Co-reporter:Yi Zou, Fang Wang, Yan Wang, Qirui Sun, Yue Hu, Yuezhen Li, Wen Liu, Wenjie Guo, Zhangjian Huang, Yihua Zhang, Qiang Xu, Yisheng Lai
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.09.025
•The extensive structural modification of GDC-0919 analogue was explored.•T-lymphocytes assays were performed to evaluate the capacity of the compounds in the reversal of IDO1-mediated immunosuppression.•UV spectra provided a direct evidence of our compounds binding to the active site of IDO1.•Molecular simulations were performed to predict the binding mode of our compounds.Indoleamine-2,3-dioxygenase-1 (IDO1) is an attractive target for cancer immunotherapy. Herein, a series of novel imidazoleisoindole derivatives were prepared and evaluated for their ability to inhibit IDO1. Among these, derivative 11r was the most active compound with nanomolar potency in the Hela cell-based assay, while showed negligible cellular toxicity. UV-visible spectra study demonstrated that compounds 11p and 11r bound to IDO1 and coordinated with the heme iron. Furthermore, they could significantly promote T cell proliferation, increase IFN-γ production, and reduce the numbers of Foxp3+ regulatory T cells. Finally, induced fit docking (IFD) and quantum mechanics/molecular mechanics (QM/MM) calculation were performed to understand the interactions of these compounds to IDO1 protein, which provided a comprehensive guide for further structural modification and optimization.Download high-res image (211KB)Download full-size image
Co-reporter:Yi Zou, Yan Wang, Fang Wang, Minghao Luo, Yuezhen Li, Wen Liu, Zhangjian Huang, Yihua Zhang, Wenjie Guo, Qiang Xu, Yisheng Lai
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.06.039
•A scaffold-hopping strategy combined with the average electrostatic potentials calculation was utilized to design novel benzoxazolinone derivatives.•A novel interesting scaffold for IDO1 inhibition was identified.•T-cell proliferation and Tregs assays were performed to evaluate the capacity of the compounds in the reversal of IDO1-mediated immunosuppression.•UV spectroscopic experiment provided a direct evidence of our compounds binding to the active site of IDO1.•The induced fit docking and QM/MM calculation were performed to predict the binding mode of IDO1 and its ligand.Indoleamine 2,3-dioxygenase 1 (IDO1) is frequently hijacked by tumors to escape the host immune response, and the enzyme is now firmly established as an attractive target for cancer immunotherapy. To identify novel IDO1 inhibitors suitable for drug development, a scaffold-hopping strategy combined with the average electrostatic potentials calculation was ultilized to design novel benzoxazolinone derivatives. Among these, compounds 7e, 7f and 9c exhibited the inhibitory potency in the low micromolar range and displayed negligible level of cytotoxicity against HeLa cells. Treatment with these three compounds promoted the proliferation of T lymphocyte and led to the dramatic decrease of regulatory T cells in the B16F1 cells and naïve T cells co-culture system. Subsequent spectroscopic experiments suggested that these benzoxazolinones formed a coordinate bond with the heme iron to stabilize the complex. This study suggested that the benzoxazolinone was an interesting scaffold for discovering novel IDO1 inhibitors, and these compounds are attractive candidates for further development.Download high-res image (196KB)Download full-size image
Co-reporter:Minghao Luo, Hui Wang, Yi Zou, Shengping Zhang, Jianhu Xiao, Guangde Jiang, Yihua Zhang, Yisheng Lai
Journal of Molecular Graphics and Modelling 2016 Volume 68() pp:128-139
Publication Date(Web):July 2016
DOI:10.1016/j.jmgm.2016.06.011
•A hierarchical docking-based virtual screening combined with molecular dynamic simulation was applied in DOT1L inhibitor discovery.•The key residue contributing the most to the binding of EPZ5676 to the DOT1L protein was identified.•The calculations for the binding free energy were accomplished by using the MM/PBSA method in GROMACS.•Phenoxyacetamide-based scaffold is promising for developing novel DOT1L inhibitors.•The amide moiety of phenoxyacetamide-based scaffold played a crucial role in anchoring the molecule into the DOT1L pocket.Dot1-like protein (DOT1L) is a histone methyltransferase that has become a novel and promising target for acute leukemias bearing mixed lineage leukemia (MLL) gene rearrangements. In this study, a hierarchical docking-based virtual screening combined with molecular dynamic (MD) simulation was performed to identify DOT1L inhibitors with novel scaffolds. Consequently, 8 top-ranked hits were eventually identified and were further subjected to MD simulation. It was indicated that all hits could reach equilibrium with DOT1L in the MD simulation and further binding free energy calculations suggested that phenoxyacetamide-derived hits such as L01, L03, L04 and L05 exhibited remarkably higher binding affinity compared to other hits. Among them, L03 showed both the lowest glide score (−12.281) and the most favorable binding free energy (−303.9 +/− 16.5 kJ/mol), thereby making it a promising lead for further optimization.
