Co-reporter:Bing Xue, Wei Wang, Jiang-Jiang Qin, Bhavitavya Nijampatnam, ... Eugenia Kharlampieva
Acta Biomaterialia 2017 Volume 58(Volume 58) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.actbio.2017.06.004
We report a novel delivery platform for a highly potent anticancer drug, 7-(benzylamino)-3,4-dihydro-pyrrolo[4,3,2-de]quinolin-8(1H)-one (BA-TPQ), using pH- and redox-sensitive poly(methacrylic acid) (PMAA) hydrogel cubes of micrometer size as the encapsulating matrix. The hydrogels are obtained upon cross-linking PMAA with cystamine in PMAA/poly(N-vinylpyrrolidone) multilayers assembled within mesoporous sacrificial templates. The BA-TPQ-loaded hydrogels maintain their cubical shape and pH-sensitivity after lyophilization, which is advantageous for long-term storage. Conversely, the particles degrade in vitro in the presence of glutathione (5 mM) providing 80% drug release within 24 h. Encapsulating BA-TPQ into hydrogels significantly increases its transport via Caco-2 cell monolayers used as a model for oral delivery where the apparent permeability of BA-TPQ-hydrogel cubes was ∼2-fold higher than that of BA-TPQ. BA-TPQ-hydrogel cubes exhibit better anticancer activity against HepG2 (IC50 = 0.52 µg/mL) and Huh7 (IC50 = 0.29 µg/mL) hepatoma cells with a 40% decrease in the IC50 compared to the non-encapsulated drug. Remarkably, non-malignant liver cells have a lower sensitivity to BA-TPQ-hydrogel cubes with 2-fold increased IC50 values compared to those of cancer cells. In addition, encapsulating BA-TPQ in the hydrogels amplifies the potency of the drug via down-regulation of MDM2 oncogenic protein and upregulation of p53 (a tumor suppressor) and p21 (cell proliferation suppressor) expression in HepG2 liver cancer cells. Moreover, enhanced inhibition of MDM2 protein expression by BA-TPQ-hydrogel cubes is independent of p53 status in Huh7 cells. This drug delivery platform of non-spherical shape provides a facile method for encapsulation of hydrophobic drugs and can facilitate the enhanced efficacy of BA-TPQ for liver cancer therapy.Statement of SignificanceMany potent anticancer drugs are hydrophobic and lack tumor selectivity, which limits their application in cancer therapy. Although cubical hydrogels of poly(methacrylic acid) exhibit excellent biocompatibility and versatility, they have not been investigated for hydrophobic drug delivery due to poor mechanical stability and incompatibility between hydrophobic drugs and a hydrophilic hydrogel network. In this study, we provide a facile method to prepare a multilayer hydrogel-based platform with controlled nanostructure, cubical shape and redox-responsiveness for delivery of highly potent anticancer therapeutics, hydrophobic BA-TPQ. The BA-TPQ-hydrogel cubes have exceptional structural stability upon lyophilization which is advantageous for a long-term storage. The greatly enhanced trans-epithelial permeability and amplified anti-tumor activity of BA-TPQ are achieved by encapsulation in these hydrogel cubes. Furthermore, the anticancer BA-TPQ-hydrogel platform retains the selective activity of BA-TPQ to hepatocellular carcinoma cells. Overall, the produced BA-TPQ-hydrogel cubes demonstrate a high potential for clinical liver cancer therapy.Download high-res image (84KB)Download full-size image
