Co-reporter:Timothy D. Lash and Gregory M. Ferrence
Inorganic Chemistry September 18, 2017 Volume 56(Issue 18) pp:11426-11426
Publication Date(Web):September 5, 2017
DOI:10.1021/acs.inorgchem.7b01946
The availability of diphenyl-23-oxa-, -thia-, and -selena-21-carbaporphyrins has enabled the reactivity of these systems to be investigated and contrasted. All three heterocarbaporphyrins reacted with palladium(II) acetate in refluxing chloroform–acetonitrile to give organometallic palladium(II) derivatives in good yields. These structures are stable and give UV–vis spectra that show increasing broadening and bathochromic shifts as the size of the heteroatom increases. Nickel(II) acetate in refluxing N,N-dimethylformamide reacted with the oxa- and thiacarbaporphyrins under nitrogen to give the corresponding nickel(II) complexes, but the selenacarbaporphyrin did not metalate under these conditions. The NMR spectra for all of the metal complexes showed that they possess strong diamagnetic ring currents, although the palladium complexes gave larger downfield shifts that were slightly diminished when larger heteroatoms were present in the porphyrinoid cavity. Reactions of the oxacarbaporphyrin with nickel(II) acetate in the presence of air led to a unique oxidation reaction that afforded a weakly diatropic 21-oxycarbaporphyrin, and low yields of a related product were also obtained from the thiacarbaporphyrin.
Co-reporter:Navneet Sahota, Gregory M. Ferrence, and Timothy D. Lash
The Journal of Organic Chemistry September 15, 2017 Volume 82(Issue 18) pp:9715-9715
Publication Date(Web):August 29, 2017
DOI:10.1021/acs.joc.7b01831
Norbornenes with two ester substituents were prepared by Diels–Alder cycloadditions of cyclopentadiene with dimethyl fumarate and dimethyl 1,1-ethylenedicarboxylate. Oxidation with potassium permanganate gave good yields of related diols that were oxidatively ring-opened to afford cyclopentane dialdehydes. MacDonald-type “3 + 1” condensations with a tripyrrane, followed by oxidation with DDQ in refluxing toluene, gave carbaporphyrin or carbachlorin products in good yields. The macrocyclic products were highly diatropic and produced porphyrin-like UV–vis spectra. The carbaporphyrin was converted into silver(III) and gold(III) organometallic derivatives. Reaction with methyl iodide in the presence of potassium carbonate gave mono- and dialkylation products, and treatment of the former with Ni(OAc)2 or Pd(OAc)2 afforded nickel(II) and palladium(II) complexes. The free base carbaporphyrin and carbachlorin, and the nickel and palladium complexes, were characterized by X-ray crystallography. The carbachlorin also reacted with silver(I) acetate to give a silver(III) derivative. Carbaporphyrins and carbachlorins underwent deuterium exchange at the meso-positions with deuteriated TFA, and this observation indicates that protonation is occurring at the bridging carbons. The new route to carbaporphyrins and carbachlorins has enabled detailed studies on the properties of these systems and provides the foundations for future investigations.
Co-reporter:Ruixiao Gao, Deyaa I. AbuSalim, and Timothy D. Lash
The Journal of Organic Chemistry July 7, 2017 Volume 82(Issue 13) pp:6680-6680
Publication Date(Web):June 2, 2017
DOI:10.1021/acs.joc.7b00829
The first examples of porphyrin analogues incorporating pyrene units are reported. Acid-catalyzed condensation of a pyrene dialdehyde with a tripyrrane, followed by oxidation with DDQ, afforded a polycyclic aromatic hydrocarbon (PAH)–porphyrin hybrid in 38% yield. Pyreniporphyrin proved to be devoid of global aromatic character, but upon protonation aromatic mono- and dicationic species were generated. In the proton NMR spectrum for the dication, the internal CH was shifted upfield to approximately +3 ppm. NICS calculations and ACID plots confirmed the diatropic nature of these structures. Pyreniporphyrin reacted with palladium(II) acetate to give excellent yields of a palladium(II) complex that showed weakly diatropic properties. Treatment of the pyrene dialdehyde with phenylmagnesium bromide generated a dicarbinol that reacted with excess pyrrole in the presence of boron trifluoride etherate to give a tripyrrane analogue. Lewis acid catalyzed ring closure with a thiophene dialcohol in 2% ethanol–dichloromethane afforded a tetraphenylthiapyreniporphyrin in 31% yield. This porphyrinoid was nonaromatic in the free-base form but showed significant diatropicity upon protonation. These results demonstrate that PAH–porphyrin hybrids are easily accessible, and this strategy may allow the incorporation of even larger aromatic subunits.
Co-reporter:Timothy D. Lash
Chemical Reviews 2017 Volume 117(Issue 4) pp:
Publication Date(Web):September 22, 2016
DOI:10.1021/acs.chemrev.6b00326
Following immediately after the serendipitous discovery of N-confused porphyrins, remarkably diverse carbaporphyrinoid systems have been synthesized and investigated. By replacing a pyrrolic unit within the porphyrin framework with cyclopentadiene, indene, azulene, cycloheptatriene, or benzene, new families of porphyrin-like macrocycles were produced. True carbaporphyrins are fully aromatic structures, while benziporphyrins are essentially devoid of macrocyclic aromatic character, and azuliporphyrins fall midway between the two extremes. Monocarbaporphyrinoids are superior organometallic ligands and form stable complexes with copper(III), silver(III), gold(III), nickel(II), palladium(II), platinum(II), rhodium(III), iridium(III), and ruthenium(II). Unusual oxidation reactions have also been discovered, commonly leading to derivatization of the internal carbon atom. In addition, structural rearrangements have been uncovered that allow the conversion of azuliporphyrins into benzocarbaporphyrins, and benziporphyrins into carbaporphyrins. Although less well studied, many examples of dicarbaporphyrinoids have been reported, and these show equally intriguing characteristics. Furthermore, contracted and expanded carbaporphyrinoids have been investigated. Studies in this area provide fundamental insights into the aromatic properties, tautomerization, and reactivity of porphyrins and related macrocyclic systems.
Co-reporter:Daming Li;Timothy D. Lash
European Journal of Organic Chemistry 2017 Volume 2017(Issue 45) pp:6775-6780
Publication Date(Web):2017/12/08
DOI:10.1002/ejoc.201701257
Chlorinated cyclohexene and cyclopentene dialdehydes, which can be prepared in one step from cyclohexanone or cyclopentanone, were condensed with a tripyrrane in the presence of trifluoroacetic acid and oxidized with DDQ to afford modest yields of internally halogenated carbachlorin-type products. The new macrocycles retained strongly aromatic properties and gave UV/Vis spectra with intense Soret bands near 400 nm and moderately strong chlorin-like absorptions at approximately 660 nm. Attempts to further oxidize a 21-chlorocarbachlorin with DDQ in refluxing toluene gave a dehalogenated carbaporphyrin and two externally chlorinated products.
Co-reporter:Timothy D. Lash
Accounts of Chemical Research 2016 Volume 49(Issue 3) pp:471
Publication Date(Web):February 8, 2016
DOI:10.1021/acs.accounts.5b00523
First reported in 1997, azuliporphyrins have proven to be a truly remarkable family of porphyrin analogues. In this system, although the porphyrin framework is retained, one of the pyrrolic moieties has been replaced by an azulene unit. Azulene favors electrophilic substitution at the 1,3-positions, which are structurally analogous to the α-positions in pyrrole, and this property facilitates the construction of azulene-containing porphyrinoid systems. Azuliporphyrins were first prepared from tripyrranes and 1,3-azulenedicarbaldehyde using a “3 + 1” variant on the MacDonald reaction. Subsequently, azulenes were shown to react with acetoxymethylpyrroles under acidic conditions to generate azulitripyrranes that could be utilized in a back-to-front “3 + 1” methodology to form azuliporphyrins and related heteroporphyrinoids. In addition, the favorability of azulenes toward 1,3-substitution was applied to one-pot syntheses of tetraarylazuliporphyrins and calix[4]azulenes.Azuliporphyrins have significant diatropic character that is greatly enhanced upon protonation. They have been shown to form organometallic complexes with Ni(II), Pd(II), Pt(II), Ir(III), Rh(III), and Ru(II) and undergo selective oxidations at the internal carbon with copper(II) or silver(I) salts to afford 21-oxyazuliporphyrins. In addition, oxidative ring contractions readily occur under basic conditions in the presence of peroxides to give benzocarbaporphyrins, and this reactivity provides access to tetraarylbenzocarbaporphyrins and their organometallic derivatives. A diazulenylmethane dialdehyde has been shown to react with dipyrrylmethanes in the presence of HCl or HBr to give diazuliporphyrins that were isolated in a monoprotonated form, and metalation with palladium(II) acetate afforded a stable zwitterionic palladium(II) complex.Equally intriguing dicarbaporphyrinoids incorporating indene and azulene rings have been reported, and these systems exhibit significant aromatic character. Recent studies have demonstrated that calixazulenes form supramolecular complexes with quaternary ammonium salts and afford a 1:1 complex with C60. In addition, conjugated structures have been prepared from calixazulenes that are structurally related to quatyrin, the theoretically important hydrocarbon analogue of the porphyrins. Examples of expanded azuliporphyrinoids have also been described. These azulene-containing porphyrinoids exhibit unique and complex reactivity that compares favorably with better studied porphyrin analogue systems such as the N-confused porphyrins, and azuliporphyrin derivatives show promise in the development of new catalytic systems.
Co-reporter:Leah M. Stateman and Timothy D. Lash
Organic Letters 2015 Volume 17(Issue 18) pp:4522-4525
Publication Date(Web):September 1, 2015
DOI:10.1021/acs.orglett.5b02219
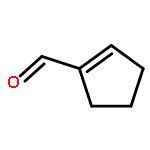
A carbatripyrrin intermediate was prepared from commercially available technical-grade indene and 2-pyrrolecarbaldehyde in three steps and 50% overall yield. This novel conjugated structure reacted with pyrroledialdehydes and 2,5-furandicarbaldehyde in the presence of TFA to give good yields of carbaporphyrins and an oxacarbaporphyrin, respectively, and unlike currently available methodologies, no oxidation step was required. The carbatripyrrin also condensed with heterocyclic dicarbinols in the presence of BF3·Et2O to give a series of heterocarbaporphyrins.
