Co-reporter:Keisuke Morisaki, Yusuke Sasano, Takahiro Koseki, Takuro Shibuta, Naoki Kanoh, Wen-Hua Chiou, and Yoshiharu Iwabuchi
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.orglett.7b02428
A divergent entry to the chiral bicyclo[5.3.0]decane skeletons relevant to sesqui- and higher terpenoids has been achieved. Its usefulness was demonstrated by formal synthesis of a guaiane sesquiterpenoid (−)-englerin A. The key reactions are (i) diastereoselective Nazarov cyclization for stereoselective construction of the bicyclo[5.3.0]decane skeleton, (ii) intramolecular C–H amination for tuning an oxidation state, and (iii) introduction of an alkyl group to a β-alkoxy ketone with a zinc(II) ate complex.
Co-reporter:Hisataka Kobayashi;Yusuke Sasano;Naoki Kanoh;Eunsang Kwon
European Journal of Organic Chemistry 2016 Volume 2016( Issue 2) pp:270-273
Publication Date(Web):
DOI:10.1002/ejoc.201501365
Abstract
The secondary metabolite, turkiyenine, is a probable bioactive component of the medicinal plant, Hypecoum procumbens L. Structurally, it is an isoquinoline-derived alkaloid composed of dihydrobenzofuran and 3-benzazepine-1-one moieties arranged in a unique spirocyclic structure. Yet despite the intriguing structure and medicinal relevance of benzofuran and benzazepine motifs, neither a synthetic study nor a biological evaluation of turkiyenine have been reported to date. Herein we communicate the first total synthesis of the proposed structure of turkiyenine, and on the basis of X-ray crystallography show that the proposed structure is incorrect. The key steps of our synthesis include an aza-spiro annulation onto a benzofuran using a rhodium–nitrenoid to construct a 3-alkyl-3-amino-2,3-dihydrobenzofuran, an acyl-alkylation of an aryne onto a β-ketolactam to form a 3-benzazepine-1-one, and chemoselective reduction of an amide in the presence of a ketone.
Co-reporter:Aki Kohyama, Naoki Kanoh, Eunsang Kwon, Yoshiharu Iwabuchi
Tetrahedron Letters 2016 Volume 57(Issue 5) pp:517-519
Publication Date(Web):3 February 2016
DOI:10.1016/j.tetlet.2015.12.049
The tricyclic polar segment of fusarisetin A, designed for preparing analogues for structure–activity relationship studies of the aliphatic segment thereof, has been constructed in an enantiocontrolled manner, featuring the Yamamoto asymmetric epoxidation of a homoallylic alcohol, C3-selective ring-opening of a 3,4-epoxy alcohol, stereocontrolled merger of a γ-lactone with Garner’s counterpart, and ruthenium-catalyzed ring-closing metathesis.
Co-reporter:Shun-ichiro Uesugi, Tsubasa Watanabe, Takamichi Imaizumi, Yu Ota, Keisuke Yoshida, Haruna Ebisu, Takumi Chinen, Yoko Nagumo, Masatoshi Shibuya, Naoki Kanoh, Takeo Usui, and Yoshiharu Iwabuchi
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12333-12350
Publication Date(Web):November 6, 2015
DOI:10.1021/acs.joc.5b02256
Irciniastatin A (a.k.a. psymberin) and irciniastatin B are members of the pederin natural product family, which have potent antitumor activity and structural complexity. Herein, we describe a full account of our total synthesis of (+)-irciniastatin A and (−)-irciniastatin B. Our synthesis features the highly regioselective Eu(OTf)3-catalyzed, DTBMP-assisted epoxide ring opening reaction with MeOH, which enabled a concise synthesis of the C1–C6 fragment, extensive use of AZADO (2-azaadamantane N-oxyl) and its related nitroxyl radical/oxoammonium salt-catalyzed alcohol oxidation throughout the synthesis, and a late-stage assembly of C1–C6, C8–C16, and C17–C25 fragments. In addition, for the synthesis of (−)-irciniastatin B, we achieved the C11-selective control of the oxidation stage via regioselective deprotection and AZADO-catalyzed alcohol oxidation. The synthetic irciniastatins showed high levels of cytotoxic activity against mammalian cells. Furthermore, chemical footprinting experiments using synthetic compounds revealed that the binding site of irciniastatins is the E-site of the ribosome.
Co-reporter:Dr. Yusuke Sasano;Naoki Kogure;Tomohiro Nishiyama;Shota Nagasawa ;Dr. Yoshiharu Iwabuchi
Chemistry – An Asian Journal 2015 Volume 10( Issue 4) pp:1004-1009
Publication Date(Web):
DOI:10.1002/asia.201403245
Abstract
The oxidation of alcohols into their corresponding carbonyl compounds is one of the most fundamental transformations in organic chemistry. In our recent report, 2-azaadamantane N-oxyl (AZADO)/copper catalysis promoted the highly chemoselective aerobic oxidation of unprotected amino alcohols into amino carbonyl compounds. Herein, we investigated the extension of the promising AZADO/copper-catalyzed aerobic oxidation of alcohols to other types of alcohol. During close optimization of the reaction conditions by using various alcohols, we found that the optimum combination of nitroxyl radical, copper salt, and solution concentration was dependent on the type of substrate. Various alcohols, including highly hindered and heteroatom-rich ones, were efficiently oxidized into their corresponding carbonyl compounds under mild conditions with lower amounts of the catalysts.
Co-reporter:Ryusuke Doi, Masatoshi Shibuya, Tsukasa Murayama, Yoshihiko Yamamoto, and Yoshiharu Iwabuchi
The Journal of Organic Chemistry 2015 Volume 80(Issue 1) pp:401-413
