Co-reporter:Taikangxiang Yun, Tan Qin, Ying Liu, Luhua Lai
European Journal of Medicinal Chemistry 2016 Volume 124() pp:229-236
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.08.043
•A series of acylthioureas were designed and synthesized as Plk1 inhibitors.•Compounds with halogen in sulfamoylphenyl group showed better binding affinities.•The best compound 3v binds to Plk1 PBD with a Kd of 2.3 ± 0.1 μM.•Compound 3v also inhibits the kinase activity of full-length Plk1.Thiourea derivatives have drawn much attention for their latent capacities of biological activities. In this study, we designed acylthiourea compounds as polo-like kinase 1 (Plk1) polo-box domain (PBD) inhibitors. A series of acylthiourea derivatives without pan assay interference structure (PAINS) were synthesized. Four compounds with halogen substituents exhibited binding affinities to Plk1 PBD in low micromole range. The most potent compound (3v) showed selectivity over other subtypes of Plk PBDs and inhibited the kinase activity of full-length Plk1.
Co-reporter:Taikangxiang Yun;Tan Qin; Ying Liu; Luhua Lai
ChemMedChem 2016 Volume 11( Issue 7) pp:713-717
Publication Date(Web):
DOI:10.1002/cmdc.201600051
Abstract
Polo-like kinase 1 (Plk1) is an evolutionarily conserved serine/threonine kinase, and its N-terminal kinase domain (KD) controls cell signaling through phosphorylation. Inhibitors of Plk1 are potential anticancer drugs. Most known Plk1 KD inhibitors are ATP-competitive compounds, which may suffer from low selectivity. In this study we discovered novel non-ATP-competitive Plk1 KD inhibitors by virtual screening and experimental studies. Potential binding sites in Plk1 KD were identified by using the protein binding site detection program Cavity. The identified site was subjected to molecular-docking-based virtual screening. The activities of top-ranking compounds were evaluated by in vitro enzyme assay with full-length Plk1 and direct binding assay with Plk1 KD. Several compounds showed inhibitory activity, and the most potent was found to be 3-((2-oxo-2-(thiophen-2-yl)ethyl)thio)-6-(pyridin-3-ylmethyl)-1,2,4-triazin-5(4H)-one (compound 4) with an IC50 value of 13.1±1.7 μm. Our work provides new insight into the design of kinase inhibitors that target non-ATP binding sites.
Co-reporter:Hu Meng, Ying Liu, and Luhua Lai
Accounts of Chemical Research 2015 Volume 48(Issue 8) pp:2242
Publication Date(Web):August 3, 2015
DOI:10.1021/acs.accounts.5b00226
Inflammation and other common disorders including diabetes, cardiovascular disease, and cancer are often the result of several molecular abnormalities and are not likely to be resolved by a traditional single-target drug discovery approach. Though inflammation is a normal bodily reaction, uncontrolled and misdirected inflammation can cause inflammatory diseases such as rheumatoid arthritis and asthma. Nonsteroidal anti-inflammatory drugs including aspirin, ibuprofen, naproxen, or celecoxib are commonly used to relieve aches and pains, but often these drugs have undesirable and sometimes even fatal side effects. To facilitate safer and more effective anti-inflammatory drug discovery, a balanced treatment strategy should be developed at the biological network level.In this Account, we focus on our recent progress in modeling the inflammation-related arachidonic acid (AA) metabolic network and subsequent multiple drug design. We first constructed a mathematical model of inflammation based on experimental data and then applied the model to simulate the effects of commonly used anti-inflammatory drugs. Our results indicated that the model correctly reproduced the established bleeding and cardiovascular side effects. Multitarget optimal intervention (MTOI), a Monte Carlo simulated annealing based computational scheme, was then developed to identify key targets and optimal solutions for controlling inflammation. A number of optimal multitarget strategies were discovered that were both effective and safe and had minimal associated side effects. Experimental studies were performed to evaluate these multitarget control solutions further using different combinations of inhibitors to perturb the network. Consequently, simultaneous control of cyclooxygenase-1 and -2 and leukotriene A4 hydrolase, as well as 5-lipoxygenase and prostaglandin E2 synthase were found to be among the best solutions.A single compound that can bind multiple targets presents advantages including low risk of drug–drug interactions and robustness regarding concentration fluctuations. Thus, we developed strategies for multiple-target drug design and successfully discovered several series of multiple-target inhibitors. Optimal solutions for a disease network often involve mild but simultaneous interventions of multiple targets, which is in accord with the philosophy of traditional Chinese medicine (TCM). To this end, our AA network model can aptly explain TCM anti-inflammatory herbs and formulas at the molecular level. We also aimed to identify activators for several enzymes that appeared to have increased activity based on MTOI outcomes. Strategies were then developed to predict potential allosteric sites and to discover enzyme activators based on our hypothesis that combined treatment with the projected activators and inhibitors could balance different AA network pathways, control inflammation, and reduce associated adverse effects.Our work demonstrates that the integration of network modeling and drug discovery can provide novel solutions for disease control, which also calls for new developments in drug design concepts and methodologies. With the rapid accumulation of quantitative data and knowledge of the molecular networks of disease, we can expect an increase in the development and use of quantitative disease models to facilitate efficient and safe drug discovery.