Co-reporter:Yi Zou, Jianhu Xiao, Zhengchao Tu, Yingyi Zhang, Kun Yao, Minghao Luo, Ke Ding, Yihua Zhang, Yisheng Lai
Bioorganic & Medicinal Chemistry Letters 2016 26(13) pp: 3052-3059
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmcl.2016.05.014
A series of novel 4,5,6-trisubstituted pyrimidines were designed as potent covalent Bruton’s tyrosine kinase (BTK) inhibitors based on the structure of ibrutinib by using a ring-opening strategy. Among these derivatives, compound I1 exhibited the most potent inhibitory activity with an IC50 value of 0.07 μM. The preliminary structure–activity relationship was discussed and the primary amino group at the C-4 position of pyrimidine was crucial for maintaining BTK activity. Furthermore, molecular dynamics simulations and binding free energy calculations were performed for three inhibitor-BTK complexes to determine the probable binding model, which provided a comprehensive guide for further structural modification and optimization.
Co-reporter:Xiangyang Feng, Guangde Jiang, Zilei Xia, Jiadong Hu, Xiaolong Wan, Jin-Ming Gao, Yisheng Lai, and Weiqing Xie
Organic Letters 2015 Volume 17(Issue 18) pp:4428-4431
Publication Date(Web):August 28, 2015
DOI:10.1021/acs.orglett.5b02046
The first enantioselective synthesis of (−)-conolutinine was achieved in 10 steps. The synthesis featured a catalytic asymmetric bromocyclization of tryptamine to forge the tricycle intermediate. Hydration of an alkene catalyzed by Co(acac)2 was also employed as a key step to diastereoselectively introduce the tertiary alcohol moiety. The absolute configuration of (−)-conolutinine was established to be (2S,5aS,8aS,13aR) based on this asymmetric total synthesis.
Co-reporter:Zhigao Shen, Zilei Xia, Huijun Zhao, Jiadong Hu, Xiaolong Wan, Yisheng Lai, Chen Zhu and Weiqing Xie
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 19) pp:5381-5384
Publication Date(Web):10 Apr 2015
DOI:10.1039/C5OB00546A
The first direct access to unprotected amino-pyrroloindoline via aminocyclization of tryptamine and tryptophan has been described. A variety of structurally diverse amino-pyrroloindolines are furnished by the use of O-(2,4-dinitrophenyl)hydroxylamine (DPH) as the nitrogen source in the presence of catalytic Rh2(esp)2.
Co-reporter:Jianhu Xiao, Shengping Zhang, Minghao Luo, Yi Zou, Yihua Zhang, Yisheng Lai
Journal of Molecular Graphics and Modelling 2015 60() pp: 142-154
Publication Date(Web):July 2015
DOI:10.1016/j.jmgm.2015.05.005
Co-reporter:Huan Liu, Guangde Jiang, Xixian Pan, Xiaolong Wan, Yisheng Lai, Dawei Ma, and Weiqing Xie
Organic Letters 2014 Volume 16(Issue 7) pp:1908-1911
Publication Date(Web):March 25, 2014
DOI:10.1021/ol5004109
Highly asymmetric bromocyclization of tryptophol by using chiral anionic phase-transfer catalyst and DABCO-derived brominating reagent is described. Optimization of the reaction conditions revealed that the reaction rate was accelerated together with improvement of enantioselectivity by addition of catalytic DABCO-derived brominating reagent. From tryptophol, 3-bromofuroindoline could be directly obtained in excellent enantioselectivities by employing this novel methodology.
Co-reporter:Shengping Zhang, Jiani Tan, Zhonghui Lai, Ying Li, Junxia Pang, Jianhu Xiao, Zhangjian Huang, Yihua Zhang, Hui Ji, and Yisheng Lai
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 6) pp:1785-1797
Publication Date(Web):May 24, 2014
DOI:10.1021/ci5002058