Publication Date(Web):December 4, 2014
DOI:10.1021/jo502426p
The development of 1,5-dimethyl-9-azanoradamantane N-oxyl (DMN-AZADO; 1,5-dimethyl-Nor-AZADO, 2) as an efficient catalyst for the selective oxidation of primary alcohols in the presence of secondary alcohols is described. The compact and rigid structure of the azanoradamantane nucleus confers potent catalytic ability to DMN-AZADO (2). A variety of hindered primary alcohols such as neopentyl primary alcohols were efficiently oxidized by DMN-AZADO (2) to the corresponding aldehydes, whereas secondary alcohols remained intact. DMN-AZADO (2) also has high catalytic efficiency for one-pot oxidation from primary alcohols to the corresponding carboxylic acids in the presence of secondary alcohols and for oxidative lactonization from diols.
Co-reporter:Keiichi Murakami ; Yusuke Sasano ; Masaki Tomizawa ; Masatoshi Shibuya ; Eunsang Kwon
Journal of the American Chemical Society 2014 Volume 136(Issue 50) pp:17591-17600
Publication Date(Web):November 20, 2014
DOI:10.1021/ja509766f
The development and characterization of enantioselective organocatalytic oxidative kinetic resolution (OKR) of racemic secondary alcohols using chiral alkoxyamines as precatalysts are described. A number of chiral alkoxyamines have been synthesized, and their structure–enantioselectivity correlation study in OKR has led us to identify a promising precatalyst, namely, 7-benzyl-3-n-butyl-4-oxa-5-azahomoadamantane, which affords various chiral aliphatic secondary alcohols (ee up to >99%, krel up to 296). In a mechanistic study, chlorine-containing oxoammonium species were identified as the active species generated in situ from the alkoxyamine precatalyst, and it was revealed that the chlorine atom is crucial for high reactivity and enantioselectivity. The present OKR is the first successful example applicable to various unactivated aliphatic secondary alcohols, including heterocyclic alcohols with high enantioselectivity, the synthetic application of which is demonstrated by the synthesis of a bioactive compound.
Co-reporter:Shun-ichiro Uesugi, Tsubasa Watanabe, Takamichi Imaizumi, Masatoshi Shibuya, Naoki Kanoh, and Yoshiharu Iwabuchi
Organic Letters 2014 Volume 16(Issue 17) pp:4408-4411
Publication Date(Web):August 27, 2014
DOI:10.1021/ol502264y
In our study of the total synthesis of (+)-irciniastatin A, we found a need to develop a method that enables a C3-selective nucleophilic ring opening of 2,3-epoxy alcohol by MeOH, by which we found that the use of combined catalytic amounts of Eu(OTf)3 and 2,6-di-tert-butyl-4-methylpyridine (DTBMP) enables the intended transformation to obtain 3-methoxy-1,2-diol efficiently. Promising features of a protocol that effects a highly regioselective nucleophilic ring opening of 2,3- and 3,4-epoxy alcohols using various nucleophiles including alcohols, thiols, and unprotected amines are described.
Co-reporter:Yusuke Sasano;Shota Nagasawa;Mai Yamazaki;Dr. Masatoshi Shibuya;Dr. Jaiwook Park;Dr. Yoshiharu Iwabuchi
Angewandte Chemie International Edition 2014 Volume 53( Issue 12) pp:3236-3240
Publication Date(Web):
DOI:10.1002/anie.201309634
Abstract
The direct oxidation of unprotected amino alcohols to their corresponding amino carbonyl compounds has often posed serious challenges in organic synthesis and has constrained chemists to adopting an indirect route, such as a protection/deprotection strategy, to attain their goal. Described herein is a highly chemoselective aerobic oxidation of unprotected amino alcohols to their amino carbonyl compounds in which 2-azaadamantane N-oxyl (AZADO)/copper catalysis is used. The catalytic system developed leads to the alcohol-selective oxidation of various unprotected amino alcohols, carrying a primary, secondary, or tertiary amino group, in good to high yield at ambient temperature with exposure to air, thus offering flexibility in the synthesis of nitrogen-containing compounds.
Co-reporter:Masatoshi Shibuya, Shota Nagasawa, Yuji Osada, and Yoshiharu Iwabuchi
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:10256-10268
Publication Date(Web):October 6, 2014
DOI:10.1021/jo501862k
The mechanism of an NOx-assisted, nitroxide(nitroxyl radical)-catalyzed aerobic oxidation of alcohols was investigated using a set of sterically and electronically modified nitroxides (i.e., TEMPO, AZADO (1), 5-F-AZADO (2), 5,7-DiF-AZADO (3), 5-MeO-AZADO (4), 5,7-DiMeO-AZADO (5), oxa-AZADO (6), TsN-AZADO (7), and DiAZADO (8)). The motivation for the present study stemmed from our previous observation that the introduction of an F atom at a remote position from the nitroxyl radical moiety on the azaadamantane nucleus effectively enhanced the catalytic activity under typical NOx-mediated aerobic-oxidation conditions. The kinetic profiles of the azaadamantane-N-oxyl-[AZADO (1)-, 5-F-AZADO (2)-, and 5,7-DiF-AZADO (3)]-catalyzed aerobic oxidations were closely investigated, revealing that AZADO (1) showed a high initial reaction rate compared to 5-F-AZADO (2) and 5,7-DiF-AZADO (3); however, AZADO-catalyzed oxidation exhibited a marked slowdown, resulting in ∼90% conversion, whereas 5-F-AZADO-catalyzed oxidation smoothly reached completion without a marked slowdown. The reasons for the marked slowdown and the role of the fluoro group are discussed. Oxa-AZADO (6), TsN-AZADO (7), and DiAZADO (8) were designed and synthesized to confirm their comparable catalytic efficiency to that of 5-F-AZADO (2), providing supporting evidence for the electronic effect on the catalytic efficiency of the heteroatoms under NOx-assisted aerobic-oxidation conditions.