Co-reporter:Timothy D. Lash, Deyaa I. AbuSalim and Gregory M. Ferrence
Chemical Communications 2015 vol. 51(Issue 88) pp:15952-15955
Publication Date(Web):10 Sep 2015
DOI:10.1039/C5CC06890H
Base-catalyzed condensation of dicyclopentadienylmethane with a dipyrrylmethane dialdehyde gave a dicarbachlorin with an internal CH2 group. This unusual porphyrinoid retained highly diatropic characteristics and exhibited a porphyrin-like UV-vis spectrum.
Co-reporter:Timothy D. Lash; Denise A. Colby; Jessica A. El-Beck; Deyaa I. AbuSalim;Gregory M. Ferrence
Inorganic Chemistry 2015 Volume 54(Issue 18) pp:9174-9187
Publication Date(Web):September 1, 2015
DOI:10.1021/acs.inorgchem.5b01587
Oxidation of tetraarylazuliporphyrins with silver(I) acetate in refluxing chloroform–acetonitrile afforded good yields of 21-oxyazuliporphyrins. Although hydroxyazuliporphyrin tautomers can be considered for this system, spectroscopic results and density functional theory calculations indicate that the keto form is favored, and this was confirmed by single-crystal X-ray diffraction. Oxyazuliporphyrins formally possess a 24π electron delocalization pathway, but the proton NMR spectra are consistent with macrocycles that have diatropic ring currents. Nucleus independent chemical shift and anisotropy of induced current density calculations also confirmed the diatropic nature of these macrocycles, although these results indicated that the seven-membered ring is antiaromatic. However, while the NMR spectra showed the azulene protons at atypically high field values, the results are consistent with a nonaromatic cycloheptatrienyl unit. Protonation gave dicationic products that exhibited enhanced diatropic character. Oxyazuliporphyrins readily form metalated derivatives with Ni(II), Pd(II), and Pt(II), and these complexes exhibited significant diatropic character even though the macrocycle is highly distorted. X-ray diffraction characterization of palladium(II) and platinum(II) complexes demonstrated that these derivatives are structurally virtually identical to a previously reported copper(II) oxyazuliporphyrin.
Co-reporter:Leah M. Stateman, Gregory M. Ferrence, and Timothy D. Lash
Organometallics 2015 Volume 34(Issue 15) pp:3842-3848
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.organomet.5b00433
The organometallic chemistry of azuliporphyrins has been extended to the first syntheses of rhodium(III) derivatives. Reaction of [Rh(CO)2Cl]2 with an azuliporphyrin in refluxing xylenes gave rhodium(III) azuliporphyrins in 62–67% yield. These derivatives incorporate solvent molecules as axial ligands and thereby introduce two carbon–rhodium bonds in a three-component reaction. The methylbenzyl ligands that are derived from xylenes overlie the π system of the porphyrinoid macrocycle, and proton NMR spectra show that these units are strongly shielded. The o-, m-, and p-xylene-derived complexes were characterized by X-ray crystallography, and this confirmed the presence of the central rhodium atom and the axial benzylic ligands. Additionally, when the azuliporphyrin was reacted with [Rh(CO)2Cl]2 in refluxing acetonitrile and then treated with acetone and basic alumina, a rhodium(III) azuliporphyrin with an acetone-derived axial ligand was generated. These results demonstrate that azuliporphyrins are superior organometallic ligands and therefore merit further investigation.
Co-reporter:Deyaa I. AbuSalim and Timothy D. Lash
The Journal of Physical Chemistry A 2015 Volume 119(Issue 46) pp:11440-11453
Publication Date(Web):October 28, 2015
DOI:10.1021/acs.jpca.5b08682
The conformations of five doubly neo-confused porphyrin structures and fifty-four neo-confused N-confused porphyrin (neo-C-NCP) tautomers were minimized using DFT-B3LYP/6-311++G(d,p). The relative stabilities of the tautomers for each series were computed using M06-2X and B3LYP-D functionals, and the bond lengths and bond angles were calculated. Nucleus-independent chemical shifts and anisotropy of induced current density plots were used to provide further insights into the diatropic properties of each species. The results demonstrate that porphyrinoid structures with two modified rings of this type are far less stable than porphyrins, N-confused porphyrins, or neo-confused porphyrins, but the fully conjugated tautomers retain highly diatropic characteristics. Nevertheless, the lowest-energy neo-C-NCP tautomers have similar relative energies to known doubly N-confused porphyrins, and the results suggest that these types of porphyrin isomers may be synthetically accessible.
Co-reporter:Deyaa I. AbuSalim ; Gregory M. Ferrence ;Timothy D. Lash
Journal of the American Chemical Society 2014 Volume 136(Issue 18) pp:6763-6772
Publication Date(Web):April 16, 2014
DOI:10.1021/ja502795x
An adj-dicarbaporphyrin was prepared by carrying out a base-catalyzed MacDonald reaction between bis(3-indenyl)methane and a dipyrrylmethane dialdehyde. The porphyrinoid system exhibited highly diatropic characteristics, and the proton NMR spectrum gave resonances at −5.74 and −6.24 ppm for the internal NH and CH protons, respectively. The UV–vis spectrum was also porphyrin-like, giving a Soret band at 455 nm and a series of Q bands at longer wavelengths. Addition of trifluoroacetic acid gave a C-protonated monocation, and at higher acid concentrations a dicationic species was observed. Addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded a monodeprotonated porphyrinoid anion. All of these species retained highly diatropic characteristics. Density functional theory calculations showed that a nonplanar tautomer with four internal hydrogens was favored, in agreement with the spectroscopic data. Nucleus-independent chemical shift calculations also confirmed the aromatic characteristics of the free-base, cationic, and anionic structures. The dicarbaporphyrin reacted with palladium(II) acetate in refluxing acetonitrile to give an unusual tripalladium sandwich complex consisting of two dianionic palladium(II) dicarbaporphyrin units surrounding a palladium(IV) cation with unique η5 interactions involving meso-carbon atoms.
Co-reporter:Deyaa I. AbuSalim and Timothy D. Lash
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 43) pp:8719-8736
Publication Date(Web):18 Sep 2014
DOI:10.1039/C4OB01659A
The conformations of a series of benziporphyrins, naphthiporphyrins, oxybenziporphyrins, and related structures were minimized using DFT-B3LYP/6-311++G(d,p). The relative stabilities of the tautomers for each series were calculated using M06-2X and B3LYP-D functionals, and bond lengths were obtained. The diatropic properties of each species were assessed by calculating nucleus independent chemical shifts (NICS) and the related NICSzz values. Although benziporphyrin and naphthiporphyrin tautomers essentially exhibit no aromatic properties, mono- and diprotonated species show weakly diatropic characteristics in agreement with experimental observations. Benziphlorins, intermediates in the MacDonald “3 + 1” synthesis of benziporphyrins, were also examined and tautomers with methylene units adjacent to the benzene ring were shown to be far more stable than tautomers with a CH2 bridge between two pyrrolic units. 2-Hydroxybenziporphyrin was shown to be significantly less stable than the aromatic tautomer oxybenziporphyrin, although diprotonation leads to a species with somewhat reduced diatropicity. Related systems were also analyzed and the results demonstrate that benziporphyrins and naphthiporphyrins range from structures with no measurable macrocyclic aromaticity to strongly aromatic systems that exhibit large diamagnetic ring currents.
Co-reporter:Timothy D. Lash, Jessica A. El-Beck and Gregory M. Ferrence
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 2) pp:316-329
Publication Date(Web):12 Nov 2013
DOI:10.1039/C3OB41992D
A series of hetero-azuliporphyrins have been prepared by the “3 + 1” variant on the MacDonald condensation. Azulitripyrranes with tert-butyl and phenyl substituents reacted with thiophene or selenophene dialdehydes in the presence of TFA to give, following an oxidation step, thia- and selena-azuliporphyrins in 45–55% yield. Two of these compounds gave crystals suitable for X-ray crystallographic analysis and the data were consistent with the presence of a 17-atom delocalization pathway. The hetero-azuliporphyrins have significant diatropic character that is enhanced by the presence of an electron-donating tert-butyl substituent. The aromatic character is further increased in polar solvents such as DMSO, which are believed to stabilize dipolar resonance contributors with 18π electron delocalization pathways. Protonation also greatly increases the diatropic characteristics of these macrocycles. The porphyrinoids underwent an oxidative ring contraction with t-BuOOH–KOH to give moderate yields of benzoheterocarbaporphyrins. Reaction of azulitripyrranes with 2,5-furandicarbaldehyde afforded oxa-azuliporphyrins, a class of carbaporphyrinoids that had previously been inaccessible. These “missing links” in the study of heteroazuliporphyrins were isolated as the dihydrochloride salts. Protonated oxa-azuliporphyrins are stable aromatic compounds, but the free base forms underwent rapid decomposition in solution.
Co-reporter:Timothy D. Lash
Chemistry – An Asian Journal 2014 Volume 9( Issue 3) pp:682-705
Publication Date(Web):
DOI:10.1002/asia.201301594
Abstract
The cavities of carbaporphyrinoid systems provide unique environments for the formation of organometallic species. These systems commonly act as either dianionic or trianionic ligands, and may stabilize unusual oxidation states such as silver(III). Although the metalation of N-confused porphyrins has been explored in great detail, the formation of metallo-derivatives of other carbaporphyrinoids remains far less well explored. Nevertheless, exciting advances have been made on the metalation of carbaporphyrins, azuliporphyrins, benziporphyrins and related macrocycles.
Co-reporter:Ruoshi Li, Aaron D. Lammer, Gregory M. Ferrence, and Timothy D. Lash
The Journal of Organic Chemistry 2014 Volume 79(Issue 9) pp:4078-4093
Publication Date(Web):April 3, 2014
DOI:10.1021/jo500580e
Neo-confused porphyrins represent a unique family of porphyrin isomers that retain overall aromatic characteristics by virtue of a 17-atom 18π electron delocalization pathway. These porphyrin analogues have a pyrrolic subunit linked in a 1,3-fashion so that a nitrogen atom is directly connected to a meso-bridging carbon. Pyrrole-3-carbaldehydes were shown to react with sodium hydride and 5-acetoxymethylpyrrole-2-carbaldehydes in DMF to give the crucial neo-confused dipyrrolic dialdehyde intermediates. MacDonald “2 + 2” condensation of the dialdehydes with a dipyrrylmethane afforded a dihydroporphyrinoid, and subsequent oxidation with 0.2% aqueous ferric chloride generated a series of fully conjugated neo-confused porphyrins. Unusual dihydroporphyrin byproducts were also identified. Reaction of neo-confused porphyrins with nickel(II) or palladium(II) acetate in refluxing acetonitrile gave excellent yields of the corresponding organometallic derivatives. Proton NMR spectroscopy demonstrates that the diatropic character of this system is diminished compared to regular porphyrins, although neo-confused porphyrins retain porphyrin-like UV–vis spectra. Protonation led to the sequential formation of mono- and dicationic species. Proton NMR spectra for the dications showed the presence of enhanced diamagnetic ring currents.
