Co-reporter:Shradha V. Patil, James M. Tanko
Tetrahedron 2016 Volume 72(Issue 48) pp:7849-7858
Publication Date(Web):1 December 2016
DOI:10.1016/j.tet.2016.05.046
Direct functionalization of rudimentary and cheap starting materials to yield complex value added products is of great interest to synthetic chemists. Particularly, direct functionalization of cheap commodity molecules that have been traditionally considered inert to known reactions are of widespread interest. We have previously demonstrated the functionalization of benzylic C–H bonds via an allyl transfer reaction using various allyl-phthalimido-N-oxyl substrates. In this work, we demonstrate the extension of our mild, metal-free, and neutral allyl transfer methodology to the direct functionalization of ethers. The C–H bond in α position to the ether oxygen in various acyclic and cyclic ethers was functionalized in high yields demonstrating wide substrate scope for this transformation. Furthermore, selective mono-functionalization of symmetrical cyclic ethers and regioselective functionalization of unsymmetrical ethers was achieved, thus demonstrating further utility of this reaction. Finally, kinetic chain length measurements were performed, which provided valuable insights into the initial rates and efficiency of the chain process involving the phthalimido-N-oxyl (PINO) radical.
Co-reporter:Lukas Rycek, John J. Hayward, Marwa Abdel Latif, James Tanko, Razvan Simionescu, and Tomas Hudlicky
The Journal of Organic Chemistry 2016 Volume 81(Issue 22) pp:10930-10941
Publication Date(Web):September 30, 2016
DOI:10.1021/acs.joc.6b01990
A second-generation approach to the synthesis of hydromorphone by oxidative dearomatization/Diels–Alder cycloaddition was investigated. Detailed analysis of the stereochemical outcome of the [4 + 2] cycloaddition was performed first on a truncated model system as well as on the material leading to ent-hydromorphone. The stereochemical assignments were made by NMR and X-ray methods. The second-generation synthesis of hydromorphone was completed in both enantiomeric series. Improvements in the dearomatization conditions were attained using hypervalent iodine reagents instead of Pb(OAc)4. Electrochemical methods of oxidative dearomatization were also investigated. New conditions enabling the rearomatization of ring A from the methoxyketal were developed, and a formal synthesis of the natural enantiomer of hydromorphone was completed. Experimental and spectral data are provided for all new compounds.
Co-reporter:Shradha Patil;Liang Chen ;James M. Tanko
European Journal of Organic Chemistry 2014 Volume 2014( Issue 3) pp:502-505
Publication Date(Web):
DOI:10.1002/ejoc.201301530
Abstract
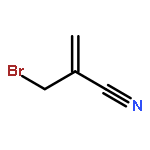
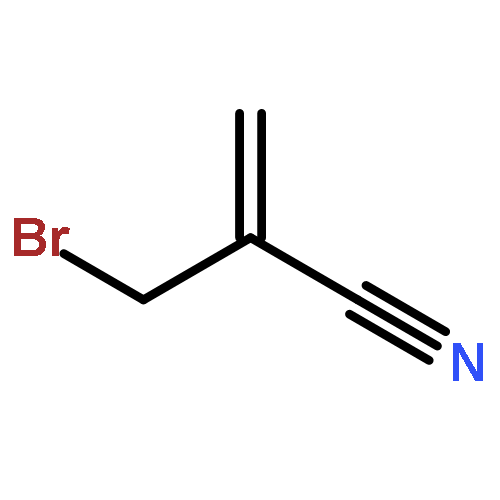
The development of a new chemical process that effects the conversion RH + C=C–C–X R–C–C=C + HX, in which X is the phthalimido-N-oxyl radical (PINO·), is reported. The reaction yields are high, mass balances are excellent, and C–H bond functionalization and C–C bond formation are achieved in a single transformation. The byproduct of the reaction, N-hydroxyphthalimide, precipitates from solution and can be easily removed by simple filtration (and recycled). The kinetic chain lengths are shorter and the reaction times are longer (relative to those of the analogous reactions of allyl bromides), most likely because PINO· is a less-reactive hydrogen-atom abstractor. There appears to be no significant difference in efficiency in the addition–elimination steps. Competition experiments reveal that Br· and PINO· are comparable in leaving group ability.