Co-reporter:Yusuke Sasano;Shota Nagasawa;Mai Yamazaki;Dr. Masatoshi Shibuya;Dr. Jaiwook Park;Dr. Yoshiharu Iwabuchi
Angewandte Chemie 2014 Volume 126( Issue 12) pp:3300-3304
Publication Date(Web):
DOI:10.1002/ange.201309634
Abstract
The direct oxidation of unprotected amino alcohols to their corresponding amino carbonyl compounds has often posed serious challenges in organic synthesis and has constrained chemists to adopting an indirect route, such as a protection/deprotection strategy, to attain their goal. Described herein is a highly chemoselective aerobic oxidation of unprotected amino alcohols to their amino carbonyl compounds in which 2-azaadamantane N-oxyl (AZADO)/copper catalysis is used. The catalytic system developed leads to the alcohol-selective oxidation of various unprotected amino alcohols, carrying a primary, secondary, or tertiary amino group, in good to high yield at ambient temperature with exposure to air, thus offering flexibility in the synthesis of nitrogen-containing compounds.
Co-reporter:Muneo Kawasumi and Yoshiharu Iwabuchi
Organic Letters 2013 Volume 15(Issue 7) pp:1788-1790
Publication Date(Web):March 26, 2013
DOI:10.1021/ol4005239
The concise synthesis of 5-(4-hydroxybutyl)-2(5H)furanone has been accomplished from 9-oxabicyclo[4.2.1]non-7-en-1-ol on the basis of HTIB [PhI(OH)OTs, a.k.a. Koser’s reagent]-mediated novel oxidative fragmentation. Chiral (−)-(R)-5-(4-hydroxy-butyl)-2(5H)-furanone (>99% ee) was used for the formal total synthesis of (+)-dubiusamine A (1).
Co-reporter:Yusuke Sasano;Keiichi Murakami;Tomohiro Nishiyama;Dr. Eunsang Kwon;Dr. Yoshiharu Iwabuchi
Angewandte Chemie 2013 Volume 125( Issue 48) pp:12856-12859
Publication Date(Web):
DOI:10.1002/ange.201307144
Co-reporter:Yusuke Sasano;Keiichi Murakami;Tomohiro Nishiyama;Dr. Eunsang Kwon;Dr. Yoshiharu Iwabuchi
Angewandte Chemie International Edition 2013 Volume 52( Issue 48) pp:12624-12627
Publication Date(Web):
DOI:10.1002/anie.201307144
Co-reporter:Masatoshi Shibuya, Takuro Shibuta, Hayato Fukuda, and Yoshiharu Iwabuchi
Organic Letters 2012 Volume 14(Issue 19) pp:5010-5013
Publication Date(Web):September 19, 2012
DOI:10.1021/ol3021435
A mild and user-friendly one-pot oxidative cleavage of vicinal diols to their corresponding (di)carboxylic acids using AZADOs and PhI(OAc)2 is described. 1,2-Diols and 2,3-diols as well as 1,2,3-triol gave one- or two-carbon-unit-shorter carboxylic acids. Internal vicinal diols also smoothly underwent one-pot oxidative cleavage to afford the corresponding dicarboxylic acids. Cyclic vicinal diols are converted to their corresponding open-form dicarboxylic acids.
Co-reporter:Masatoshi Shibuya, Ryu̅suke Doi, Takuro Shibuta, Shun-ichiro Uesugi, and Yoshiharu Iwabuchi
Organic Letters 2012 Volume 14(Issue 19) pp:5006-5009
Publication Date(Web):September 19, 2012
DOI:10.1021/ol3021429
The organocatalytic one-pot oxidative cleavage of terminal 1,2-diols to one-carbon-unit-shorter carboxylic acids is described. The combination of 1-Me-AZADO (cat.), NaOCl (cat.), and NaClO2 caused smooth one-pot oxidative cleavage under mild conditions. A broad range of substrates including carbohydrates and N-protected amino diols were converted without epimerization. Terminal triols and tetraols respectively underwent cleavage of their C-2 and C-3 moieties to afford their corresponding two- and three-carbon-unit-shorter carboxylic acids.
Co-reporter:Aki Kohyama, Michihiro Fukuda, Shunsuke Sugiyama, Hiroyuki Yamakoshi, Naoki Kanoh, Chikashi Ishioka, Hiroyuki Shibata and Yoshiharu Iwabuchi
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 45) pp:NaN10687-10687
Publication Date(Web):2016/10/24
DOI:10.1039/C6OB01771A
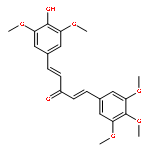
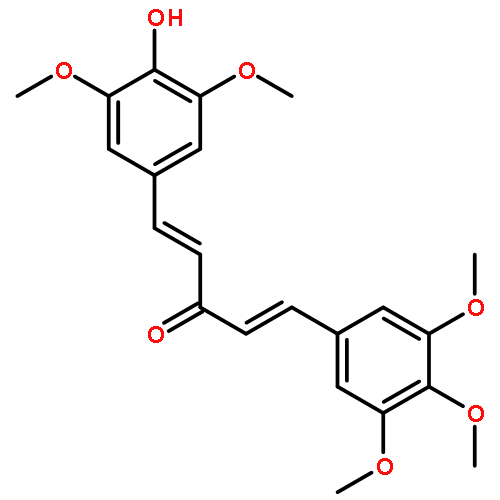
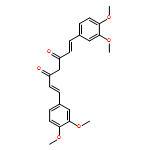
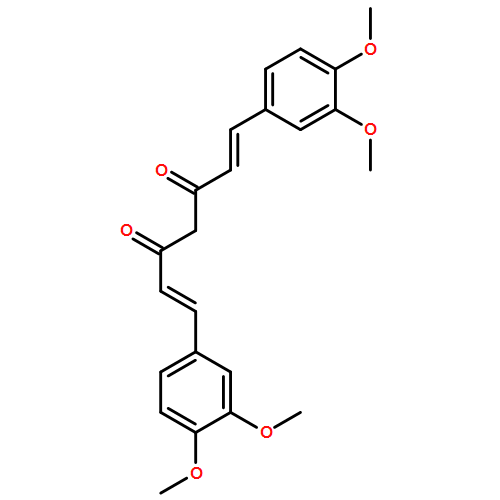
C5-curcuminoids [a.k.a. bis(arylmethylidene)acetones] are curcumin analogues bearing a reactive cross-conjugated dienone structure essential for eliciting cytotoxicity. To gain insight into the mode of action of C5-curcuminoids, we investigated the reversibility of the thia-Michael reaction of 1,5-bis(3,5-bis(methoxymethoxy)phenyl)-1,4-pentadiene-3-one, named GO-Y030 which is the most potent cytotoxic C5-curcuminoid, using spectroscopic methods. A panel of GO-Y030-bis-thiol-adducts were synthesized and the structure–reactivity relationship regarding the retro thia-Michael reaction as well as the cell growth inhibitory activity against human colon cancer HCT116 were evaluated. Some C5-curcuminoid thiol-adducts exhibited comparable cytotoxicity with GO-Y030, demonstrating their potential use as prodrugs. These results imply that C5-curcuminoids elicit cytotoxicity by covalently interacting with various biothiols via a reversible thia-Michael reaction.