Co-reporter:Daming Li and Timothy D. Lash
The Journal of Organic Chemistry 2014 Volume 79(Issue 15) pp:7112-7121
Publication Date(Web):July 7, 2014
DOI:10.1021/jo501287q
Acid-catalyzed condensation of 3-ethoxymethylenecyclopentene-1-carbaldehyde with a tripyrrane, followed by oxidation with aqueous ferric chloride solutions, afforded moderate yields of a carbachlorin. This porphyrinoid exhibited a porphyrin-like UV–vis spectrum with a slightly intensified peak at 650 nm. The proton NMR spectrum showed that the carbachlorin is highly diatropic, and this has been confirmed by nucleus independent chemical shift (NICS) calculations. Oxidation of the carbachlorin with DDQ gave the first example of a carbaporphyrin that is unsubstituted on the carbocyclic ring. Reaction of the carbachlorin with silver(I) acetate gave the corresponding silver(III) organometallic complex. When the carbachlorin was refluxed with methyl iodide and potassium carbonate in acetone, the 22-methyl derivative was formed. Treatment of the N-alkylation product with palladium(II) acetate afforded an unstable palladium(II) carbachlorin that was partially converted into a palladium(II) carbaporphyrin via an oxidation–methyl group migration process. Improved yields of the carbaporphyrin complex were obtained when the reaction mixture was stirred with aqueous ferric chloride solutions. These results open up the field of carbachlorin and carbaporphyrin chemistry for further study.
Co-reporter:Timothy D. Lash, Gean C. Gilot, and Deyaa I. AbuSalim
The Journal of Organic Chemistry 2014 Volume 79(Issue 20) pp:9704-9716
Publication Date(Web):September 17, 2014
DOI:10.1021/jo5018553
Previous attempts to prepare tropone-fused carbaporphyrins by reacting peroxides with azuliporphyrins under basic conditions afforded benzocarbaporphyrins instead. In this study, a methoxyazulitripyrrane condensed with a pyrrole dialdehyde in the presence of TFA, followed by oxidation with ferric chloride, to give a tropone-fused carbaporphyrin following a spontaneous demethylation. The porphyrinoid gave a modified UV–vis spectrum showing multiple bands in the Soret region, and the proton NMR spectrum showed that it has a reduced diamagnetic ring current in comparison to other carbaporphyrins. The tropone-fused derivative failed to react with tert-butyl hydroperoxide and potassium hydroxide, demonstrating that this type of structure is not an intermediate in the formation of benzocarbaporphyrins. However, the reaction with silver(I) acetate gave the corresponding silver(III) complex. Condensation of the methoxyazulitripyrrane with 2,5-thiophenedicarbaldehyde gave a related tropone-fused thiacarbaporphyrin together with a methoxythiaazuliporphyrin. Treatment of the carbaporphyrins with DBU resulted in the formation of anionic species, while addition of acid afforded dicationic structures. DFT studies were performed on a series of tautomers, protonated species, and anionic structures related to these tropone-fused carbaporphyrins, and NICS calculations were carried out. These results allowed favorable conjugation pathways to be identified. In addition, these studies predicted that protonation initially occurs on the carbonyl moiety rather than on the expected interior pyrrolenine nitrogen atom.
Co-reporter:Stacy C. Fosu, Gregory M. Ferrence, and Timothy D. Lash
The Journal of Organic Chemistry 2014 Volume 79(Issue 22) pp:11061-11074
Publication Date(Web):October 16, 2014
DOI:10.1021/jo502063w
Dimethoxybenzitripyrranes were prepared in excellent yields by reacting benzene dicarbinols with BF3·Et2O and excess pyrrole in refluxing 1,2-dichloroethane. Reaction with a pyrrole dialdehyde in the presence of TFA, followed by oxidation with DDQ, afforded good yields of meso-diphenyldimethoxybenziporphyrins. These dimethoxyporphyrinoids exhibited weakly diatropic properties that were enhanced upon protonation. The dimethoxybenzitripyrranes also reacted with a thiophene dicarbinol to give dimethoxythiabenziporphyrins, and the nonplanar nature of this system was demonstrated by X-ray crystallography. The dimethoxybenziporphyrins reacted with palladium(II) acetate to give the related organometallic derivatives, but the thiabenziporphyrins underwent a demethylation to afford palladium(II) thiaoxybenziporphyrins. Related palladium(II) complexes were also prepared from previously reported thiacarbaporphyrinoids. The X-ray structure for one of the complexes showed that the six-membered ring is very distorted and the thiophene ring is strongly tilted out of the plane of the macrocycle. The dimethoxybenzitripyrranes also reacted with dimethoxybenzene dicarbinols to give the first examples of dibenziporphyrins, thereby further demonstrating the versatility of this synthetic methodology.
Co-reporter:Ruoshi Li, Gregory M. Ferrence and Timothy D. Lash
Chemical Communications 2013 vol. 49(Issue 68) pp:7537-7539
Publication Date(Web):12 Jul 2013
DOI:10.1039/C3CC44248A
MacDonald “2 + 2” condensation of a 1,2′-dipyrrylmethane dialdehyde with a 2,2′-dipyrrylmethane afforded a neo-confused porphyrin in 55% yield. In addition, a novel dihydroporphyrin with two appended pyrrolic units was isolated and structurally characterized.
Co-reporter:Alexandra M. Young and Timothy D. Lash
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 39) pp:6841-6848
Publication Date(Web):03 Sep 2013
DOI:10.1039/C3OB41506F
Reaction of 2,6-pyridinedicarbaldehyde with a series of tripyrranes in the presence of trifluoroacetic acid, followed by oxidation of a pyriphlorin intermediate with silver(I) acetate, gave 6-oxopyriphlorins in moderate to good yields. The oxophlorin analogues were nonaromatic porphyrinoids that gave bright green colored solutions. Protonation with TFA afforded species that were attributed to mono- and dicationic structures. The proton NMR spectra in TFA–CDCl3 showed downfield shifts to the meso-protons and upfield shifts to the interior NH resonances that indicated the presence of a weakly diatropic dicationic species. Reaction of a 6-oxopyriphlorin with nickel(II) acetate in DMF or palladium(II) acetate in acetonitrile afforded the corresponding metallo-derivatives.
Co-reporter:Deyaa I. AbuSalim and Timothy D. Lash
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 48) pp:8306-8323
Publication Date(Web):31 Oct 2013
DOI:10.1039/C3OB42063A
The conformations of eight neo-confused porphyrin (neo-CP) structures and seven related benzo-neo-confused porphyrins (B-neo-CPs) were minimized using DFT-B3LYP/6-311++G(d,p). In addition, in order to provide contrasts to the B-neo-CP tautomers, a series of twelve benzoporphyrin (BP) tautomers and twelve benzocarbaporphyrins (BCPs) were also analyzed, and two N-confused porphyrin (NCP) tautomers were also considered. The relative stability of the tautomers for each series was computed, and the bond lengths and bond angles were calculated. Surprisingly, all of the neo-CP and B-neo-CP tautomers were near planar. The fully conjugated members of these series showed a significant degree of bond length alternation, unlike porphyrins or the aromatic forms of NCP, BP or BCP. The most stable tautomer of neo-CP was calculated to be 27.04 kcal mol−1 higher in energy than porphyrin and 9.95 kcal mol−1 higher in energy than the most favored NCP tautomer. The most stable tautomer of B-neo-CP was similarly shown to be 26.40 kcal mol−1 higher in energy than benzoporphyrin. The favored tautomers also place the internal NH opposite to the neo-confused ring to minimize steric and lone pair–lone pair interactions, and to provide improved hydrogen bonding interactions. Nucleus independent chemical shifts (NICS) demonstrated that the fully conjugated neo-CP and B-neo-CP tautomers exhibit a significant amount of diatropic character, but these values are somewhat lower than the results obtained for porphyrins, BPs or BCPs. BCPs have three internal hydrogens, but the favored tautomer places the two pyrrolic hydrogens on each side of the indene unit to maximize hydrogen bonding interactions. Tautomers with an internal CH2 unit are also feasible, but these are energetically less favorable, have reduced diatropicity, and show significant bond length alternation. The results indicate that 18π electron delocalization pathways that pass through a fused benzene ring are less favorable than alternative delocalization pathways that bypass this unit.
Co-reporter:Timothy D. Lash, Ashley M. Toney, Kylie M. Castans, and Gregory M. Ferrence
The Journal of Organic Chemistry 2013 Volume 78(Issue 18) pp:9143-9152
Publication Date(Web):August 14, 2013
DOI:10.1021/jo401365p
Benzitripyrranes were prepared by reacting diphenyl-substituted benzenedicarbinols with excess pyrrole in the presence of BF3·Et2O. These dipyrrolic compounds underwent acid-catalyzed condensations with a pyrrole dialdehyde to afford good yields of diphenylbenziporphyrins, and further reaction with palladium(II) acetate gave stable organometallic derivatives. The X-ray crystal structure of a palladium(II) benziporphyrin showed that the system deviates significantly from planarity. Although the benzitripyrranes failed to give stable macrocyclic products with furan or thiophene dialdehydes, they afforded tetraphenyl heterobenziporphyrins upon reaction with diphenyl-substituted furan- or thiophenedicarbinols and BF3·Et2O. Benziporphyrins and their heteroanalogues showed no indication of a diamagnetic ring current by proton NMR spectroscopy, but addition of TFA gave rise to the formation of weakly diatropic dications.
Co-reporter:Deyaa I. AbuSalim, Michelle L. Merfeld, and Timothy D. Lash
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10360-10368
Publication Date(Web):September 24, 2013
DOI:10.1021/jo401756q
A series of diformylbenzophenones were generated by sequentially reacting protected bromobenzaldehydes with n-butyllithium and ethyl N,N-dimethylcarbamate. The acetal protective groups were cleaved with refluxing formic acid. Vilsmeier–Haack formylation of 2,2′,4,4′-tetramethoxybenzophenone also afforded a related dialdehyde. MacDonald “2 + 2” condensation of three benzophenone dialdehydes with a dipyrrylmethane gave oxophlorin analogues constructed from two benzene and two pyrrole rings. The free base oxodibenziphlorins were rather unstable in solution, and in most cases these porphyrinoids were isolated as the corresponding trifluoroacetate salts. The spectroscopic properties of 6-oxo-adj-dibenziphlorins are consistent with a nonaromatic ring system. DFT calculations indicated that the macrocycles considerably diverge from planarity, particularly when methoxy substituents are present on the arene rings.