The NEDD8-activating enzyme (NAE) is an emerging target for cancer therapy, which regulates the degradation and turnover of a variety of cancer-related proteins by activating the cullin-RING E3 ubiquitin ligases. Among a limited number of known NAE inhibitors, the covalent inhibitors have demonstrated the most potent efficacy through their covalently linked adducts with NEDD8. Inspired by this unique mechanism, in this study, a novel combined strategy of virtual screening (VS) was adopted with the aim to identify diverse covalent inhibitors of NAE. To be specific, a docking-enabled pharmacophore model was first built from the possible active conformations of chosen covalent inhibitors. Meanwhile, a dynamic structure-based phamacophore was also established based on the snapshots derived from molecular dynamic simulation. Subsequent screening of a focused ZINC database using these pharmacophore models combined with covalent docking discovered three novel active compounds. Among them, compound LZ3 exhibited the most potent NAE inhibitory activity with an IC50 value of 1.06 ± 0.18 μM. Furthermore, a cell-based washout experiment proved the proposed covalent binding mechanism for compound LZ3, which confirmed the successful application of our combined VS strategy, indicating it may provide a viable solution to systematically discover novel covalent ligands.
Co-reporter:Yi Zou, Fang Wang, Yan Wang, Wenjie Guo, Yihua Zhang, Qiang Xu, Yisheng Lai
European Journal of Medicinal Chemistry (5 May 2017) Volume 131() pp:
Publication Date(Web):5 May 2017
DOI:10.1016/j.ejmech.2017.03.021
•It was the first study that characterized the interactions between the inhibitors and IDO1 through an integrated computational approach.•The induced fit docking (IFD) method which considered the flexibility of protein enhanced the accuracy of predicted binding mode of IDO1.•Molecular dynamics (MD) simulation was performed to recognize the crucial residues for ligand binding.•The quantitative research by quantum mechanics analysis provided in-depth explanations on the critical role of those key residues.•Several visualization methods such as ESP and nonbonding interaction analysis were used for the display of the quantum mechanics analysis.Indoleamine 2,3-dioxygenase 1 (IDO1) is regarded as an attractive target for cancer immunotherapy. To rationalize the detailed interactions between IDO1 and its inhibitors at the atomic level, an integrated computational approach by combining molecular mechanics and quantum mechanics methods was employed in this report. Specifically, the binding modes of 20 inhibitors was initially investigated using the induced fit docking (IFD) protocol, which outperformed other two docking protocols in terms of correctly predicting ligand conformations. Secondly, molecular dynamics (MD) simulations and MM/PBSA free energy calculations were employed to determine the dynamic binding process and crucial residues were confirmed through close contact analysis, hydrogen-bond analysis and binding free energy decomposition calculations. Subsequent quantum mechanics and nonbonding interaction analysis were carried out to provide in-depth explanations on the critical role of those key residues, and Arg231 and 7-propionate of the heme group were major contributors to ligand binding, which lowed a great amount of interaction energy. We anticipate that these findings will be valuable for enzymatic studies and rational drug design.
Co-reporter:Xiaolong Wan, Jiadong Hu, Dongyang Xu, Yang Shang, Yanxia Zhen, Chenchen Hu, Fan Xiao, Yu-Peng He, Yisheng Lai, Weiqing Xie
Tetrahedron Letters (22 March 2017) Volume 58(Issue 12) pp:
Publication Date(Web):22 March 2017
DOI:10.1016/j.tetlet.2017.01.066
•R2O7 is capable of promoting dehydration of monoallylic diols.•Short reaction time and no need of cautious exclusion of water and oxygen.•High E-olefin selectivity.•Wide substrate scope.A Re2O7 catalyzed cycloetherification of monoallylic diols is described. The reaction features short reaction time, mild reaction conditions and exclusive E selectivity. A wide range of monoallylic alcohols with alkyl or aryl substituents on olefin smoothly undergo ring closure to deliver corresponding oxa-heterocycles. The reaction is also operationally simple and not sensitive to air and moisture.Download high-res image (46KB)Download full-size image
Co-reporter:Zhigao Shen, Zilei Xia, Huijun Zhao, Jiadong Hu, Xiaolong Wan, Yisheng Lai, Chen Zhu and Weiqing Xie
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 19) pp:NaN5384-5384
Publication Date(Web):2015/04/10
DOI:10.1039/C5OB00546A
The first direct access to unprotected amino-pyrroloindoline via aminocyclization of tryptamine and tryptophan has been described. A variety of structurally diverse amino-pyrroloindolines are furnished by the use of O-(2,4-dinitrophenyl)hydroxylamine (DPH) as the nitrogen source in the presence of catalytic Rh2(esp)2.