![2-Butenoic acid, 4-[bis(phenylmethyl)amino]-, methyl ester, (2E)-](http://img.cochemist.com/ccimg/479700/479672-18-9.png)
![2-Butenoic acid, 4-[bis(phenylmethyl)amino]-, methyl ester, (2E)-](http://img.cochemist.com/ccimg/479700/479672-18-9_b.png)





![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-, (2S)-](/data/chemimg/105300/187458-29-3.png)
![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-, (2S)-](/data/chemimg/105300/187458-29-3_b.png)
![2-Butanone, 4-[4-(2-pyrimidinyl)-1-piperazinyl]-](http://img.cochemist.com/ccimg/185400/185310-86-5.png)
![2-Butanone, 4-[4-(2-pyrimidinyl)-1-piperazinyl]-](http://img.cochemist.com/ccimg/185400/185310-86-5_b.png)









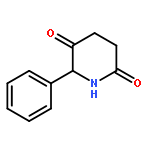
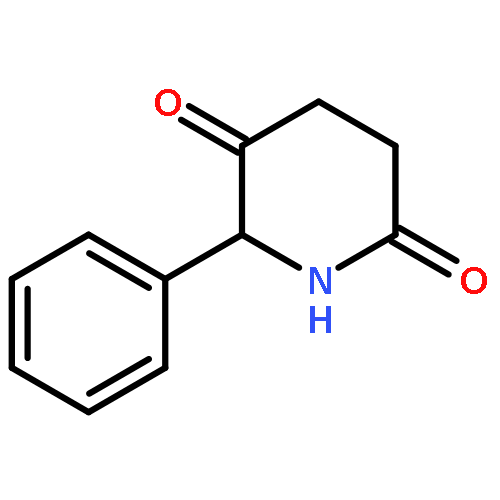


![Propanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/144700/144681-67-4.png)
![Propanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/144700/144681-67-4_b.png)










![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4_b.png)