Co-reporter:Deyaa I. AbuSalim and Timothy D. Lash
The Journal of Organic Chemistry 2013 Volume 78(Issue 22) pp:11535-11548
Publication Date(Web):October 28, 2013
DOI:10.1021/jo4021198
Monocarbaporphyrins, dicarbaporphyrins, tricarbaporphyrins, and quatyrins can all potentially exist in a number of tautomeric forms, but little is known about these species. In addition, no examples of tricarbaporphyrins or quatyrins are known at this time. In order to get information on the relative stabilities of carbaporphyrin tautomers, a series of aromatic and nonaromatic structures were assessed using computational methods. The conformations of 41 carbaporphyrin structures, together with four tautomers of porphyrin and the related antiaromatic species didehydroquatyrin, were minimized using DFT-B3LYP/6-311++G(d,p). The relative stabilities of the tautomers for each series were computed, and the bond lengths and bond angles were calculated. Many aromatic carbaporphyrin structures have very crowded cavities with up to six hydrogens being present. However, this was not sufficient to destabilize the aromatic structures relative to the nonaromatic forms. Furthermore, nucleus-independent chemical shifts (NICS) demonstrated that all of the porphyrinoids with 18-π-electron delocalization pathways are highly diatropic. adj-Dicarbaporphyrin and tricarbaporphyrin favor tautomers with an internal methylene group, while two interior methylenes are present in the favored tautomers for quatyrin. The results provide valuable information that will be helpful in designing synthetic routes to higher carbaporphyrinoid systems.
Co-reporter:Timothy D. Lash, Aaron D. Lammer, Aparna S. Idate, Denise A. Colby, and Kristen White
The Journal of Organic Chemistry 2012 Volume 77(Issue 5) pp:2368-2381
Publication Date(Web):January 30, 2012
DOI:10.1021/jo2026977
A “2 + 2” strategy for synthesizing adj-dicarbaporphyrinoid systems has been developed. In a model study, an azulenylmethylpyrrole dialdehyde was condensed with a dipyrrylmethane in the presence of HCl, followed by oxidation with ferric chloride, to give a modest yield of an azuliporphyrin. Fulvene aldehydes were prepared by reacting an indene-derived enamine with azulene aldehydes in the presence of Bu2BOTf, and azulene dialdehydes similarly reacted to give fulvene dialdehydes. The dialdehydes were condensed with dipyrrylmethanes in TFA/dichloromethane to afford good to excellent yields of dicarbaporphyrinoids with adjacent indene and azulene subunits. These 22-carbaazuliporphyrins exhibited significant diatropic character, and this property was magnified upon protonation. These characteristics are attributed to tropylium-containing resonance contributors that possess 18π electron delocalization pathways. Protonation studies demonstrated that C-protonation readily occurred at the interior indene carbon, but deuterium exchange also occurred at the internal azulene CH as well as at the meso-positions with TFA-d. Reaction of a carbaazuliporphyrin with silver(I) acetate in methanol or ethanol solutions also gave unusual nonaromatic dialkoxy derivatives.
Co-reporter:Timothy D. Lash and Katrina M. Bergman
The Journal of Organic Chemistry 2012 Volume 77(Issue 21) pp:9774-9783
Publication Date(Web):October 2, 2012
DOI:10.1021/jo301945f
Tripyrranes with tert-butyl and phenyl substituents have been prepared and used to synthesize oxybenziporphyrins, oxypyriporphyrins, benzocarbaporphyrins, and azuliporphyrins with phenyl and tert-butyl substituents via a “3 + 1” methodology. The proton NMR spectra for the tripyrrane dibenzyl esters indicate that these tripyrrolic systems take on a helical conformation that favors macrocycle formation, and the NMR data can be a useful predictor on the efficiency of the “3 + 1” synthesis. Nevertheless, a tetraphenyltripyrrane proved to be susceptible to acidolytic cleavage under the usual reaction conditions and gave poor yields of porphyrinoid products. This problem could be overcome to a certain extent by carrying out the reactions in neat TFA. The presence of these substituents led to significant changes in the spectroscopic properties and diatropic character of the new porphyrinoid structures.
Co-reporter:Dr. Timothy D. Lash;Aaron D. Lammer ;Dr. Gregory M. Ferrence
Angewandte Chemie International Edition 2012 Volume 51( Issue 43) pp:10871-10875
Publication Date(Web):
DOI:10.1002/anie.201206385
Co-reporter:Dr. Timothy D. Lash;Aaron D. Lammer ;Dr. Gregory M. Ferrence
Angewandte Chemie 2012 Volume 124( Issue 43) pp:11029-11033
Publication Date(Web):
DOI:10.1002/ange.201206385
Co-reporter:Timothy D. Lash
Organic Letters 2011 Volume 13(Issue 17) pp:4632-4635
Publication Date(Web):August 3, 2011
DOI:10.1021/ol2018483
Reaction of a benzocarbaporphyrin with methyl or ethyl iodide and potassium carbonate in refluxing acetone primarily afforded the 22-alkylation products. Subsequent metalation with palladium(II) acetate in refluxing acetonitrile gave the palladium(II) organometallic derivatives where the alkyl group had migrated to the 21-position.
Co-reporter:Alexandra M. Young, Amber L. Von Ruden and Timothy D. Lash
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 18) pp:6293-6305
Publication Date(Web):15 Jun 2011
DOI:10.1039/C1OB05603D
A series of porphyrin analogues with pyrazole rings replacing one of the usual pyrrole subunits have been synthesized. This was accomplished by reacting 1-phenyl, 1-methyl and 1-ethyl pyrazole-1,3-dicarbaldehydes with a tripyrrane in the presence of TFA, followed by an oxidation step. The initially formed phlorin product was sufficiently stable for the N-phenyl system to be isolated and characterized, although the related N-alkyl phlorin analogues were less stable. Attempts to dehydrogenate the intermediary phlorins with DDQ resulted in decomposition, but the N-alkylphlorins could be oxidized with 0.2% aqueous ferric chloride solutions. Although the phenyl-substituted phlorin could not be oxidized under these conditions, it did afford the pyrazoloporphyrin upon treatment with silver acetate under acidic conditions. Oxidations with silver acetate also afforded oxophlorin analogues where the oxo-linkage was selectively formed at the 5-position. The pyrazole-containing porphyrin analogues are cross-conjugated and exhibit only a small degree of diatropic character. The internal CH resonances were observed between 5.27 and 5.87 ppm, while the external meso-protons fell into a range of 6.84–7.88 ppm. The borderline overall aromatic character was attributed to dipolar resonance contributors. Protonation considerably increased the diatropicity and the diprotonated dications formed from these porphyrin analogues gave the internal CH resonance at upfield values of 2.65–3.20 ppm. The aromatic character was enhanced by the presence of an electron-donating alkyl substituent on the nitrogen compared to the phenyl-substituted species. The pyrazoloporphyrins reacted with nickel(II) acetate in DMF, or palladium(II) acetate in acetonitrile, to give the corresponding organometallic derivatives. The metal complexes showed increased diatropic character but protonation afforded nonaromatic cations. The oxophlorin analogues were also nonaromatic in the free base and protonated forms. This work extends our understanding of carbaporphyrinoid systems and provides the first detailed studies on pyrazole-containing porphyrin analogues.
Co-reporter:Timothy D. Lash, Teresa R. Lamm, J. Andy Schaber, Wen-hsiang Chung, Eric K. Johnson, Marjorie A. Jones
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 4) pp:1492-1504
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmc.2010.12.053
Analogues of coproporphyrinogen-III have been prepared with acetate or butyrate groups attached to the C and D pyrrolic subunits. The corresponding porphyrin methyl esters were synthesized by first generating a,c-biladienes by reacting a dipyrrylmethane with pyrrole aldehydes in the presence of HBr. Cyclization with copper(II) chloride in DMF, followed by demetalation with 15% H2SO4–TFA and reesterification, gave the required porphyrins in excellent yields. Hydrolysis with 25% hydrochloric acid and reduction with sodium-amalgam gave novel diacetate and dibutyrate porphyrinogens 9. Diacetate 9a was incubated with chicken red cell hemolysates (CRH), but gave complex results due to the combined action of two of the enzymes present in these preparations. Separation of uroporphyrinogen decarboxylase (URO-D) from coproporphyrinogen oxidase (CPO) allowed the effects of both enzymes on the diacetate substrate to be assessed. Porphyrinogen 9a proved to be a relatively poor substrate for CPO compared to the natural substrate coproporphyrinogen-III, and only the A ring propionate moiety was processed to a significant extent. Similar results were obtained for incubations of 9a with purified human recombinant CPO. Diacetate 9a was also a substrate for URO-D and a porphyrinogen monoacetate was the major product in this case; however, some conversion of a second acetate unit was also evident. The dibutyrate porphyrinogen 9b was only recognized by the enzyme CPO, but proved to be a modest substrate for incubations with CRH. However, 9b was an excellent substrate for purified human recombinant CPO. The major product for these incubations was a monovinylporphyrinogen, but some divinyl product was also generated in incubations using purified recombinant human CPO. The incubation products were converted into the corresponding porphyrin methyl esters, and these were characterized by proton NMR spectroscopy and mass spectrometry. The results extend our understanding of substrate recognition and catalysis for this intriguing enzyme and have allowed us to extend the active site model for CPO. In addition, the competitive action of both URO-D and CPO on the same diacetate porphyrinogen substrate provides additional perspectives on the potential existence of abnormal pathways for heme biosynthesis.Coproporphyrinogen analogues with acetate or butyrate groups in place of the usual propionate moieties on rings C and D have been used to probe the substrate binding requirements for coproporphyrinogen oxidase.