![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8.png)
![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8_b.png)
























![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)





























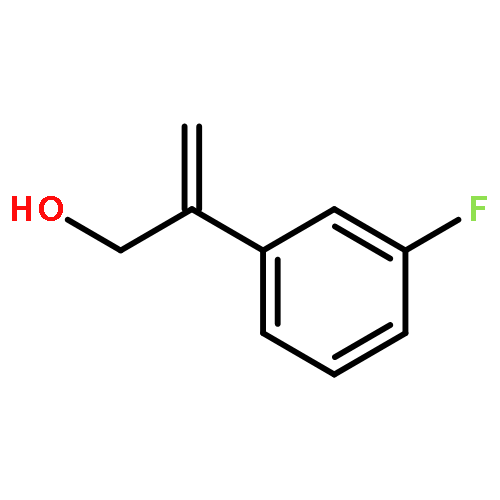


















![Methyl 7-methyl-2-propyl-1H-benzo[d]imidazole-5-carboxylate](http://img.cochemist.com/ccimg/152700/152628-00-7.png)
![Methyl 7-methyl-2-propyl-1H-benzo[d]imidazole-5-carboxylate](http://img.cochemist.com/ccimg/152700/152628-00-7_b.png)
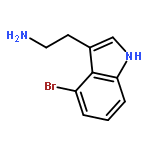
![1H-Indole, 5-bromo-3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-](http://img.cochemist.com/ccimg/148400/148366-44-3.png)
![1H-Indole, 5-bromo-3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-](http://img.cochemist.com/ccimg/148400/148366-44-3_b.png)






















![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6.png)
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6_b.png)
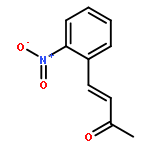

![3-Buten-2-one, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/115700/115665-92-4.png)
![3-Buten-2-one, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/115700/115665-92-4_b.png)
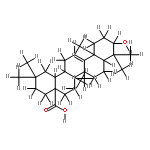


![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5.png)
![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5_b.png)
![Benzo[c]thiophen-1(3H)-one, 3-propyl-](http://img.cochemist.com/ccimg/97900/97873-41-1.png)
![Benzo[c]thiophen-1(3H)-one, 3-propyl-](http://img.cochemist.com/ccimg/97900/97873-41-1_b.png)



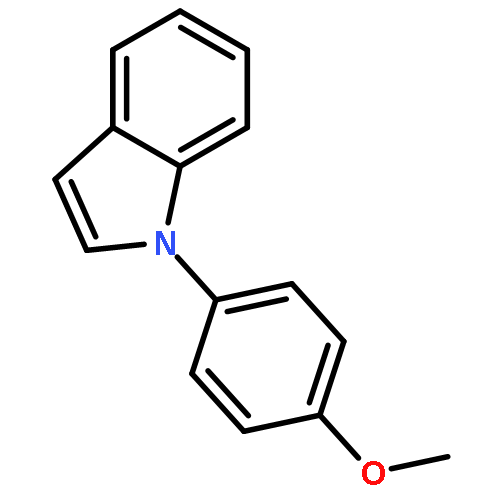



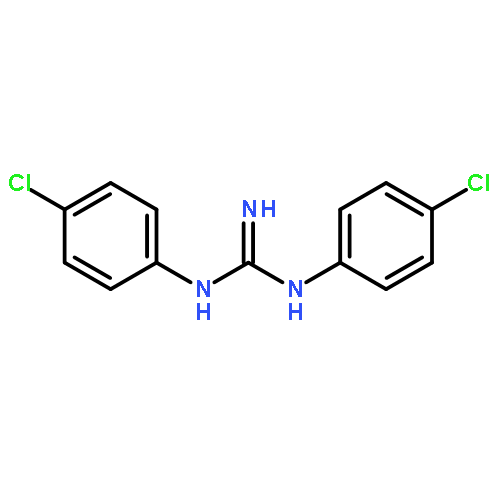
![[4,4'-Bi-1,3-benzodioxole]-5,5'-dimethanol, 7,7'-dimethoxy-](http://img.cochemist.com/ccimg/79300/79279-08-6.png)
![[4,4'-Bi-1,3-benzodioxole]-5,5'-dimethanol, 7,7'-dimethoxy-](http://img.cochemist.com/ccimg/79300/79279-08-6_b.png)