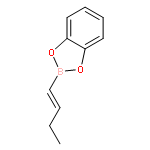
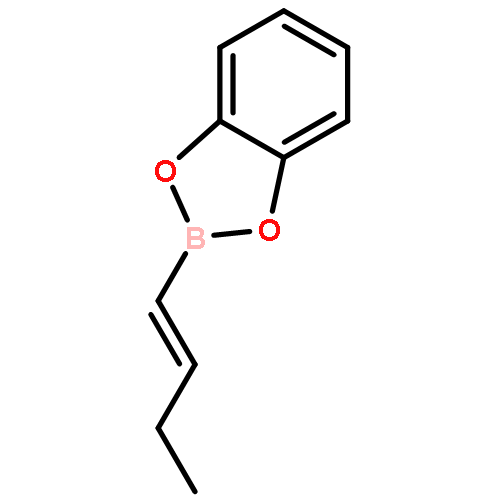
![Pyridine,1,2,3,4-tetrahydro-1-(phenylmethyl)-5-[1-(phenylmethyl)-2-piperidinyl]-](http://img.cochemist.com/ccimg/102700/102666-89-7.png)
![Pyridine,1,2,3,4-tetrahydro-1-(phenylmethyl)-5-[1-(phenylmethyl)-2-piperidinyl]-](http://img.cochemist.com/ccimg/102700/102666-89-7_b.png)





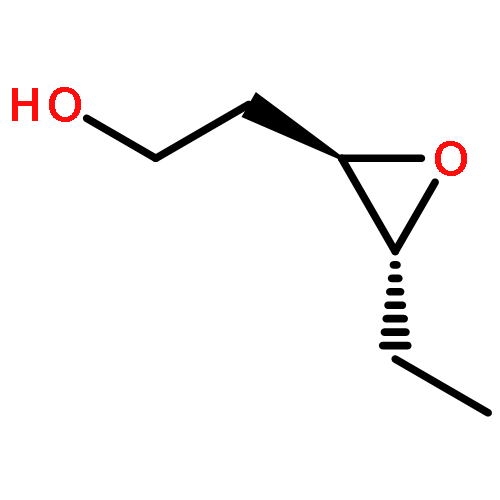
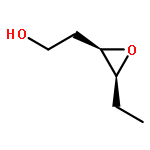
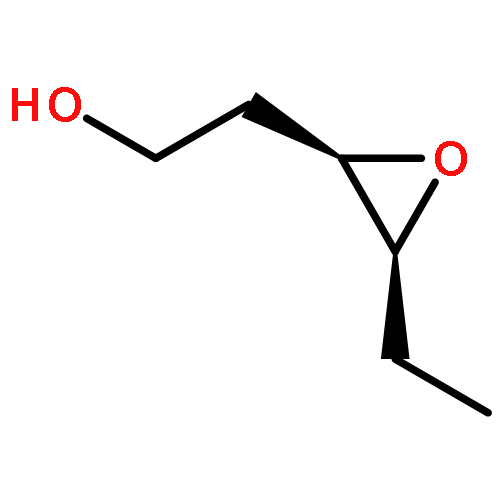


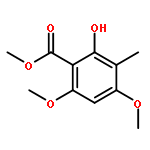
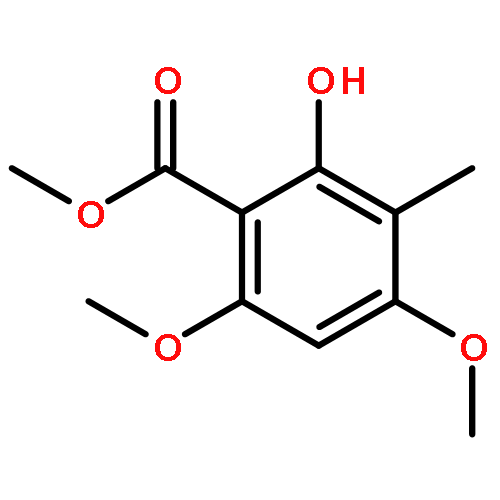
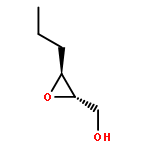



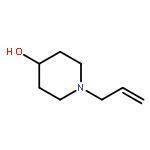
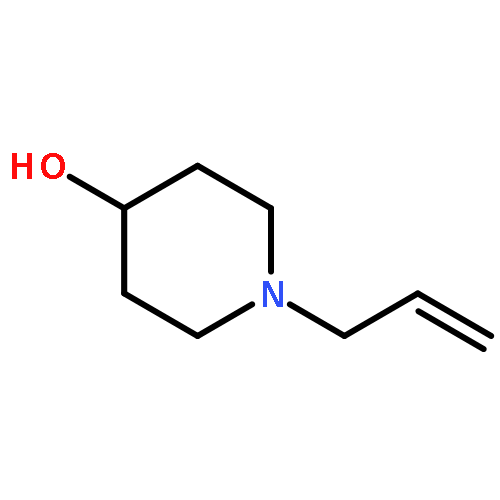
![8-Oxabicyclo[5.1.0]octan-2-ol, (1R,2R,7S)-rel-](http://img.cochemist.com/ccimg/65700/65697-35-0.png)
![8-Oxabicyclo[5.1.0]octan-2-ol, (1R,2R,7S)-rel-](http://img.cochemist.com/ccimg/65700/65697-35-0_b.png)






![Acetic acid, [(4-methoxyphenyl)methoxy]-, ethyl ester](http://img.cochemist.com/ccimg/58000/57938-80-4.png)
![Acetic acid, [(4-methoxyphenyl)methoxy]-, ethyl ester](http://img.cochemist.com/ccimg/58000/57938-80-4_b.png)