Co-reporter:Dr. Timothy D. Lash;Aaron D. Lammer ;Dr. Gregory M. Ferrence
Angewandte Chemie 2011 Volume 123( Issue 41) pp:9892-9895
Publication Date(Web):
DOI:10.1002/ange.201104826
Co-reporter:Timothy D. Lash, Breland E. Smith, Michael J. Melquist, and Bradley A. Godfrey
The Journal of Organic Chemistry 2011 Volume 76(Issue 13) pp:5335-5345
Publication Date(Web):May 20, 2011
DOI:10.1021/jo2006895
Indene-fused porphyrins have been synthesized starting from 2-indanone. Knorr-type reaction of oximes derived from benzyl or tert-butyl acetoacetate with 2-indanone and zinc dust in propionic acid gave good yields of indenopyrroles. Treatment with N-chlorosuccinimide then gave 8-chloro derivatives, and these reacted with 5-unsubstituted pyrroles to give dipyrroles incorporating the fused indene unit. Hydrogenolysis of the benzyl ester protective groups afforded the related dicarboxylic acids, but condensation with a dipyrrylmethane dialdehyde under MacDonald “2 + 2” reaction conditions gave poor yields of the targeted indenoporphyrins. However, when an indene-fused dipyrrole was converted into the corresponding dialdehyde with TFA–trimethyl orthoformate and then reacted with a dipyrrylmethane dicarboxylic acid, an indenoporphyrin was isolated in 26% yield. The porphyrin gave a highly modified UV–vis absorption spectrum with three strong bands showing up in the Soret region and a series of Q bands that extended beyond 700 nm. The proton NMR spectrum also showed a significantly reduced diamagnetic ring current where the meso-protons gave resonances near 9.3 ppm instead of typical porphyrin values of 10 ppm. Nickel(II), copper(II), and zinc complexes were also prepared, and these exhibited unusual UV–vis absorption spectra with bathochromically shifted Soret and Q absorptions. The diamagnetic nickel(II) and zinc complexes also showed reduced diatropic character compared to typical nickel(II) and zinc porphyrins.
Co-reporter:Timothy D. Lash, Alexandra M. Young, Jane M. Rasmussen, and Gregory M. Ferrence
The Journal of Organic Chemistry 2011 Volume 76(Issue 14) pp:5636-5651
Publication Date(Web):May 23, 2011
DOI:10.1021/jo200622s
Benziporphyrins, cross-conjugated porphyrin analogues with a benzene ring in place of one of the usual pyrrole units, have varying degrees of macrocyclic aromaticity because the 6π electron arene needs to give up its aromatic characteristics to facilitate conjugation over the entire system. As naphthalene would lose less resonance stabilization energy in giving up one of its benzene units, it was proposed that naphthiporphyrins would exhibit enhanced diatropicity compared to the related benziporphyrins. A naphthiporphyrin was prepared using the “3 + 1” variant of the MacDonald condensation by reacting 1,3-naphthalenedicarbaldehyde with a tripyrrane in the presence of TFA, followed by oxidation with DDQ. Although the free base form of naphthiporphyrin showed no overall diatropicity, the corresponding dication in TFA-CDCl3 demonstrated a significant diatropic ring current where the internal CH shifted upfield to between 4.0 and 4.6 ppm. Naphthiporphyrin was converted to the corresponding palladium(II) complexes by reaction with Pd(OAc)2 in acetonitrile, and the complex was further characterized by X-ray crystallography. Oxynaphthiporphyrins were similarly prepared by the “3 + 1” methodology from 4-methoxy-1,3-naphthalene-dicarbaldehyde, and these showed slightly enhanced diatropic character compared to oxybenziporphyrins. Reaction of oxybenziporphyrins or oxynaphthiporphyrins with silver(I) acetate afforded the corresponding silver(III) organometallic derivatives. A meso-tetraphenyl naphthiporphyrin was also synthesized in 4% yield by reacting a 1,4-naphthalene dicarbinol with 2 equiv of benzaldehyde and 3 equiv of pyrrole in the presence of BF3.Et2O, followed by oxidation with DDQ. However, this 1,4-naphthiporphyrin showed reduced diatropic character compared to the corresponding p-benziporphyrin system. The NMR spectra for the 1,4-naphthiporphyrin show that the naphthalene unit pivots over the macrocycle and this presumably leads to further steric interactions that reduce the planarity of the macrocycle. These results demonstrate that while naphthiporphyrins can show enhanced aromatic properties as predicted, other factors may overwhelm this effect.
Co-reporter:Dr. Timothy D. Lash;Aaron D. Lammer ;Dr. Gregory M. Ferrence
Angewandte Chemie International Edition 2011 Volume 50( Issue 41) pp:9718-9721
Publication Date(Web):
DOI:10.1002/anie.201104826
Co-reporter:Timothy D. Lash, Kae Miyake, Linlin Xu, and Gregory M. Ferrence
The Journal of Organic Chemistry 2011 Volume 76(Issue 15) pp:6295-6308
Publication Date(Web):June 21, 2011
DOI:10.1021/jo201098c
Tripyrrane analogues were prepared by reacting resorcinol or 2-methylresorcinol with 2 equiv of an acetoxymethylpyrrole in the presence of p-toluenesulfonic acid and calcium chloride. Following removal of the benzyl ester protective groups, the resorcinol-derived benzitripyrrane was reacted with a pyrrole dialdehyde to give an aromatic hydroxyoxybenziporphyrin. However, furan and thiophene dialdehydes gave highly insoluble products that could not be fully characterized. The methylresorcinol-derived tripyrrane analogue reacted with pyrrole, furan, thiophene, and selenophene dialdehydes to give unstable porphyrinoids that were further oxidized with [bis(trifluoroacetoxy)iodo]benzene to give stable benziporphyrin derivatives. These oxidized benziporphyrins showed strongly diatropic properties by proton NMR spectroscopy where the differences in chemical shifts (Δδ) were >18 ppm in some cases. The selenophene-derived system was further characterized by X-ray crystallography, and these results showed that one of the pyrrole subunits in this crowded structure was tilted by 21° relative to the mean macrocyclic plane. The tripyrrolic system reacted with silver(I) acetate to give the corresponding silver(III) organometallic complex. Regioselective alkylation with methyl or ethyl iodide and potassium carbonate gave diastereomeric mixtures of N-alkyl derivatives, and the N-ethyl substitution products showed highly diastereotopic characteristics.
Co-reporter:Timothy D. Lash ; Sarah A. Jones ;Gregory M. Ferrence
Journal of the American Chemical Society 2010 Volume 132(Issue 37) pp:12786-12787
Publication Date(Web):August 26, 2010
DOI:10.1021/ja105146a
McMurry coupling of a pyrrole bisacrylaldehyde afforded a dipyrrolic macrocycle that is structurally midway between 1,10-diaza[18]annulene and the porphyrins. The diprotonated dication of this system retains porphyrin-like properties and provides insights into the nature of porphyrinoid aromaticity.
Co-reporter:Virajkumar Gandhi, Michelle L. Thompson, Timothy D. Lash
Tetrahedron 2010 66(10) pp: 1787-1799
Publication Date(Web):
DOI:10.1016/j.tet.2010.01.046
Co-reporter:Timothy D. Lash, Ukti N. Mani, Anna-Sigrid I. M. Keck and Marjorie A. Jones
The Journal of Organic Chemistry 2010 Volume 75(Issue 10) pp:3183-3192
Publication Date(Web):April 13, 2010
DOI:10.1021/jo100083t
A series of vinylporphyrinogens were prepared to probe the enzyme coproporphyrinogen oxidase (CPO). Six (2-chloroethyl)porphyrins were synthesized from a common dipyrrylmethane via a,c-biladiene intermediates in excellent yields. Subsequent dehydrohalogenation with DBU in refluxing DMF then gave the required vinylporphyrin methyl esters, including harderoporphyrin-I, harderoporphyrin-III, and isoharderoporphyrin. The corresponding porphyrinogen carboxylic acids were incubated with chicken red cell hemolysates, which contain the enzyme CPO, and the products analyzed. The 17-ethyl analogue of harderoporphyrinogen-III, but not its 13-ethyl isomer, was shown to be an excellent substrate for CPO in accord with a proposed model for the active site of this enzyme. In addition, harderoporphyrinogen-VII, the monovinyl intermediate in the metabolism of coproporphyrinogen-IV, was shown to be an equally good substrate for this enzyme. However, isoharderoporphyrinogen, which lacks the correct ordering of peripheral substituents, was also a substrate for CPO. Furthermore, a nonnatural type I isomer of harderoporphyrinogen was shown to be acted on by CPO, but in this case further metabolism was noted and this afforded an unprecedented trivinyl porphyrinogen product. The corresponding porphyrin methyl ester was isolated and characterized by FAB MS and proton NMR spectroscopy. The results from these studies allow the binding requirements of CPO to be further assessed and provide a series of substrates to investigate this poorly understood enzyme.
Co-reporter:Pankaj Jain, Gregory M. Ferrence, and Timothy D. Lash
The Journal of Organic Chemistry 2010 Volume 75(Issue 19) pp:6563-6573
Publication Date(Web):September 9, 2010
DOI:10.1021/jo101310m
A series of fulvene monoaldehydes were prepared by reacting furan or thiophene carbaldehydes with an indene-derived enamine in the presence of di-n-butylboron triflate, but considerable difficulties were encountered in the preparation of fulvene dialdehydes needed for the synthesis of novel porphyrin analogues. These problems were overcome by reacting protected iodofulvenes with magnesium ate complexes at low temperatures, followed by addition of DMF and hydrolysis. The thiophene-containing fulvene gave good yields of the dialdehyde at −78 °C or −100 °C, but the furan system gave a major byproduct formally derived from valeraldehyde under the higher temperature conditions. This compound was fully characterized by NMR spectroscopy, mass spectrometry, and X-ray crystallography. However, this side reaction could be completely avoided at −100 °C, and the required furan-containing fulvene dialdehyde was isolated in 46% yield. The furan-derived dialdehyde reacted with a dipyrrylmethane in the presence of trifluoroacetic acid to give the 22-oxa-21-carbaporphyrin 19 in excellent yields (73−79%). However, the thiophene-containing fulvene dialdehyde failed to give any of the anticipated macrocyclic product. An unstable acyclic intermediate was isolated and partially characterized, but this species could not be induced to cyclize. Steric factors may play a role, but X-ray crystallography confirmed that the fulvene dialdehyde precursor does have the correct geometry to facilitate the formation of the porphyrinoid macrocycle. The new oxacarbaporphyrin was fully characterized and could easily be converted into the corresponding mono- and dicationic species. The second protonation involves addition onto the internal indene carbon and proton NMR spectroscopy for the sample in HCl−TFA demonstrates that it retains strongly diatropic characteristics. The free base oxacarbaporphyrin reacted with Pd(OAc)2 in DMF to give the corresponding palladium(II) organometallic derivative 27. The proton NMR spectrum for this complex also shows the retention of a strong, albeit slightly reduced, diatropic ring current. The free base oxacarbaporphyrin and the palladium derivative were both structurally characterized by X-ray crystallography. The bond lengths for 19 and 27 were consistent with the presence of significant 18π-electron delocalization pathways.