Co-reporter:Bhavitavya Nijampatnam, Luke Casals, Ruowen Zheng, Hui Wu, Sadanandan E. Velu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3508-3513
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.06.033
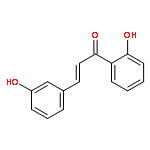
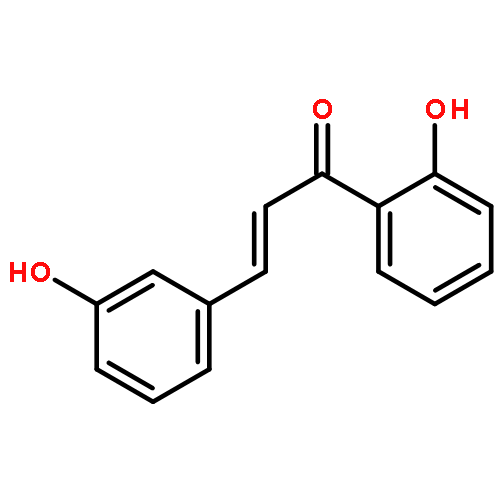
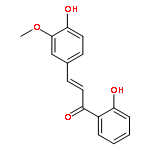
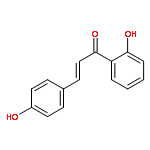
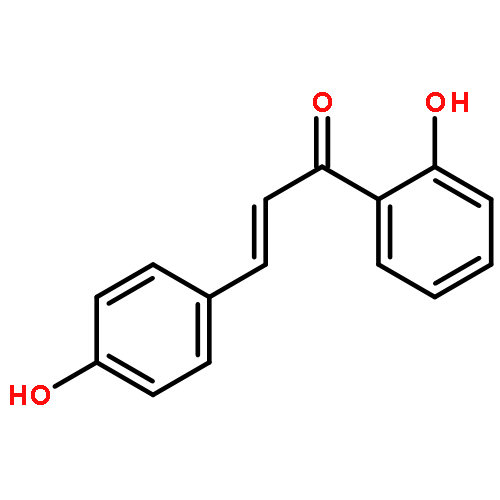
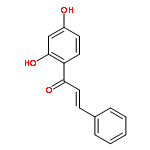
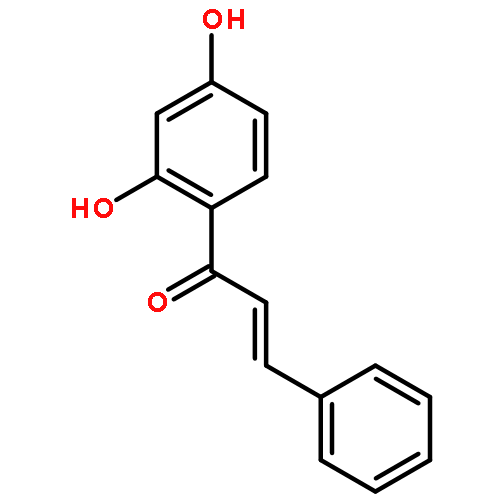
•Low micromolar inhibitors of S. mutans biofilm and Gtf are identified.•The lead compounds are E and Z isomers of hydroxychalcones.•The Z isomers of chalcones produced more potent anti-biofilm activities.•Chalcones with a 3-OH group on ring A showed better selectivity for biofilm inhibition.•These compounds do not affect bacterial growth.Streptococcus mutans has been implicated as the major etiological agent in the initiation and the development of dental caries due to its robust capacity to form tenacious biofilms. Ideal therapeutics for this disease will aim to selectively inhibit the biofilm formation process while preserving the natural bacterial flora of the mouth. Several studies have demonstrated the efficacies of flavonols on S. mutans biofilms and have suggested the mechanism of action through their effect on S. mutans glucosyltransferases (Gtfs). These enzymes metabolize sucrose into water insoluble and soluble glucans, which are an integral measure of the dental caries pathogenesis. Numerous studies have shown that flavonols and polyphenols can inhibit Gtf and biofilm formation at millimolar concentrations. We have screened a group of 14 hydroxychalcones, synthetic precursors of flavonols, in an S. mutans biofilm assay. Several of these compounds emerged to be biofilm inhibitors at low micro-molar concentrations. Chalcones that contained a 3-OH group on ring A exhibited selectivity for biofilm inhibition. Moreover, we synthesized 6 additional analogs of the lead compound and evaluated their potential activity and selectivity against S. mutans biofilms. The most active compound identified from these studies had an IC50 value of 44 μM against biofilm and MIC50 value of 468 μM against growth displaying >10-fold selectivity inhibition towards biofilm. The lead compound displayed a dose dependent inhibition of S. mutans Gtfs. The lead compound also did not affect the growth of two commensal species (Streptococcus sanguinis and Streptococcus gordonii) at least up to 200 μM, indicating that it can selectively inhibit cariogenic biofilms, while leaving commensal and/or beneficial microbes intact. Thus non-toxic compounds have the potential utility in public oral health regimes.