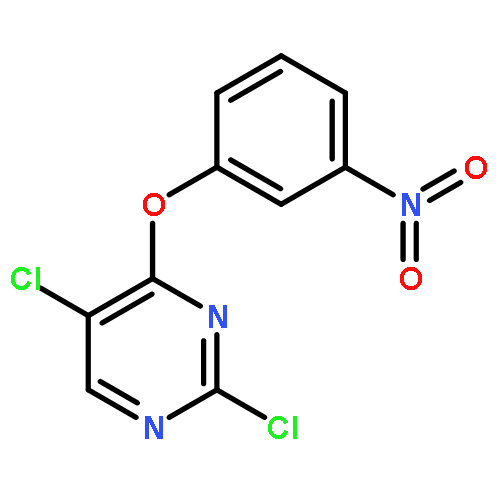






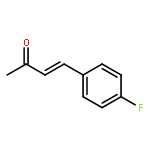
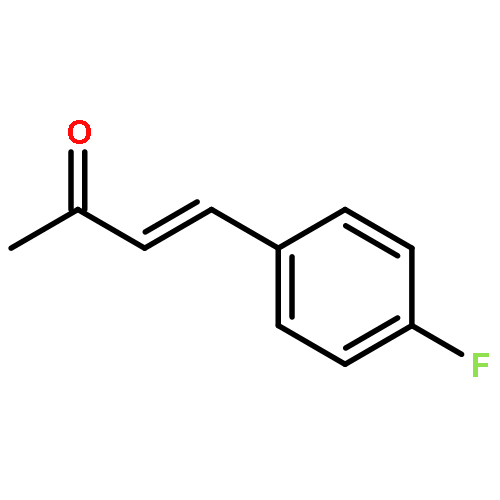

![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)




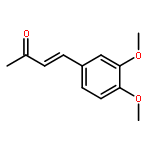
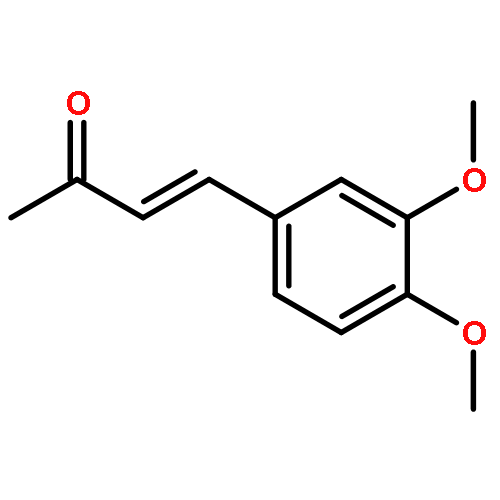





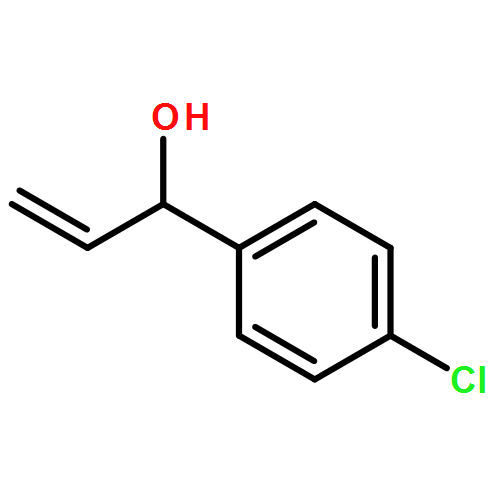
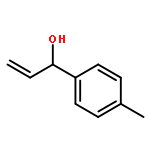
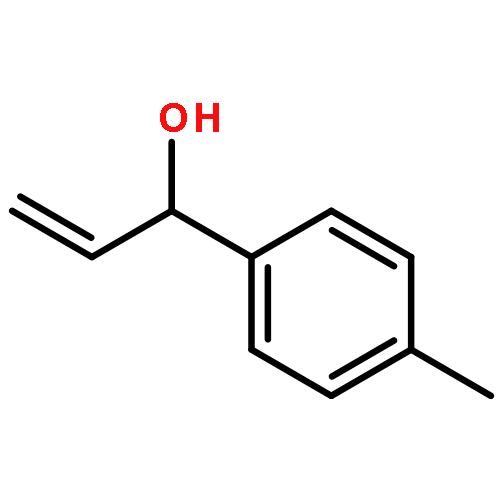