Co-reporter:Erchang Shang, Yaxia Yuan, Xinyi Chen, Ying Liu, Jianfeng Pei, and Luhua Lai
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 4) pp:1235-1241
Publication Date(Web):March 10, 2014
DOI:10.1021/ci500021v
The discovery of multitarget drugs has recently attracted much attention. Most of the reported multitarget ligands have been serendipitous discoveries. Although a few methods have been developed for rational multitarget drug discovery, there is a lack of elegant methods for de novo multitarget drug design and optimization, especially for multiple targets with large differences in their binding sites. In this paper, we report the first de novo multitarget ligand design method, with an iterative fragment-growing strategy. Using this method, dual-target inhibitors for COX-2 and LTA4H were designed, with the most potent one inhibiting PGE2 and LTB4 production in the human whole blood assay with IC50 values of 7.0 and 7.1 μM, respectively. Our strategy is generally applicable in rational and efficient multitarget drug design, especially for the design of highly integrated inhibitors for proteins with dissimilar binding pockets.
Co-reporter:Jianshu Hu;Wei Zhu;Hu Meng;Xin Wang;Chun Hu
Chemical Biology & Drug Design 2014 Volume 84( Issue 6) pp:642-647
Publication Date(Web):
DOI:10.1111/cbdd.12356
A series of novel 1,4-dihydrothieno[3′,2′:5,6]thiopyrano[4,3-c]-pyrazole-3-carboxamide derivatives were synthesized and evaluated for their inhibitory activity to human 5-lipo-oxygenase (5-LOX). Compound 7c was found to exhibit significant inhibition to human 5-LOX with IC50 value of 5.7 ± 0.9 μm. Compound 7c was further studied using molecular docking in order to delineate its structure–activity relationship and to gain insight into the design of effective 5-LOX inhibitors.
Co-reporter:Erchang Shang, Ying Liu, Yiran Wu, Wei Zhu, Chong He, Luhua Lai
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 8) pp:2396-2402
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmc.2014.03.008
Human 5-lipoxygenase (5-LOX) is a well-validated target for anti-inflammatory therapy. Development of novel 5-LOX inhibitors with higher activities is highly demanded. In previous study, we have built a model for the active conformation of human 5-LOX, and identified naphthalen-1-yl 3,5-dinitrobenzoate (JMC-4) as a 5-LOX inhibitor by virtual screening. In the present work, 3,5-dinitrobenzoate-based 5-lipoxygenase inhibitors were developed. Twenty aryl 3,5-dinitrobenzoates, N-aryl 3,5-dinitrobenzamides and analogues were designed and synthesized. Several of them were found with significantly increased activities according to cell-free assay and human whole blood assay. The structure–activity relationship study may provide useful insights for designing effective 5-LOX inhibitors.In the present work, 3,5-dinitrobenzoate-based 5-lipoxygenase inhibitors were developed. Twenty derivatives were designed and synthesized. Several of them were found with significantly increased activity according to cell-free assay and human whole blood assay. The structure–activity relationship study may provide useful insights for designing effective 5-LOX inhibitors.
Co-reporter:Erchang Shang, Yiran Wu, Pei Liu, Ying Liu, Wei Zhu, Xiaobing Deng, Chong He, Shan He, Cong Li, Luhua Lai
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 12) pp:2764-2767
Publication Date(Web):15 June 2014
DOI:10.1016/j.bmcl.2014.04.006
A series of 6-nitro-3-(m-tolylamino) benzo[d]isothiazole 1,1-dioxide analogues were synthesized and evaluated for their inhibition activity against 5-lipoxygenase (5-LOX) and microsomal prostaglandin E2 synthase (mPGES-1). These compounds can inhibit both enzymes with IC50 values ranging from 0.15 to 23.6 μM. One of the most potential compounds, 3g, inhibits 5-LOX and mPGES-1 with IC50 values of 0.6 μM, 2.1 μM, respectively.
Co-reporter:Zheng Chen ; Yiran Wu ; Ying Liu ; Suijia Yang ; Yunjie Chen ;Luhua Lai
Journal of Medicinal Chemistry 2011 Volume 54(Issue 10) pp:3650-3660
Publication Date(Web):May 4, 2011
DOI:10.1021/jm200063s