Co-reporter:Wei Wang;Bhavitavya Nijampatnam
Frontiers of Chemical Science and Engineering 2016 Volume 10( Issue 1) pp:1-15
Publication Date(Web):2016 March
DOI:10.1007/s11705-016-1562-6
Natural products and their derivatives represent a rich source for the discovery and development of new cancer therapeutic drugs. Bioactive components derived from natural sources including marine compounds have been shown to be effective agents in the clinic or in preclinical settings. In the present review, we present a story of discovery, synthesis and evaluation of three synthetic tricyclic pyrroloquinone (TPQ) alkaloid analogs as cancer therapeutic agents. Chemical synthesis of these compounds (BA-TPQ, TBA-TPQ, and TCBA-TPQ) has been accomplished and the mechanisms of action (MOA) and structure-activity relationships (SAR) have been investigated. In the past, the complexity of chemical synthesis and the lack of well-defined MOA have dampened the enthusiasm for the development of some makaluvamines. Recent discovery of novel molecular targets for these alkaloids (unrelated to inhibition of Topoisomerase II) warrant further consideration as clinical candidates in the future. In addition to the establishment of novel synthetic approaches and demonstration of in vitro and in vivo anticancer activities, we have successfully demonstrated that these makaluvamines attack several key molecular targets, including the MDM2-p53 pathway, providing ample opportunities of modulating the compound structure based on SAR and the use of such compounds in combination therapy in the future.
Co-reporter:Su Xu, Thao Nguyen, Irene Pomilio, Maria C. Vitale, Sadanandan E. Velu
Tetrahedron 2014 70(35) pp: 5928-5933
Publication Date(Web):
DOI:10.1016/j.tet.2014.06.021
Co-reporter:Dwayaja H. Nadkarni, Srinivasan Murugesan, Sadanandan E. Velu
Tetrahedron 2013 69(20) pp: 4105-4113
Publication Date(Web):
DOI:10.1016/j.tet.2013.03.052
Co-reporter:Bala Chandra Chenna, Jason R. King, Bidhan A. Shinkre, Amanda L. Glover, Aaron L. Lucius, Sadanandan E. Velu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 9) pp:3752-3761
Publication Date(Web):September 2010
DOI:10.1016/j.ejmech.2010.05.024
Synthetic methods have been developed for lead Sortase A inhibitors identified from previous studies. Several derivatives of the lead inhibitor were synthesized to derive preliminary structure activity relationships (SAR). Different regions of the lead inhibitor that are critical for the enzyme activity have been determined by systematic SAR studies. The E stereochemistry of the lead compound was found to be critical for its activity. Replacement of the E double bond with Z double bond or a rigid triple bond reduced the enzyme inhibitory activity in most cases. Reduction of the double bond to a C–C single bond resulted in complete loss of activity. Amide carbonyl and NH groups were also found to be crucial for the activity of this class of inhibitors, as well. The morpholine ring oxygen atom was also found to be an important factor for the activity of the lead inhibitor. Preliminary SAR studies led to the identification of compounds with improved enzyme inhibition. The most active compound was found to have an IC50 value of 58 μM against the enzyme.Synthesis and structure activity relationship studies of a lead structural template of Staphylococcus aureus Sortase A inhibitor are described.
Co-reporter:Yun J. Lee;Jason R. King;Bala Chandra Chenna
Journal of Chemical Crystallography 2009 Volume 39( Issue 12) pp:
Publication Date(Web):2009 December
DOI:10.1007/s10870-009-9588-y
Details of the synthesis and crystal structure determination of (E)-2-(7-(3-(thiophen-2-yl)acrylamido)-2,3-dihydro-5-oxobenzo[e][1,4]oxazepin-1(5H)-yl)ethyl acetate are presented. The compound crystallizes in the triclinic P−1 space group (a = 8.3377(17), b = 9.792(2), c = 12.469(3) Å, α = 96.39(3)°, β = 108.50(3)°, γ = 97.68(3)°, V = 943.9(3) Å3, Z = 2). Interesting features of the structure include intermolecular hydrogen bonding between the amide proton on one molecule and the carbonyl oxygen of the dihydrooxazepinone ring on an adjacent molecule, the boat conformation of the dihydrooxazepinone ring, and π-π stacking between thiophene and phenyl rings on adjacent molecules with a distance between centroids of 3.79(2) Å.