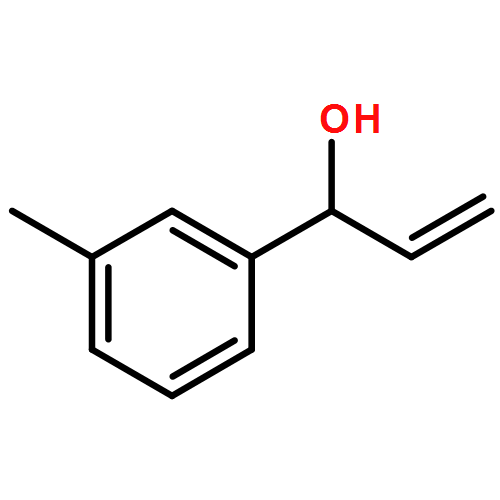
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/58700/58635-45-3.png)
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/58700/58635-45-3_b.png)


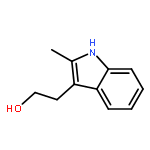





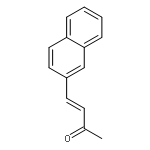
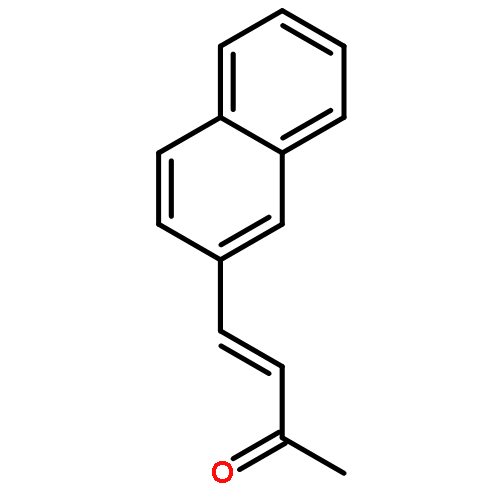


![BENZOIC ACID, 4-[1-(HYDROXYMETHYL)ETHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/46400/46333-28-2.png)
![BENZOIC ACID, 4-[1-(HYDROXYMETHYL)ETHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/46400/46333-28-2_b.png)









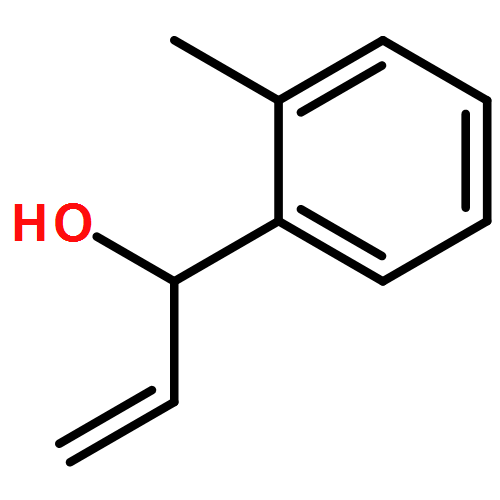




![CARBAMIC ACID, [2-(1H-INDOL-3-YL)ETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/38800/38750-13-9.png)
![CARBAMIC ACID, [2-(1H-INDOL-3-YL)ETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/38800/38750-13-9_b.png)


![Benzenesulfonamide, N-[2-(1H-indol-3-yl)ethyl]-4-nitro-](http://img.cochemist.com/ccimg/33300/33284-09-2.png)
![Benzenesulfonamide, N-[2-(1H-indol-3-yl)ethyl]-4-nitro-](http://img.cochemist.com/ccimg/33300/33284-09-2_b.png)
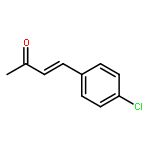
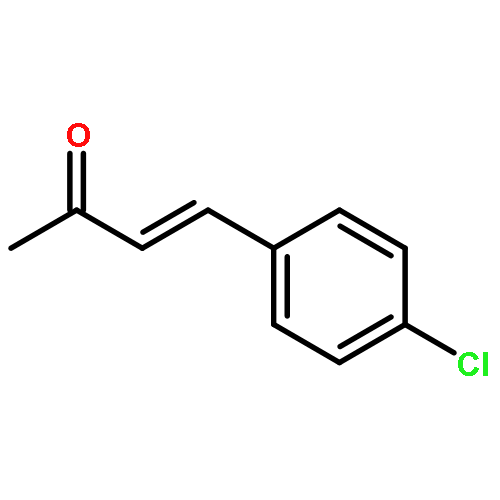