Co-reporter:Breland E. Smith, Timothy D. Lash
Tetrahedron 2010 66(25) pp: 4413-4422
Publication Date(Web):
DOI:10.1016/j.tet.2010.04.069
Co-reporter:Zhenjun Zhang, Gregory M. Ferrence and Timothy D. Lash
Organic Letters 2009 Volume 11(Issue 6) pp:1249-1252
Publication Date(Web):February 26, 2009
DOI:10.1021/ol802945s
Acid-catalyzed condensations between a dipyrrylmethane and a pyrrole bisacrylaldehyde gave an unusual stretched sapphyrin structure together with a hexaphyrin byproduct. A diazulihexaphyrin was prepared similarly. The highly diatropic sapphyrin analogue was isolated as a dication and was characterized by X-ray crystallography.
Co-reporter:Timothy D. Lash, Jessica A. El-Beck and Denise A. Colby
The Journal of Organic Chemistry 2009 Volume 74(Issue 22) pp:8830-8833
Publication Date(Web):October 26, 2009
DOI:10.1021/jo901959k
Substituted calix[4]azulenes were prepared by reacting 6-alkylazulenes with paraformaldehyde in the presence of florisil. Hydride abstraction of a calix[4]azulene with Ph3CPF6 afforded a tetraazulene analogue of the porphodimethenes.
Co-reporter:Timothy D. Lash, Valerie R. Yant
Tetrahedron 2009 65(46) pp: 9527-9535
Publication Date(Web):
DOI:10.1016/j.tet.2009.09.060
Co-reporter:Randall N. Davis, Timothy D. Lash
Tetrahedron 2009 65(48) pp: 9935-9943
Publication Date(Web):
DOI:10.1016/j.tet.2009.10.013
Co-reporter:Timothy D. Lash, Alexandra M. Young, Amber L. Von Ruden and Gregory M. Ferrence
Chemical Communications 2008 (Issue 47) pp:6309-6311
Publication Date(Web):07 Nov 2008
DOI:10.1039/B816057K
N-substituted pyrazoledialdehydes are shown to react with a tripyrrane under ‘3 + 1’ conditions to give aza-analogues of the N-confused porphyrins; these novel porphyrinoids show borderline aromatic properties and readily afford organometallic derivatives with Ni(OAc)2 and Pd(OAc)2.
Co-reporter:Timothy D. Lash
European Journal of Organic Chemistry 2007 Volume 2007(Issue 33) pp:5461-5481
Publication Date(Web):30 AUG 2007
DOI:10.1002/ejoc.200700478
Synthetic methods have been developed for the efficient synthesis of meso-unsubstituted carbaporphyrinoid systems, including oxybenziporphyrins, benzocarbaporphyrins, tropiporphyrins, and azuliporphyrins. In addition, meso-tetrarylporphyrinoids are available by using modified Lindsey–Rothemund reaction conditions. These porphyrin analogs show a broad spectrum of aromatic properties that range from nonaromatic benziporphyrins to highly diatropic systems such as benzocarbaporphyrins. Carbaporphyrinoid systems exhibit unusual reactivity and are often capable of generating organometallic derivatives under mild conditions. Many of the trends observed in the carbaporphyrinoid series were also evident for an expanded series of carbasapphyrins as well. Syntheses of dicarbaporphyrinoids have been investigated but it remains to be seen whether these studies will eventually lead to the formation of hydrocarbon analogs of the porphyrins.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Timothy D. Lash;Jessica A. El-Beck
European Journal of Organic Chemistry 2007 Volume 2007(Issue 24) pp:3981-3990
Publication Date(Web):25 JUL 2007
DOI:10.1002/ejoc.200700526
6-tert-Butyl- and 6-phenylazulene reacted with pyrrole and benzaldehyde in a molar ratio of 1:3:4 in the presence of BF3·Et2O in chloroform, followed by oxidation with DDQ, to give 23-substituted tetraphenylazuliporphyrins in 15–20 % yield. Slightly higher yields of the related meso-tetrakis(4-chlorophenyl)azuliporphyrins were obtained using 4-chlorobenzaldehyde. The presence of an electron-donating tert-butyl substituent increased the diatropic character of the azuliporphyrin system as determined by the proton NMR chemical shifts for the internal CH resonance, while intermediary results were noted for 23-phenylazuliporphyrins. Addition of TFA afforded dications with increased aromatic ring currents, but electron-donating substituents (tBu > Ph) again produced a larger upfield shift for the internal CH signal due to stabilization of the tropylium character that is required so that the system can attain carbaporphyrin-type aromaticity. The substituted azuliporphyrins reacted with nickel(II) acetate or palladium(II) acetate to give the corresponding organometallic derivatives. In addition, oxidations with tBuOOH and KOH afforded benzocarbaporphyrin products in approximately 50 % yield. The presence of tert-butyl or phenyl substituents did not block these oxidative ring contraction processes, and the rate of reaction was slightly increased compared to 23-unsubstituted azuliporphyrins. The major products were 22-tert-butyl or phenyl-substituted benzocarbaporphyrins and minor products with an additional formyl substituent were also isolated. These products are consistent with an initial nucleophilic addition occurring at the position adjacent to the R group on the azulene ring. Detailed mechanisms are proposed to explain these observations. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Jessica A. El-Beck;Timothy D. Lash
European Journal of Organic Chemistry 2007 Volume 2007(Issue 24) pp:
Publication Date(Web):2 AUG 2007
DOI:10.1002/ejoc.200790057
The cover picture shows the oxidative ring contraction of tetraphenylazuliporphyrins to give benzocarbaporphyrins. Bulky substituents on the azulene moiety influence the regioselectivity but do not inhibit this unusual chemistry. Details are discussed in the article by J. A. El-Beck and T. D. Lash on p. 3981 ff.
Co-reporter:Kae Miyake and Timothy D. Lash
Chemical Communications 2004 (Issue 2) pp:178-179
Publication Date(Web):08 Dec 2003
DOI:10.1039/B313229N
Acid catalyzed condensation of resorcinol or 2-methylresorcinol with 2 equiv. of an acetoxymethylpyrrole gave bis(pyrrolylmethyl)benzene derivatives in moderate yields; these afforded a series of novel aromatic benziporphyrins using the MacDonald “3 + 1” methodology.
Benzo[1,2-c:3,4-c']bis[1,2,5]selenadiazole, [1,2,5]selenadiazolo-[3,4-e]-2,1,3-benzothiadiazole, furazanobenzo-2,1,3-thiadiazole, furazanobenzo-2,1,3-selenadiazole and related heterocyclic systems
Co-reporter:Catherine M. Cillo;Timothy D. Lash
Journal of Heterocyclic Chemistry 2004 Volume 41(Issue 6) pp:955-962
Publication Date(Web):11 MAR 2009
DOI:10.1002/jhet.5570410616
The first synthesis of benzo[1,2-c:3,4-c']bis[1,2,5]selenadiazole has been developed starting from commercially available 4-nitrobenzo-2,1,3-selenadiazole. Improved syntheses of the related heterocycles [1,2,5]selenadiazolo[3,4-e]-2,1,3-benzothiadiazole, furazanobenzo-2,1,3-thiadiazole and furazanobenzo-2,1,3-selenadiazole are also reported.
Co-reporter:Denise A. Colby;Gregory M. Ferrence Dr.;Timothy D. Lash Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 11) pp:
Publication Date(Web):27 JAN 2004
DOI:10.1002/anie.200353189
Abnormal upfield shifts for the external azulene protons are seen in the 1H NMR spectra on oxidation of azuliporphyrins with copper(II) acetate to give highly distorted copper(II) complexes (see structure, brown Cu, blue N, red O, green H). These undergo demetalation with 10 % trifluoroacetic acid/chloroform to give unique cross-conjugated ketones.
Co-reporter:Denise A. Colby;Gregory M. Ferrence Dr.;Timothy D. Lash Dr.
Angewandte Chemie 2004 Volume 116(Issue 11) pp:
Publication Date(Web):27 JAN 2004
DOI:10.1002/ange.200353189
Anomale Hochfeldverschiebungen der äußeren Azulen-Protonen wurden bei der Oxidation von Azuliporphyrinen mit Kupfer(II)-acetat zu hochgradig verzerrten Kupfer(II)-Komplexen beobachtet (siehe Struktur; braun Cu, blau N, rot O, grün H). Demetallierung mit 10 % Trifluoressigsäure in Chloroform führte zu ungewöhnlichen kreuzkonjugierten Ketonen.
Co-reporter:Timothy D. Lash;Denise A. Colby;Gregory M. Ferrence
European Journal of Organic Chemistry 2003 Volume 2003(Issue 23) pp:
Publication Date(Web):19 NOV 2003
DOI:10.1002/ejoc.200300530
Azulene has been shown to react with pyrrole and a series of aromatic aldehydes in the presence of boron trifluoride etherate to give meso-tetraarylazuliporphyrins 6. Good yields of azuliporphyrins were obtained for benzaldehyde, 4-chlorobenzaldehyde, 4-bromobenzaldehyde, and 4-iodobenzaldehyde, and under dilute conditions p-tolualdehyde gave respectable yields. In each case, substantial amounts of meso-tetraarylporphyrins were also formed and a minor fraction of carbaporphyrin by-products could be detected, but otherwise no other macrocyclic products could be identified. 4-Nitrobenzaldehyde gave relatively poor yields of the corresponding azuliporphyrin, while p-anisaldehyde only gave trace amounts of product. Pentafluorobenzaldehyde gave variable results, although in this case a large number of additional by-products were identified including N-fused pentaphyrin, hexaphyrin, and higher order porphyrinoids, but no expanded azulene-containing macrocycles could be detected. Azuliporphyrins undergo reversible nucleophilic substitution on the seven-membered ring with pyrrolidine, benzenethiol, hydrazine, or benzylamine to give carbaporphyrin adducts. This property appears to facilitate an oxidative ring contraction of azuliporphyrins 6 with tert-butyl hydroperoxide in the presence of potassium hydroxide to produce mixtures of benzocarbaporphyrins 19 and 20. Tetraaryl-benzocarbaporphyrins exhibit slightly reduced diatropic ring currents compared to their meso-unsubstituted counterparts, although their UV/Vis spectra are very porphyrin-like and exhibit strong Soret bands near 450 nm. The benzocarbaporphyrins undergo reversible protonation to give monocationic and dicationic species. The latter involves C-protonation to generate an internal CH2 within the macrocyclic cavity. X-ray crystallography of tetraphenylbenzocarbaporphyrin 19a confirms that the preferred tautomer has the two NHs on either side of the indene subunit, in agreement with previous theoretical and spectroscopic studies. In addition, the presence of phenyl substituents at the 5,20-positions was found to tilt the indene moiety substantially by 27.4(1)° relative to the [18]annulene substructure. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:Dachun Liu and Timothy D. Lash
Chemical Communications 2002 (Issue 20) pp:2426-2427
Publication Date(Web):23 Sep 2002
DOI:10.1039/B206276N
Modified tripyrranes incorporating furan and thiophene rings were found to condense with benzene, pyridine and indene dialdehydes to give a series of novel porphyrin analogues, including thia- and oxa-carbaporphyrins; the latter readily forms nickel(II) and palladium(II) organometallic complexes.