Dual target inhibitors against COX-2 and LTA4H were designed by adding functional groups from a marketed COX-2 inhibitor, Nimesulide, to an existing LTA4H inhibitor 1-(2-(4-phenoxyphenoxy) ethyl) pyrrolidine. A series of phenoxyphenyl pyrrolidine compounds were synthesized and tested for their inhibition activities using enzyme assays and human whole blood assay. Introduction of small electron withdrawing groups like NO2 and CF3 in the ortho-position of the terminal phenyl ring was found to change the original single target LTA4H inhibitor to dual target LTA4H and COX-2 inhibitors. Compound 5a and 5m showed dual LTA4H and COX-2 inhibition activities in the enzyme assays and the HWB assay with IC50 values in the micromolar to submicromolar range. As their activities in HWB assay were comparable to the two starting single target inhibitors, the two compounds are promising for further studies. The strategy used in the current study may be generally applicable to other dual target drug designs.
Co-reporter:Yiran Wu, Zheng Chen, Ying Liu, Lanlan Yu, Lu Zhou, Suijia Yang, Luhua Lai
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 11) pp:3361-3366
Publication Date(Web):1 June 2011
DOI:10.1016/j.bmc.2011.04.039
A series of novel fused heterocycle methyl esters were designed and synthesized as human nonpancreatic secretory phospholipase A2 (hnps-PLA2) competitive inhibitors. Among the 22 synthesized compounds, 17 quinoline-4-methyl esters displayed hnps-PLA2 inhibition activity in the in vitro bioassay. The IC50 value for the best compound 3o was 1.5 μM. The structure-inhibition–activity relationships of the compounds were studied using molecular docking.
Co-reporter:Lu Zhou ; Chao Fang ; Ping Wei ; Shiyong Liu ; Ying Liu ;Luhua Lai
Journal of Medicinal Chemistry 2008 Volume 51(Issue 12) pp:3360-3366
Publication Date(Web):June 4, 2008
DOI:10.1021/jm7010707
A series of novel bis-indole compounds, 1,ω-bis(((3-acetamino-5-methoxy-2-methylindole)-2-methylene)phenoxy)alkane, have been designed and synthesized on the basis of the enzyme structure of human nonpancreatic secretory phospholipase A2 (hnps PLA2). Their inhibition activities against hnps PLA2 were improved compared to that of the monofunctional protocompound. These bivalent ligands not only inhibited hnps PLA2 but also drove the dimerization of hnps PLA2. Their dimerization ability correlated with the linker length and position. Further study on the potent compound 5 (1,5-bis(((3-acetamino-5-methoxy-2-methylindole)-2-methylene)phenoxy)pentane, IC50 = 24 nM) revealed that cooperative binding interactions between the two enzyme molecules also contributed to the stability of the ternary complex. The combination of bivalent ligands and hnps PLA2 can be used as a novel chemically induced dimerization (CID) system for designing regulatory inhibitors.
Co-reporter:Yu-Xiu Liu, Deng-Guo Wei, Ye-Rong Zhu, Shao-Hua Liu, Yong-Lin Zhang, Qi-Qi Zhao, Bao-Li Cai, Yong-Hong Li, Hai-Bin Song, Ying Liu, Yong Wang, Run-Qiu Huang and Qing-Min Wang
Journal of Agricultural and Food Chemistry 2008 Volume 56(Issue 1) pp:204-212
Publication Date(Web):December 4, 2007
DOI:10.1021/jf072851x
A series of novel 2-cyanoacrylates containing different aromatic rings were synthesized, and their structures were characterized by 1H NMR, elemental analysis, and single-crystal X-ray diffraction analysis. Their herbicidal activities against four weeds and inhibition of photosynthetic electron transport against isolated chloroplasts (the Hill reaction) were evaluated. Both in vivo and in vitro data showed that the compounds containing benzene, pyridine, and thiazole moieties gave higher activities than those containing pyrimidine, pyridazine, furan, and tetrahedronfuran moieties. To further explore the comprehensive structure–activity relationship on the basis of in vitro data, comparative molecular field analysis (CoMFA) was performed, and the results showed that a bulky and electronegative group around the para-position of the aromatic rings would have the potential for higher activity, which offered important structural insights into designing highly active compounds prior to the next synthesis.
Co-reporter:Xiaolu Jiang, Lu Zhou, Dengguo Wei, Hu Meng, Ying Liu, Luhua Lai
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 24) pp:6549-6552
Publication Date(Web):15 December 2008
DOI:10.1016/j.bmcl.2008.10.044
The synthesis and biological evaluation of a series of diphenyl ether derivatives were described. The compounds can either activate or inhibit the aminopeptidase activity of leukotriene A4 hydrolase, while at the same time do not influence the hydrolase activity. Further enzyme kinetics and molecular modeling investigation on these novel chemical activators revealed their possible activation mechanism. These compounds can be used as probes to regulate the aminopeptidase activity of leukotriene A4 hydrolase.Diphenyl ether and derivatives can either activate or inhibit the aminopeptidase activity of leukotriene A4 hydrolase, by binding at the hydrophobic pocket of LTA4H.
Co-reporter:Liu Ying;Zhang Xiao-Hong;Jin Gui-Yu
Chinese Journal of Chemistry 2005 Volume 23(Issue 2) pp:
Publication Date(Web):1 MAR 2005
DOI:10.1002/cjoc.200590182
Novel fused heterotricyclic compounds 3–6 containing two different dipyrazolopyrimidine framework were prepared from 1a–1d. The reaction of 1 with K2CO3 in DMSO or NaH in DMF led to the formation of 2 or 3, respectively, which reacted further to afford 6 or 5, respectively