Co-reporter:Srinivasan Murugesan, Dwayaja H. Nadkarni, Sadanandan E. Velu
Tetrahedron Letters 2009 50(25) pp: 3074-3076
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.021
Co-reporter:Bidhan A. Shinkre, Kevin P. Raisch, Liming Fan, Sadanandan E. Velu
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 5) pp:2541-2549
Publication Date(Web):1 March 2008
DOI:10.1016/j.bmc.2007.11.051
Analogs of marine alkaloid, makaluvamine, bearing substituted benzyl and substituted phenethyl side chains have been synthesized and their antiproliferative activities have been evaluated. 4-Methyl, 4-chloro, and 4-fluoro substituted benzyl analogs possessed pronounced antiproliferative effects on the breast cancer cell line, MCF-7 at IC50 values of 2.3 μM, 1.8 μM, and 2.8 μM, respectively. 4-Methyl, 4-chloro, and 3,4-methylenedioxy derivatives showed the best activity against MCF-7 among the phenethyl analogs with IC50 values of 2.3 μM, 2.8 μM, and 2.4 μM, respectively. In general, methoxy substitutions resulted in slight loss in activity in both benzyl and phenethyl series. Benzyl, 4-fluorobenzyl, 3,4-dimethoxyphenethyl, and 3,4-methylenedioxyphenethyl analogs were tested by NCI in their 60 cell lines in vitro human cancer cell screen. All four compounds showed excellent inhibition against several tested cancer cell lines. Benzyl and 4-fluorobenzyl analogs were relatively more active than 3,4-dimethoxy phenethyl and 3,4-methylenedioxy phenethyl analogs. In NCI assays, the best Log GI50 values were shown by the fluorobenzyl analog against the renal cancer cell line RXF-393 (<−8.0 M) and dimethoxy phenethyl analog against the CNS cancer cell line, SF-268 (<−8.0 M). The best Log LC50 value was shown by the fluorobenzyl analog against the breast cancer cell line MCF-7 (−6.01 M).
Co-reporter:Bala Chandra Chenna, Bidhan A. Shinkre, Jason R. King, Aaron L. Lucius, Sthanam V.L. Narayana, Sadanandan E. Velu
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 1) pp:380-385
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmcl.2007.10.051
In-silico virtual screening of bacterial surface enzyme Staphylococcus aureus Sortase A against commercial compound libraries using FlexX software package has led to the identification of novel inhibitors. Inhibition of enzyme catalytic activity was determined by monitoring the steady state cleavage of a model peptide substrate. Preliminary structure activity relationship studies on the lead compound resulted in the identification of compounds with improved activity. The most active compound has an IC50 value of 58 μM against the enzyme.A novel class of inhibitors of Staphylococcus aureus Sortase A is discovered by in-silico virtual screening and structure activity relationship studies.