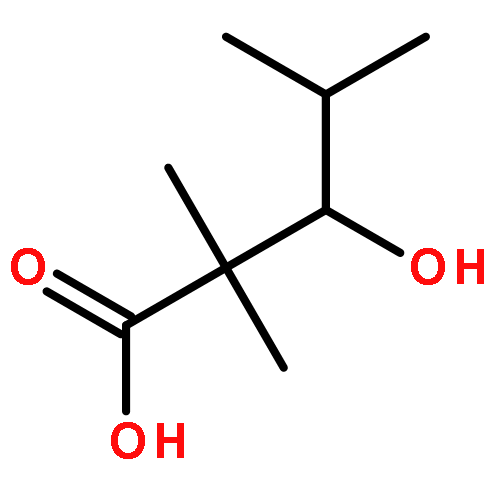
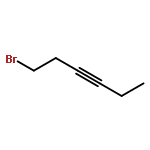

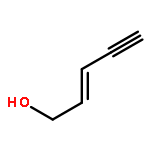
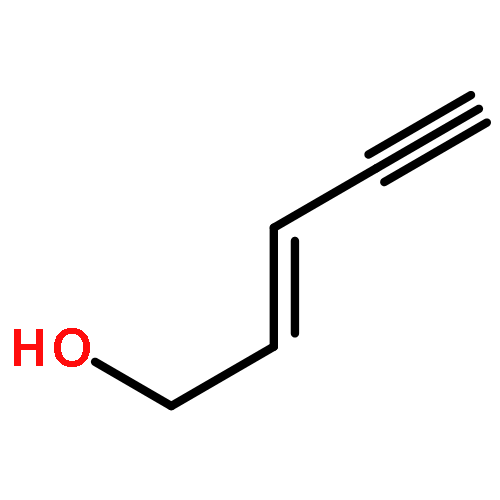


![1-Propanone, 3-(dimethylamino)-1-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/31900/31878-58-7.png)
![1-Propanone, 3-(dimethylamino)-1-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/31900/31878-58-7_b.png)
![9-Oxabicyclo[6.1.0]nonan-2-ol, (1R,2R,8S)-rel-](http://img.cochemist.com/ccimg/31900/31821-36-0.png)
![9-Oxabicyclo[6.1.0]nonan-2-ol, (1R,2R,8S)-rel-](http://img.cochemist.com/ccimg/31900/31821-36-0_b.png)

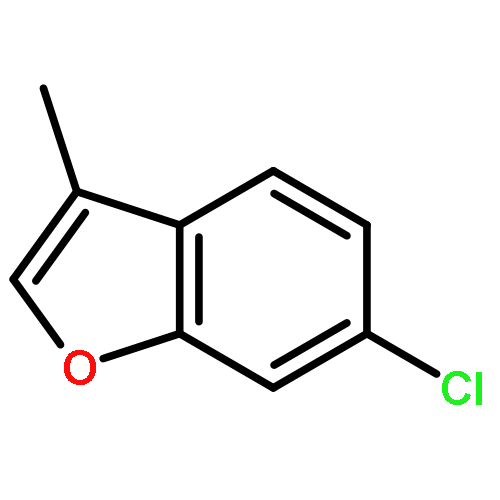
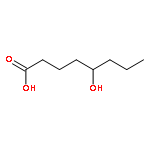
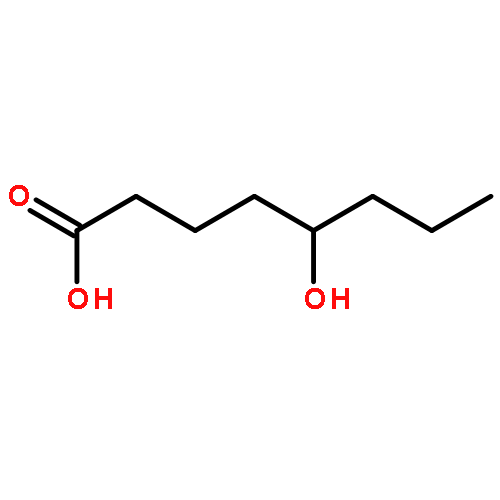
![7-Oxabicyclo[4.1.0]heptan-2-ol,(1R,2R,6S)-rel-](http://img.cochemist.com/ccimg/26900/26828-72-8.png)
![7-Oxabicyclo[4.1.0]heptan-2-ol,(1R,2R,6S)-rel-](http://img.cochemist.com/ccimg/26900/26828-72-8_b.png)






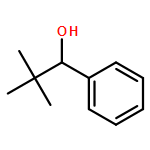
![4-Piperidinol, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/22000/21937-59-7.png)
![4-Piperidinol, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/22000/21937-59-7_b.png)
![Ethanone, 1-[5-(acetyloxy)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/21300/21222-04-8.png)
![Ethanone, 1-[5-(acetyloxy)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/21300/21222-04-8_b.png)