Co-reporter:Shelley R. Graham, Gregory M. Ferrence and Timothy D. Lash
Chemical Communications 2002 (Issue 8) pp:894-895
Publication Date(Web):25 Mar 2002
DOI:10.1039/B200131B
Brief treatment of azuliporphyrins 5 with nickel(II) or palladium(II) acetate in refluxing DMF afforded excellent yields of the related chelates; these novel organometallic compounds retain cross-conjugated borderline aromatic structures as judged by UV-Vis and NMR spectroscopy, and X-ray crystallography.
Co-reporter:Denise A. Colby;Timothy D. Lash Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 23) pp:
Publication Date(Web):27 NOV 2002
DOI:10.1002/1521-3765(20021202)8:23<5397::AID-CHEM5397>3.0.CO;2-D
Electrophilic substitution of azulene has recently been shown to provide the means by which carbon–carbon bonds can be generated to form novel macrocyclic systems such as calixazulenes. These studies inspired us to develop a “one-pot” Rothemund-type synthesis of meso-tetraphenylazuliporphyrin. Azuliporphyrins, a group of cross-conjugated carbaporphyrinoids that exhibit intriguing chemistry and metallation properties, have previously only been available by multistep syntheses. In this work, azulene, pyrrole and benzaldehyde were shown to react in a 1:3:4 ratio in the presence of boron trifluoride etherate to give meso-tetraphenylazuliporphyrin 7 a. The free base shows only a minor diatropic ring current, but addition of TFA generates the related dication which shows greatly enhanced diatropicity where the internal CH shifts from δ=+3.35 to −0.5 ppm. Addition of pyrrolidine to 7 a gave rise to a carbaporphyrin adduct which showed a porphyrin-like UV/Vis spectrum and the internal CH shifted further upfield to give a resonance near δ=−5.7 ppm. Treatment of 7 a with tert-butyl hydroperoxide in the presence of potassium hydroxide afforded a mixture of benzocarbaporphyrins 9 a–c. These tetraphenylcarbaporphyrins were fully aromatic by NMR spectroscopy and gave typical porphyrin-type UV/Vis spectra with a strong Soret band near 446 nm. This new methodology makes these important porphyrin analogues readily available for further study.
Co-reporter:Shelley R. Graham;Denise A. Colby;Timothy D. Lash Dr.
Angewandte Chemie 2002 Volume 114(Issue 8) pp:
Publication Date(Web):16 APR 2002
DOI:10.1002/1521-3757(20020415)114:8<1429::AID-ANGE1429>3.0.CO;2-3
Ein Back-to-Front-Ansatz für die Synthese von Azuliporphyrinen aus einem Azulipyrran 1 wurde entwickelt. Mit dieser Methode gelang erstmals die rationale Synthese des Dicarbaporphyrinoids 2.
Co-reporter:Shelley R. Graham;Denise A. Colby;Timothy D. Lash Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 8) pp:
Publication Date(Web):16 APR 2002
DOI:10.1002/1521-3773(20020415)41:8<1371::AID-ANIE1371>3.0.CO;2-Q
A back-to-front synthesis of azuliporphyrins from an azulitripyrrane (1) has been developed. This methodology has allowed the first rational synthesis of the dicarbaporphyrinoid system 2.
Co-reporter:Timothy D. Lash Dr.
Angewandte Chemie 2000 Volume 112(Issue 10) pp:
Publication Date(Web):15 MAY 2000
DOI:10.1002/(SICI)1521-3757(20000515)112:10<1833::AID-ANGE1833>3.0.CO;2-5
Co-reporter:Venkata A. K. Adiraju, Gregory M. Ferrence and Timothy D. Lash
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 44) pp:NaN10533-10533
Publication Date(Web):2016/10/18
DOI:10.1039/C6OB02052F
Azuliporphyrins are intriguing porphyrin analogues that incorporate an azulene ring in place of a pyrrolic unit. This system undergoes regioselective oxidation reactions and favors the formation of stable organometallic derivatives. Reaction of meso-unsubstituted azuliporphyrins with Co2(CO)8 or CoCl2·6H2O gave 21-oxyazuliporphyrins, while Cu(OAc)2 produced the corresponding copper(II) complexes. Treatment of an oxyazuliporphyrin with Ni(OAc)2 or Pd(OAc)2 afforded analogous nickel(II) and palladium(II) derivatives. Silver(I) acetate in pyridine reacted with azuliporphyrins to give moderate yields of silver(III) benzocarbaporphyrins, and the prevalence of structures with a formyl moiety at the sterically crowded 21-position suggested that the ring contraction reactions were triggered in part by intramolecular attack from an axial peroxide ligand. Related thiaazuliporphyrins reacted with palladium(II) acetate to give palladium(II) benzothiacarbaporphyrins but this chemistry did not give rise to structures with 21-formyl groups, suggesting that the ring contraction reactions occurred by a different mechanistic pathway. These results demonstrate the existence of a rich tapestry of oxidation and metalation reactions for azuliporphyrin systems.
Co-reporter:Timothy D. Lash, Alexandra M. Young, Amber L. Von Ruden and Gregory M. Ferrence
Chemical Communications 2008(Issue 47) pp:NaN6311-6311
Publication Date(Web):2008/11/07
DOI:10.1039/B816057K
N-substituted pyrazoledialdehydes are shown to react with a tripyrrane under ‘3 + 1’ conditions to give aza-analogues of the N-confused porphyrins; these novel porphyrinoids show borderline aromatic properties and readily afford organometallic derivatives with Ni(OAc)2 and Pd(OAc)2.
Co-reporter:Deyaa I. AbuSalim and Timothy D. Lash
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 43) pp:NaN8736-8736
Publication Date(Web):2014/09/18
DOI:10.1039/C4OB01659A
The conformations of a series of benziporphyrins, naphthiporphyrins, oxybenziporphyrins, and related structures were minimized using DFT-B3LYP/6-311++G(d,p). The relative stabilities of the tautomers for each series were calculated using M06-2X and B3LYP-D functionals, and bond lengths were obtained. The diatropic properties of each species were assessed by calculating nucleus independent chemical shifts (NICS) and the related NICSzz values. Although benziporphyrin and naphthiporphyrin tautomers essentially exhibit no aromatic properties, mono- and diprotonated species show weakly diatropic characteristics in agreement with experimental observations. Benziphlorins, intermediates in the MacDonald “3 + 1” synthesis of benziporphyrins, were also examined and tautomers with methylene units adjacent to the benzene ring were shown to be far more stable than tautomers with a CH2 bridge between two pyrrolic units. 2-Hydroxybenziporphyrin was shown to be significantly less stable than the aromatic tautomer oxybenziporphyrin, although diprotonation leads to a species with somewhat reduced diatropicity. Related systems were also analyzed and the results demonstrate that benziporphyrins and naphthiporphyrins range from structures with no measurable macrocyclic aromaticity to strongly aromatic systems that exhibit large diamagnetic ring currents.
Co-reporter:Timothy D. Lash, Jessica A. El-Beck and Gregory M. Ferrence
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 2) pp:NaN329-329
Publication Date(Web):2013/11/12
DOI:10.1039/C3OB41992D
A series of hetero-azuliporphyrins have been prepared by the “3 + 1” variant on the MacDonald condensation. Azulitripyrranes with tert-butyl and phenyl substituents reacted with thiophene or selenophene dialdehydes in the presence of TFA to give, following an oxidation step, thia- and selena-azuliporphyrins in 45–55% yield. Two of these compounds gave crystals suitable for X-ray crystallographic analysis and the data were consistent with the presence of a 17-atom delocalization pathway. The hetero-azuliporphyrins have significant diatropic character that is enhanced by the presence of an electron-donating tert-butyl substituent. The aromatic character is further increased in polar solvents such as DMSO, which are believed to stabilize dipolar resonance contributors with 18π electron delocalization pathways. Protonation also greatly increases the diatropic characteristics of these macrocycles. The porphyrinoids underwent an oxidative ring contraction with t-BuOOH–KOH to give moderate yields of benzoheterocarbaporphyrins. Reaction of azulitripyrranes with 2,5-furandicarbaldehyde afforded oxa-azuliporphyrins, a class of carbaporphyrinoids that had previously been inaccessible. These “missing links” in the study of heteroazuliporphyrins were isolated as the dihydrochloride salts. Protonated oxa-azuliporphyrins are stable aromatic compounds, but the free base forms underwent rapid decomposition in solution.
Co-reporter:Ruoshi Li, Gregory M. Ferrence and Timothy D. Lash
Chemical Communications 2013 - vol. 49(Issue 68) pp:NaN7539-7539
Publication Date(Web):2013/07/12
DOI:10.1039/C3CC44248A
MacDonald “2 + 2” condensation of a 1,2′-dipyrrylmethane dialdehyde with a 2,2′-dipyrrylmethane afforded a neo-confused porphyrin in 55% yield. In addition, a novel dihydroporphyrin with two appended pyrrolic units was isolated and structurally characterized.