![Piperazine,1-[4-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/187700/187669-62-1.png)
![Piperazine,1-[4-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/187700/187669-62-1_b.png)




![[5,6]Fullerene-C -I -octadecol](http://img.cochemist.com/ccimg/182100/182024-42-6.png)
![[5,6]Fullerene-C -I -octadecol](http://img.cochemist.com/ccimg/182100/182024-42-6_b.png)


![2-Propenoic acid, 3-[3,4-bis(2-propenyloxy)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/179200/179128-84-8.png)
![2-Propenoic acid, 3-[3,4-bis(2-propenyloxy)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/179200/179128-84-8_b.png)


![[1,1'-Biphenyl]-4-ol, 3,5-dinitrobenzoate](http://img.cochemist.com/ccimg/178300/178274-12-9.png)
![[1,1'-Biphenyl]-4-ol, 3,5-dinitrobenzoate](http://img.cochemist.com/ccimg/178300/178274-12-9_b.png)


![4-[3-(Furan-2-carbonyl)-thioureido]-benzenesulfonamide](http://img.cochemist.com/ccimg/160800/160788-28-3.png)
![4-[3-(Furan-2-carbonyl)-thioureido]-benzenesulfonamide](http://img.cochemist.com/ccimg/160800/160788-28-3_b.png)



































![2-Propenoic acid, 3-[3,4-bis(acetyloxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/88700/88623-81-8.png)
![2-Propenoic acid, 3-[3,4-bis(acetyloxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/88700/88623-81-8_b.png)
![b-D-Glucopyranoside,2-(3,4-dihydroxyphenyl)ethyl, 4-[(2E)-3-(3,4-dihydroxyphenyl)-2-propenoate]](http://img.cochemist.com/ccimg/84800/84744-28-5.png)
![b-D-Glucopyranoside,2-(3,4-dihydroxyphenyl)ethyl, 4-[(2E)-3-(3,4-dihydroxyphenyl)-2-propenoate]](http://img.cochemist.com/ccimg/84800/84744-28-5_b.png)