Co-reporter:Shradha Patil, Liang Chen, James M. Tanko
Tetrahedron Letters 2014 Volume 55(Issue 51) pp:7029-7033
Publication Date(Web):17 December 2014
DOI:10.1016/j.tetlet.2014.10.129
Previously, we reported allyl transfer reactions of allyl bromide and allyl phthalimido-N-oxyl substrates with hydrocarbons that result in CC bond formation. In both cases, efficient chain transfer processes along with high reaction yields were observed. Since PINO chemistry leads to an environmentally friendly method of hydrocarbon functionalization, additional studies were performed in order to improve the process. To expand the utility of this reaction, we carried out experiments to optimize reaction conditions and tested the effect of Lewis acids and low temperature initiators. Although allyl-PINO substrates reacted slightly slower than the bromides, the reactions were cleaner with little or no side products. The chain lengths for these reactions were compromised at lower temperatures, attributable to the high activation energy required for the hydrogen atom abstraction by PINO. The addition of a Lewis acid catalyst (AlCl3) improves the product yield and reaction rate, possibly due to the formation of a PINO/AlCl3 complex which lowers the activation energy for hydrogen abstraction step.Graphical abstract
Co-reporter:Michelle L. Grimm, William J. Allen, Meghan Finn, Neal Castagnoli Jr., James M. Tanko
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 4) pp:1458-1463
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmc.2011.01.002
A photochemical model study of benzophenone triplet (3BP) with the MAO-B substrate 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP (1)] and two of it’s derivatives, 1-cyclopropyl-4-phenyl-1,2,3,6-tetrahydropyridine (2) and (±)-[trans-2-phenylcyclopropyl-4-phenyl-1,2,3,6-tetrahydropyridine (3) were performed. Literature precedent and calculations reported herein suggest that the barrier to ring opening for aminyl radical cations derived from N-cyclopropyl derivatives of tertiary amines (such as MPTP) will be low. The LFP results reported herein demonstrate that pathways for the reaction of 3BP with 1, 2, and 3 are very similar. In each instance, disappearance of 3BP is accompanied solely by appearance of bands corresponding to the diphenylhydroxylmethyl radical and neutral radical derived from MPTP and it’s two derivatives 2 and 3. These results suggest that the reaction between benzophenone triplet and tertiary aliphatic amines proceed via a simple hydrogen atom transfer reaction. Additionally these model examinations provide evidence that oxidations of N-cyclopropyl derivatives of MPTP catalyzed by MAO-B may not be consistent with a pure SET pathway.
Co-reporter:Susan Mitroka ; Stephanie Zimmeck ; Diego Troya ;James M. Tanko
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:2907-2913
Publication Date(Web):February 10, 2010
DOI:10.1021/ja903856t
The hydroxyl radical (HO•) is a highly reactive oxygen-centered radical whose bimolecular rate constants for reaction with organic compounds (hydrogen atom abstraction) approach the diffusion-controlled limit in aqueous solution. The results reported herein show that hydroxyl radical is considerably less reactive in dipolar, aprotic solvents such as acetonitrile. This diminished reactivity is explained on the basis of a polarized transition state for hydrogen abstraction, in which the oxygen of the hydroxyl radical becomes highly negative and can serve as a hydrogen bond acceptor. Because acetonitrile cannot participate as a hydrogen bond donor, the transition state cannot be stabilized by hydrogen bonding, and the reaction rate is lower; the opposite is true when water is the solvent. This hypothesis explains hydroxyl radical reactivity both in solution and in the gas phase and may be the basis for a “containment strategy” used by Nature when hydroxyl radical is produced endogenously.
Co-reporter:Xiangzhong Li, Michelle L. Grimm, Kazuo Igarashi, Neal Castagnoli, Jr. and J. M. Tanko
Chemical Communications 2007 (Issue 25) pp:2648-2650
Publication Date(Web):03 Apr 2007
DOI:10.1039/B702157G
Using direct and indirect electrochemical methods, the rate constant for ring opening of the radical cation generated from N-cyclopropyl-N-methylaniline was found to be 4.1 × 104 s−1.
Co-reporter:James M. Tanko;Mitra Sadeghipour
Angewandte Chemie 1999 Volume 111(Issue 1‐2) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19990115)111:1/2<219::AID-ANGE219>3.0.CO;2-N
Br\.-Radikale sind die Kettenträger einer neuartigen selektiven Allylierung von Kohlenwasserstoffen (und anderen Substraten): Sie abstrahieren unter Bildung von R\. ein Wasserstoffatom vom Kohlenwasserstoff RH. Die Addition von R\. an ein substituiertes Allylbromid (unter Bildung eines β-Bromradikals) mit anschließender β-Spaltung führt zur Rückbildung von Br\. und zur Bildung des Produkts. Die Reaktionsausbeuten und die kinetischen Kettenlängen dieser Reaktion sind dann am höchsten, wenn das Allylbromid einen radikalstabilisierenden Substituenten trägt.
Co-reporter:James. M. Tanko;Mitra Sadeghipour
Angewandte Chemie International Edition 1999 Volume 38(Issue 1‐2) pp:
Publication Date(Web):18 JAN 1999
DOI:10.1002/(SICI)1521-3773(19990115)38:1/2<159::AID-ANIE159>3.0.CO;2-6
The Br. radical is the chain carrier of a new highly selective allylation of hydrocarbons (and other substrates): It abstracts a hydrogen atom from R−H to yield R.. Addition of R. to substituted allyl bromide (to form a β-bromo radical) followed by β-cleavage regenerates Br. and provides the product. Reaction yields and kinetic chain lengths for this reaction are highest when the allyl bromide contains a radical-stabilizing substituent.
Co-reporter:Xiangzhong Li, Michelle L. Grimm, Kazuo Igarashi, Neal Castagnoli, Jr. and J. M. Tanko
Chemical Communications 2007(Issue 25) pp:NaN2650-2650
Publication Date(Web):2007/04/03
DOI:10.1039/B702157G
Using direct and indirect electrochemical methods, the rate constant for ring opening of the radical cation generated from N-cyclopropyl-N-methylaniline was found to be 4.1 × 104 s−1.

![Benzene, [3-(bromomethyl)-3-butenyl]-](http://img.cochemist.com/ccimg/864000/863916-51-2.png)
![Benzene, [3-(bromomethyl)-3-butenyl]-](http://img.cochemist.com/ccimg/864000/863916-51-2_b.png)