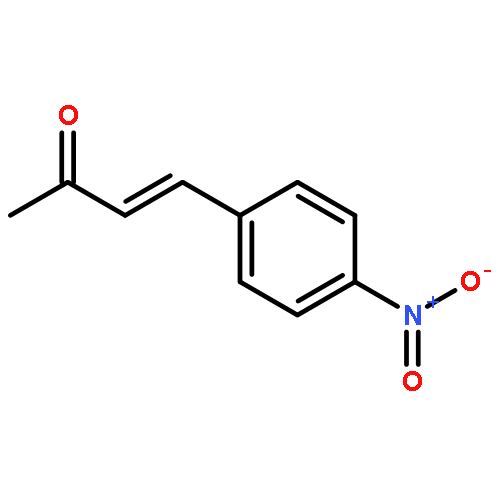
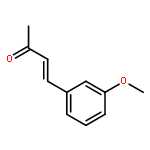
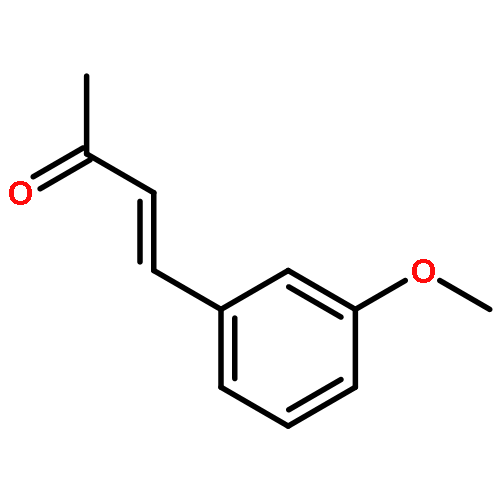
![2-[(4-methoxyphenyl)amino]-1,3-thiazol-4(5H)-one](http://img.cochemist.com/ccimg/27100/27052-12-6.png)
![2-[(4-methoxyphenyl)amino]-1,3-thiazol-4(5H)-one](http://img.cochemist.com/ccimg/27100/27052-12-6_b.png)
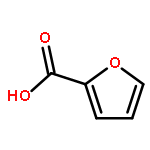















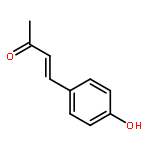
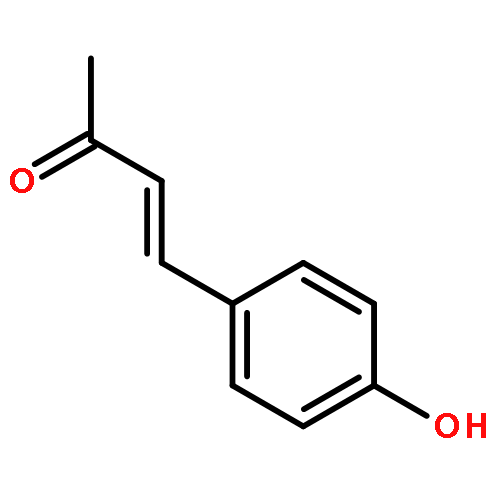




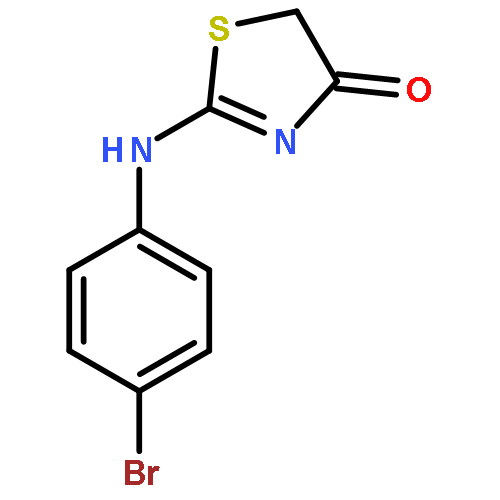
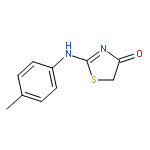
![4(5H)-Thiazolone, 2-[(4-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/21300/21262-23-7.png)
![4(5H)-Thiazolone, 2-[(4-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/21300/21262-23-7_b.png)

