![Methyl (2s,4s,5r,6r)-5-amino-4-hydroxy-2-methoxy-6-[(1r,2r)-1,2,3-trihydroxypropyl]oxane-2-carboxylate](http://img.cochemist.com/ccimg/56100/56070-37-2.png)
![Methyl (2s,4s,5r,6r)-5-amino-4-hydroxy-2-methoxy-6-[(1r,2r)-1,2,3-trihydroxypropyl]oxane-2-carboxylate](http://img.cochemist.com/ccimg/56100/56070-37-2_b.png)
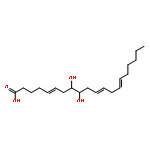
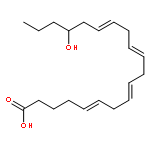
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)
![(5R,11S,13S)-13-Amino-5-methyl-5,6,7,8,9,10,11,12-octahydro-5,11-methanobenzo[10]annulen-3-ol](http://img.cochemist.com/ccimg/53700/53648-55-8.png)
![(5R,11S,13S)-13-Amino-5-methyl-5,6,7,8,9,10,11,12-octahydro-5,11-methanobenzo[10]annulen-3-ol](http://img.cochemist.com/ccimg/53700/53648-55-8_b.png)
















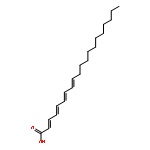
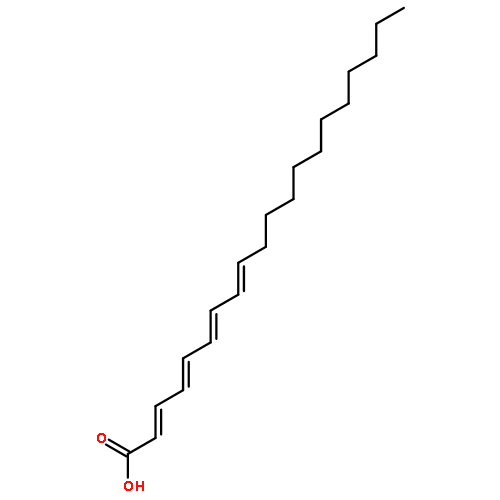
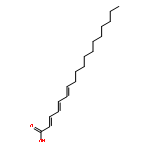














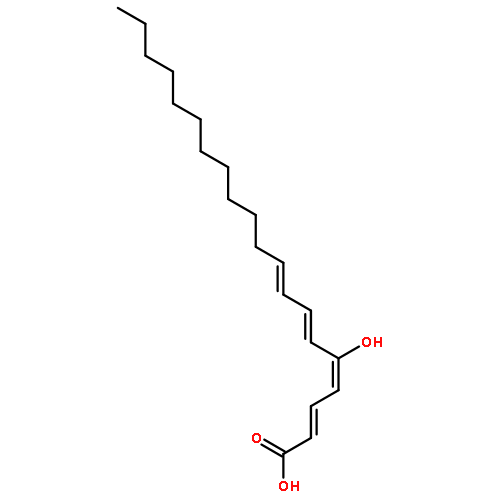
![(1,10-Phenanthroline)tris[4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedionato]europium(III)](http://img.cochemist.com/ccimg/18000/17904-86-8.png)
![(1,10-Phenanthroline)tris[4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedionato]europium(III)](http://img.cochemist.com/ccimg/18000/17904-86-8_b.png)
![Benzoic acid,3-[(phenylamino)carbonyl]-](http://img.cochemist.com/ccimg/16800/16776-94-6.png)
![Benzoic acid,3-[(phenylamino)carbonyl]-](http://img.cochemist.com/ccimg/16800/16776-94-6_b.png)
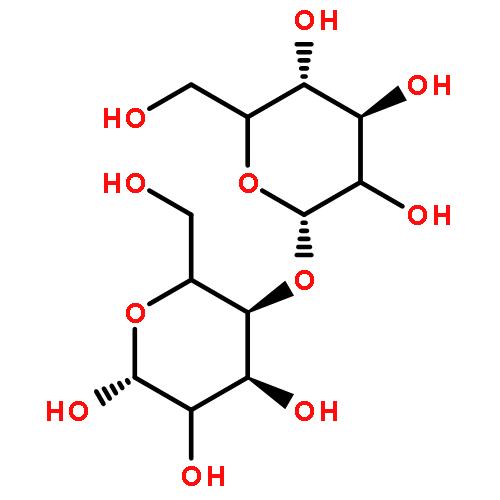
![(2S)-2-AMINO-2-METHYL-4-[(2-METHYL-2-PROPANYL)OXY]-4-OXOBUTANOIC <WBR />ACID (NON-PREFERRED NAME)](http://img.cochemist.com/ccimg/7700/7675-04-9.png)
![(2S)-2-AMINO-2-METHYL-4-[(2-METHYL-2-PROPANYL)OXY]-4-OXOBUTANOIC <WBR />ACID (NON-PREFERRED NAME)](http://img.cochemist.com/ccimg/7700/7675-04-9_b.png)




![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)






![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7.png)
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7_b.png)