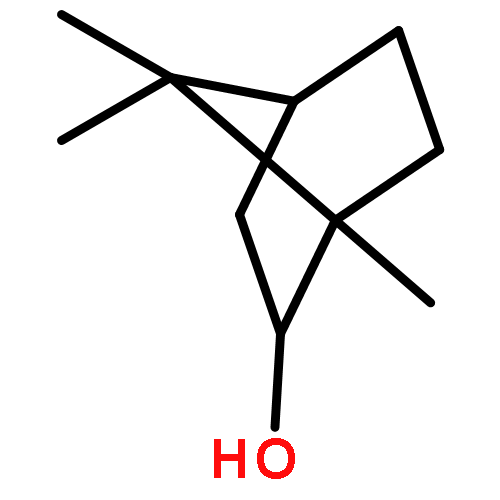
![Bicyclo[2.2.1]heptan-2-ol, 1,7,7-trimethyl-, (1S,2S,4S)-](http://img.cochemist.com/ccimg/16800/16725-71-6.png)
![Bicyclo[2.2.1]heptan-2-ol, 1,7,7-trimethyl-, (1S,2S,4S)-](http://img.cochemist.com/ccimg/16800/16725-71-6_b.png)
![1-Propanone,2-[(dimethylamino)methyl]-2-methyl-1-phenyl-](http://img.cochemist.com/ccimg/15500/15451-29-3.png)
![1-Propanone,2-[(dimethylamino)methyl]-2-methyl-1-phenyl-](http://img.cochemist.com/ccimg/15500/15451-29-3_b.png)
![Benzenemethanol, a-[2-(dimethylamino)-1,1-dimethylethyl]-](http://img.cochemist.com/ccimg/15500/15451-15-7.png)
![Benzenemethanol, a-[2-(dimethylamino)-1,1-dimethylethyl]-](http://img.cochemist.com/ccimg/15500/15451-15-7_b.png)







![Benzene, 1-methoxy-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/4100/4039-83-2.png)
![Benzene, 1-methoxy-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/4100/4039-83-2_b.png)








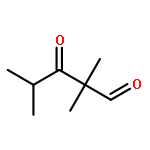
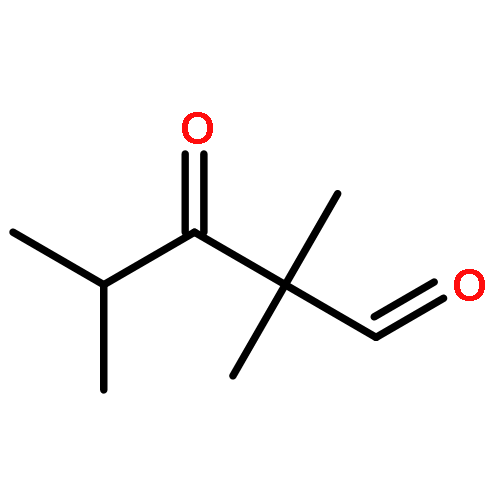


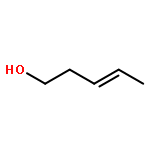
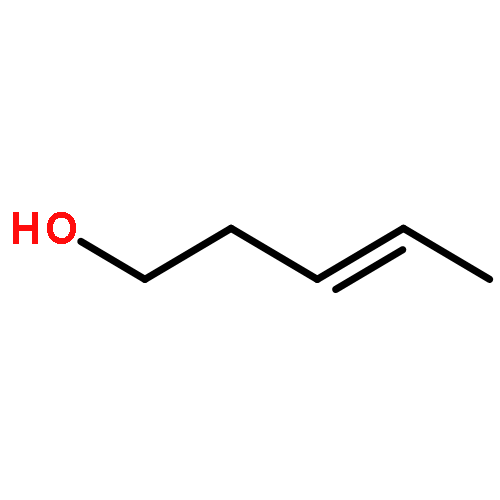



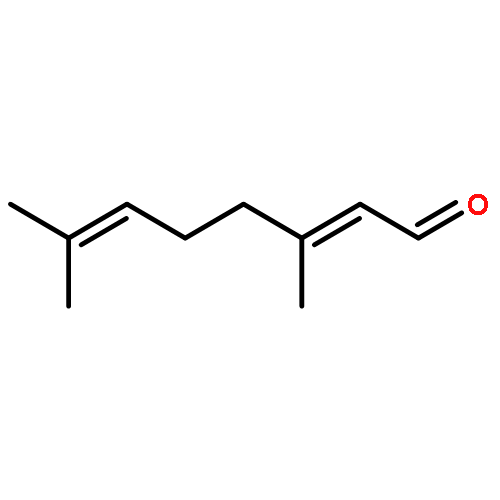
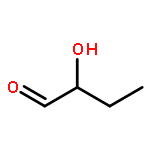
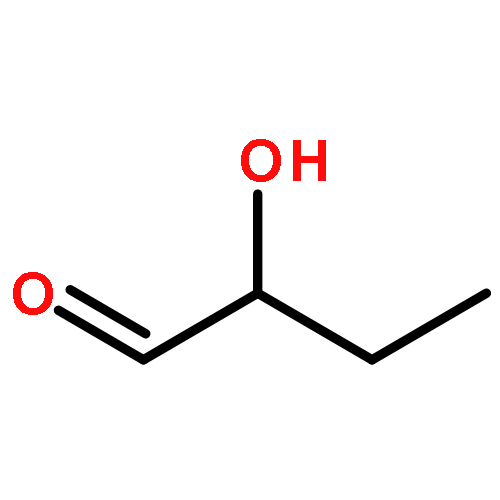