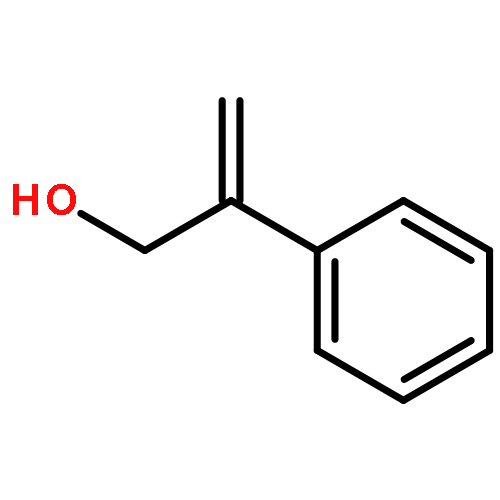
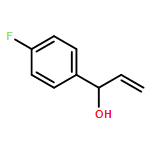
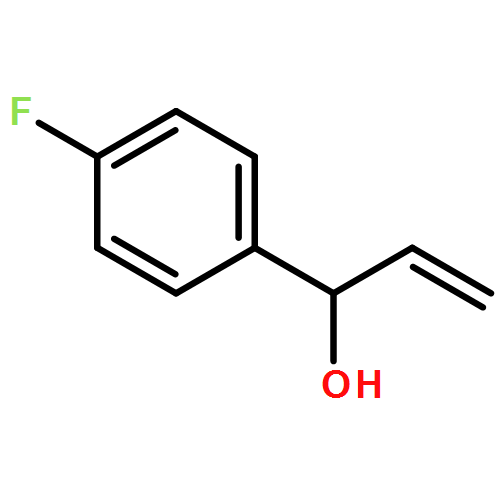

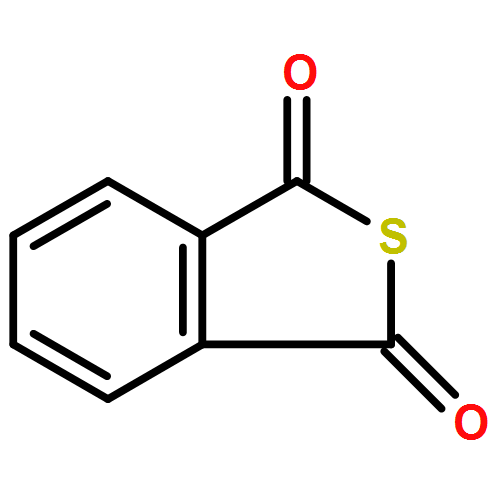
![N-[2-(1H-indol-3-yl)ethyl]-Benzamide](http://img.cochemist.com/ccimg/4800/4753-09-7.png)
![N-[2-(1H-indol-3-yl)ethyl]-Benzamide](http://img.cochemist.com/ccimg/4800/4753-09-7_b.png)






![4-[(E)-3-HYDROXYPROP-1-ENYL]PHENOL](http://img.cochemist.com/ccimg/3700/3690-05-9.png)
![4-[(E)-3-HYDROXYPROP-1-ENYL]PHENOL](http://img.cochemist.com/ccimg/3700/3690-05-9_b.png)









































![Methyl 5'-(hydroxymethyl)-7,7'-dimethoxy-[4,4'-bibenzo[d][1,3]dioxole]-5-carboxylate](http://img.cochemist.com/ccimg/118200/118159-48-1.png)
![Methyl 5'-(hydroxymethyl)-7,7'-dimethoxy-[4,4'-bibenzo[d][1,3]dioxole]-5-carboxylate](http://img.cochemist.com/ccimg/118200/118159-48-1_b.png)
![[4,4'-Bi-1,3-benzodioxole]-5,5'-dicarboxylic acid, 7,7'-dimethoxy-](/data/chemimg/916600/105868-34-6.png)
![[4,4'-Bi-1,3-benzodioxole]-5,5'-dicarboxylic acid, 7,7'-dimethoxy-](/data/chemimg/916600/105868-34-6_b.png)













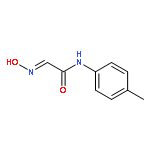
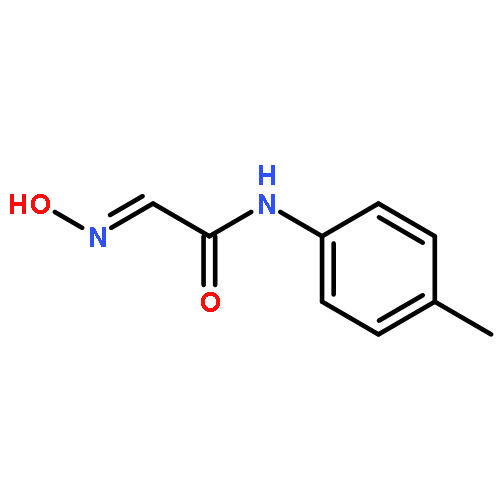
















![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)


![Ethanol, 2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/1005600/901792-84-5.png)
![Ethanol, 2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/1005600/901792-84-5_b.png)
![1-Propanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3151700/361541-87-9.png)
![1-Propanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3151700/361541-87-9_b.png)




![2-Butanol, 4-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3196400/452095-52-2.png)
![2-Butanol, 4-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3196400/452095-52-2_b.png)

