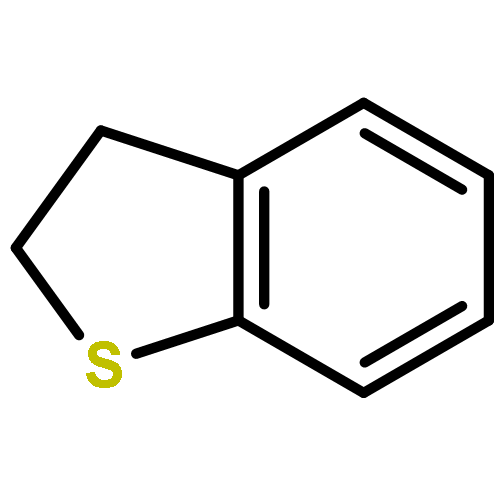
![1,3-Dihydrobenzo[c]thiophene](http://img.cochemist.com/ccimg/2500/2471-92-3.png)
![1,3-Dihydrobenzo[c]thiophene](http://img.cochemist.com/ccimg/2500/2471-92-3_b.png)