Co-reporter:Bala Chandra Chenna;Bidhan A. Shinkre;Shweta Patel
Journal of Chemical Crystallography 2008 Volume 38( Issue 3) pp:189-194
Publication Date(Web):2008 March
DOI:10.1007/s10870-007-9288-4
Synthesis, separation and X-ray crystal structures of E and Z isomers of 3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylic acid are reported. Separation of E and Z isomers from a 1:1 mixture has been carried out by selective acidification of their sodium salts carefully controlling the pH of the solution. The structures of E and Z isomers were confirmed by determining crystal structures of these isomers using single crystal X-ray diffraction. The E isomer crystallizes in the P21/c space group with a = 11.493(2) Å, b = 5.5456(11) Å, c = 24.900(5) Å, α = 90°, β = 92.36(3)°, γ = 90°, Z = 4. The Z isomer crystallizes in the P21/c space group with a = 10.192(2) Å, b = 12.893(3) Å, c = 13.948(3) Å, α = 90°, β = 92.18(3)°, γ = 90°, Z = 4. Details of the synthesis and structural characterization and X-ray crystal structure determination of the title compounds are presented.Synthesis, Separation and Crystal Structures of E and Z Isomers of 3-(2,5-Dimethoxyphenyl)-2-(4-Methoxyphenyl)Acrylic AcidBalachandra Chenna, Bidhan A. Shinkre, Shweta Patel, Samuel B. Owens Jr., Gary M. Gray, Sadanandan E. Velu*
Co-reporter:Bidhan A. Shinkre;Dwayaja H. Nadkarni
Journal of Chemical Crystallography 2008 Volume 38( Issue 3) pp:205-209
Publication Date(Web):2008 March
DOI:10.1007/s10870-007-9290-x
Details of the synthesis of the E isomer of 3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylonitrile, and the X-ray crystal structures of both the E and Z isomers of this compound are presented. The E isomer crystallizes in the P21/c space group with cell parameters, a = 8.5659(17) Å, b = 16.399(3) Å, c = 11.224(2) Å, α = 90°, β = 95.27(3)°, γ = 90°and Z = 4. The Z isomer crystallizes in the Pca21 space group with cell parameters, a = 4.1223(8) Å, b = 19.113(4) Å, c = 19.453(4) Å, α = 90°, β = 90°, γ = 90° and Z = 4.
Co-reporter:Bidhan A. Shinkre, Kevin P. Raisch, Liming Fan, Sadanandan E. Velu
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 10) pp:2890-2893
Publication Date(Web):15 May 2007
DOI:10.1016/j.bmcl.2007.02.065
Twelve analogs of makaluvamines have been synthesized. These compounds were evaluated for their ability to inhibit the enzyme topoisomerase II. Five compounds were shown to inhibit topoisomerase catalytic activity comparable to two known topoisomerase II targeting control drugs, etoposide and m-AMSA. Their cytotoxicity against human colon cancer cell line HCT-116 and human breast cancer cell lines MCF-7 and MDA-MB-468 has been evaluated. Four makaluvamine analogs exhibited better IC50 values against HCT-116 as compared to control drug etoposide. One analog exhibited better IC50 value against HCT-116 as compared to m-AMSA. All 12 of the makaluvamine analogs exhibited better IC50 values against MCF-7 and MDA-MB-468 as compared to etoposide as well as m-AMSA.Synthesis, topoisomerase II inhibition, and anticancer activities of makaluvamine analogs are presented.

![7H-Indolo[3,2-j]phenanthridine-7,13(12H)-dione, 12-(phenylmethyl)-](http://img.cochemist.com/ccimg/827400/827340-35-2.png)
![7H-Indolo[3,2-j]phenanthridine-7,13(12H)-dione, 12-(phenylmethyl)-](http://img.cochemist.com/ccimg/827400/827340-35-2_b.png)
![12,13-Dihydro-7H-indolo[3,2-j]phenanthridine-7,13-dione](http://img.cochemist.com/ccimg/254200/254114-34-6.png)
![12,13-Dihydro-7H-indolo[3,2-j]phenanthridine-7,13-dione](http://img.cochemist.com/ccimg/254200/254114-34-6_b.png)
![12,13-Dihydro-7H-indolo[3,2-j]phenanthridine-7,13-dione 5-oxide](http://img.cochemist.com/ccimg/254200/254114-33-5.png)
![12,13-Dihydro-7H-indolo[3,2-j]phenanthridine-7,13-dione 5-oxide](http://img.cochemist.com/ccimg/254200/254114-33-5_b.png)





![2,4-Quinazolinediamine,5-methyl-6-[[(3,4,5-trimethoxyphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/52200/52128-35-5.png)
![2,4-Quinazolinediamine,5-methyl-6-[[(3,4,5-trimethoxyphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/52200/52128-35-5_b.png)
![10H-Phenothiazin-3-ol,8-chloro-10-[3-(dimethylamino)propyl]-](http://img.cochemist.com/ccimg/2100/2095-62-7.png)
![10H-Phenothiazin-3-ol,8-chloro-10-[3-(dimethylamino)propyl]-](http://img.cochemist.com/ccimg/2100/2095-62-7_b.png)