Co-reporter:Deyaa I. AbuSalim and Timothy D. Lash
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 48) pp:NaN8323-8323
Publication Date(Web):2013/10/31
DOI:10.1039/C3OB42063A
The conformations of eight neo-confused porphyrin (neo-CP) structures and seven related benzo-neo-confused porphyrins (B-neo-CPs) were minimized using DFT-B3LYP/6-311++G(d,p). In addition, in order to provide contrasts to the B-neo-CP tautomers, a series of twelve benzoporphyrin (BP) tautomers and twelve benzocarbaporphyrins (BCPs) were also analyzed, and two N-confused porphyrin (NCP) tautomers were also considered. The relative stability of the tautomers for each series was computed, and the bond lengths and bond angles were calculated. Surprisingly, all of the neo-CP and B-neo-CP tautomers were near planar. The fully conjugated members of these series showed a significant degree of bond length alternation, unlike porphyrins or the aromatic forms of NCP, BP or BCP. The most stable tautomer of neo-CP was calculated to be 27.04 kcal mol−1 higher in energy than porphyrin and 9.95 kcal mol−1 higher in energy than the most favored NCP tautomer. The most stable tautomer of B-neo-CP was similarly shown to be 26.40 kcal mol−1 higher in energy than benzoporphyrin. The favored tautomers also place the internal NH opposite to the neo-confused ring to minimize steric and lone pair–lone pair interactions, and to provide improved hydrogen bonding interactions. Nucleus independent chemical shifts (NICS) demonstrated that the fully conjugated neo-CP and B-neo-CP tautomers exhibit a significant amount of diatropic character, but these values are somewhat lower than the results obtained for porphyrins, BPs or BCPs. BCPs have three internal hydrogens, but the favored tautomer places the two pyrrolic hydrogens on each side of the indene unit to maximize hydrogen bonding interactions. Tautomers with an internal CH2 unit are also feasible, but these are energetically less favorable, have reduced diatropicity, and show significant bond length alternation. The results indicate that 18π electron delocalization pathways that pass through a fused benzene ring are less favorable than alternative delocalization pathways that bypass this unit.
Co-reporter:Alexandra M. Young and Timothy D. Lash
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 39) pp:NaN6848-6848
Publication Date(Web):2013/09/03
DOI:10.1039/C3OB41506F
Reaction of 2,6-pyridinedicarbaldehyde with a series of tripyrranes in the presence of trifluoroacetic acid, followed by oxidation of a pyriphlorin intermediate with silver(I) acetate, gave 6-oxopyriphlorins in moderate to good yields. The oxophlorin analogues were nonaromatic porphyrinoids that gave bright green colored solutions. Protonation with TFA afforded species that were attributed to mono- and dicationic structures. The proton NMR spectra in TFA–CDCl3 showed downfield shifts to the meso-protons and upfield shifts to the interior NH resonances that indicated the presence of a weakly diatropic dicationic species. Reaction of a 6-oxopyriphlorin with nickel(II) acetate in DMF or palladium(II) acetate in acetonitrile afforded the corresponding metallo-derivatives.
Co-reporter:Timothy D. Lash, Deyaa I. AbuSalim and Gregory M. Ferrence
Chemical Communications 2015 - vol. 51(Issue 88) pp:NaN15955-15955
Publication Date(Web):2015/09/10
DOI:10.1039/C5CC06890H
Base-catalyzed condensation of dicyclopentadienylmethane with a dipyrrylmethane dialdehyde gave a dicarbachlorin with an internal CH2 group. This unusual porphyrinoid retained highly diatropic characteristics and exhibited a porphyrin-like UV-vis spectrum.
Co-reporter:Alexandra M. Young, Amber L. Von Ruden and Timothy D. Lash
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 18) pp:NaN6305-6305
Publication Date(Web):2011/06/15
DOI:10.1039/C1OB05603D
A series of porphyrin analogues with pyrazole rings replacing one of the usual pyrrole subunits have been synthesized. This was accomplished by reacting 1-phenyl, 1-methyl and 1-ethyl pyrazole-1,3-dicarbaldehydes with a tripyrrane in the presence of TFA, followed by an oxidation step. The initially formed phlorin product was sufficiently stable for the N-phenyl system to be isolated and characterized, although the related N-alkyl phlorin analogues were less stable. Attempts to dehydrogenate the intermediary phlorins with DDQ resulted in decomposition, but the N-alkylphlorins could be oxidized with 0.2% aqueous ferric chloride solutions. Although the phenyl-substituted phlorin could not be oxidized under these conditions, it did afford the pyrazoloporphyrin upon treatment with silver acetate under acidic conditions. Oxidations with silver acetate also afforded oxophlorin analogues where the oxo-linkage was selectively formed at the 5-position. The pyrazole-containing porphyrin analogues are cross-conjugated and exhibit only a small degree of diatropic character. The internal CH resonances were observed between 5.27 and 5.87 ppm, while the external meso-protons fell into a range of 6.84–7.88 ppm. The borderline overall aromatic character was attributed to dipolar resonance contributors. Protonation considerably increased the diatropicity and the diprotonated dications formed from these porphyrin analogues gave the internal CH resonance at upfield values of 2.65–3.20 ppm. The aromatic character was enhanced by the presence of an electron-donating alkyl substituent on the nitrogen compared to the phenyl-substituted species. The pyrazoloporphyrins reacted with nickel(II) acetate in DMF, or palladium(II) acetate in acetonitrile, to give the corresponding organometallic derivatives. The metal complexes showed increased diatropic character but protonation afforded nonaromatic cations. The oxophlorin analogues were also nonaromatic in the free base and protonated forms. This work extends our understanding of carbaporphyrinoid systems and provides the first detailed studies on pyrazole-containing porphyrin analogues.
Co-reporter:Venkata A. K. Adiraju, Gregory M. Ferrence and Timothy D. Lash
Dalton Transactions 2016 - vol. 45(Issue 35) pp:NaN13694-13694
Publication Date(Web):2016/08/10
DOI:10.1039/C6DT03093A
Treatment of a benzocarbaporphyrin with [Rh(CO)2Cl]2 in refluxing dichloromethane gave a rhodium(I) dicarbonyl complex, and further reaction in refluxing pyridine afforded an organometallic rhodium(III) derivative. The carbaporphyrin also reacted with [Ir(COD)Cl]2 and pyridine in refluxing p-xylene to generate a related iridium(III) compound. These novel metalated porphyrinoids retained strongly diatropic characteristics and were fully characterized by XRD.

![23,24,25-TRIAZAPENTACYCLO[16.3.1.13,6.18,11.113,16]PENTACOSA-1(22),2,4,6(25),7,9,11,13(23),14,16,18,20-DODECAEN-19-OL](http://img.cochemist.com/ccimg/393900/393860-70-3.png)
![23,24,25-TRIAZAPENTACYCLO[16.3.1.13,6.18,11.113,16]PENTACOSA-1(22),2,4,6(25),7,9,11,13(23),14,16,18,20-DODECAEN-19-OL](http://img.cochemist.com/ccimg/393900/393860-70-3_b.png)
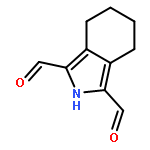
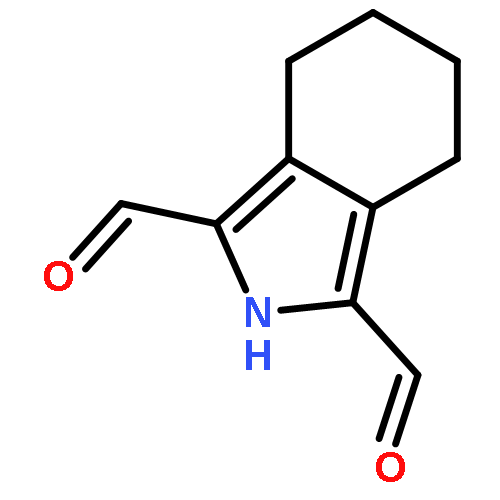
![2H-Dibenz[e,g]isoindole-1,3-dicarboxaldehyde](http://img.cochemist.com/ccimg/180200/180199-08-0.png)
![2H-Dibenz[e,g]isoindole-1,3-dicarboxaldehyde](http://img.cochemist.com/ccimg/180200/180199-08-0_b.png)
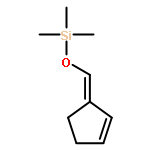

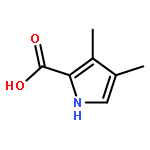
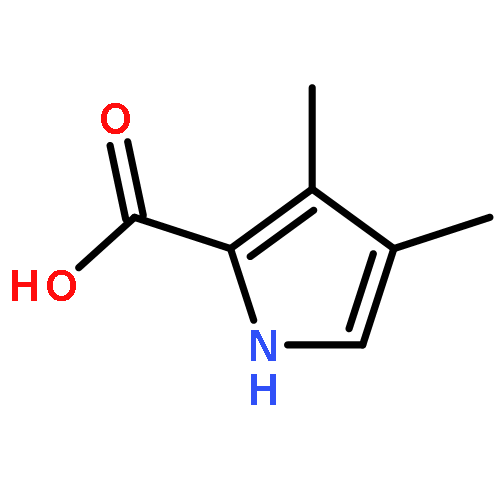






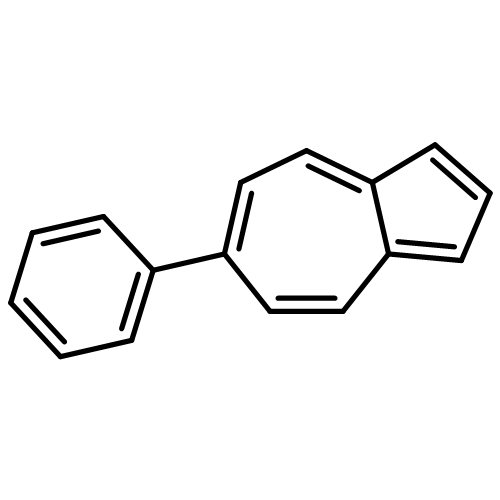
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
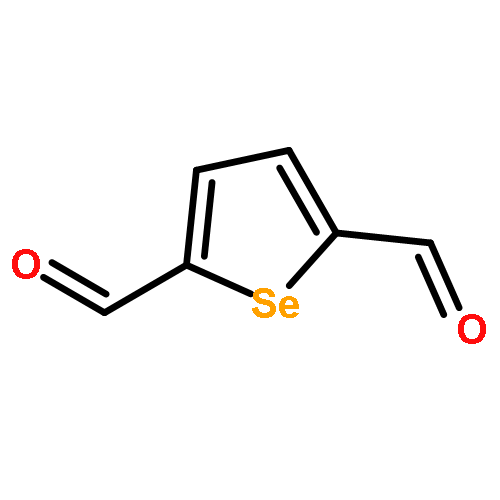
![5-[(5-CARBOXY-3-ETHYL-4-METHYL-1H-PYRROL-2-YL)METHYL]-4-ETHYL-3-METHYL-1H-PYRROLE-2-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/7700/7670-41-9.png)
![5-[(5-CARBOXY-3-ETHYL-4-METHYL-1H-PYRROL-2-YL)METHYL]-4-ETHYL-3-METHYL-1H-PYRROLE-2-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/7700/7670-41-9_b.png)