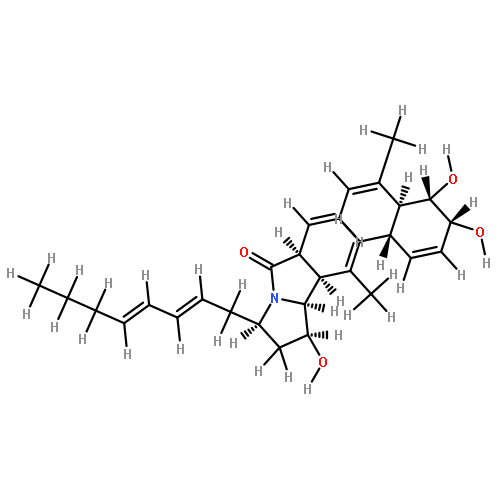
![(3E,5E,7E,9R,10S,11Z,13E,15E,17E,20R)-9,10-DIHYDROXY-7,15-DIMETHYL-20-[(2E,4E)-2,4-OCTADIEN-1-YL]AZACYCLOICOSA-3,5,7,11,13,15,17-HEPTAEN-2-ONE](/data/chemimg/417200/1257083-94-5.png)
![(3E,5E,7E,9R,10S,11Z,13E,15E,17E,20R)-9,10-DIHYDROXY-7,15-DIMETHYL-20-[(2E,4E)-2,4-OCTADIEN-1-YL]AZACYCLOICOSA-3,5,7,11,13,15,17-HEPTAEN-2-ONE](/data/chemimg/417200/1257083-94-5_b.png)
![1,4-Pentadien-3-one, 1,5-bis[3,5-bis(methoxymethoxy)phenyl]-,(1E,4E)-](http://img.cochemist.com/ccimg/917900/917813-62-8.png)
![1,4-Pentadien-3-one, 1,5-bis[3,5-bis(methoxymethoxy)phenyl]-,(1E,4E)-](http://img.cochemist.com/ccimg/917900/917813-62-8_b.png)

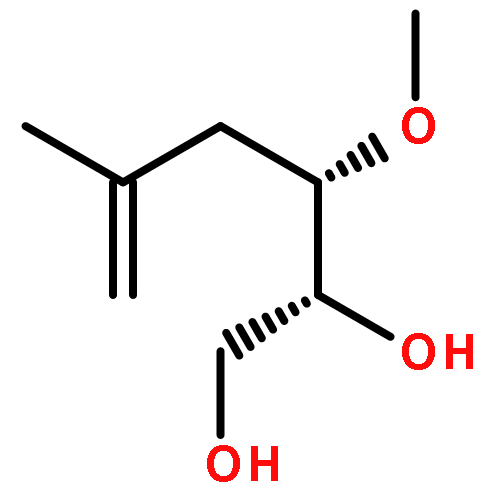
![2-Butenal, 4-[(4-methoxyphenyl)methoxy]-, (2E)-](http://img.cochemist.com/ccimg/842200/842125-04-6.png)
![2-Butenal, 4-[(4-methoxyphenyl)methoxy]-, (2E)-](http://img.cochemist.com/ccimg/842200/842125-04-6_b.png)


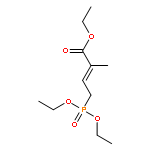
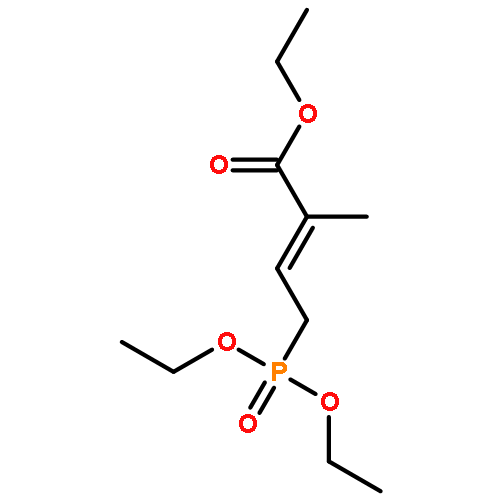
![Oxacyclohexadeca-3,5,9,13-tetraen-2-one,8-hydroxy-3,5,7-trimethyl-16-[(1S,3E,6S,7R,8S,9R,10S)-6,8,10-trihydroxy-1,7,9-trimethyl-3-undecen-1-yl]-,(3E,5E,7R,8R,9E,13E,16S)-](http://img.cochemist.com/ccimg/145200/145177-62-4.png)
![Oxacyclohexadeca-3,5,9,13-tetraen-2-one,8-hydroxy-3,5,7-trimethyl-16-[(1S,3E,6S,7R,8S,9R,10S)-6,8,10-trihydroxy-1,7,9-trimethyl-3-undecen-1-yl]-,(3E,5E,7R,8R,9E,13E,16S)-](http://img.cochemist.com/ccimg/145200/145177-62-4_b.png)


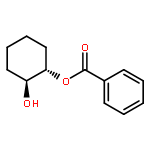
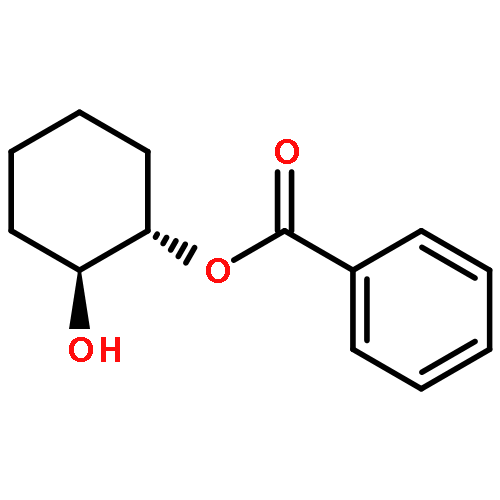
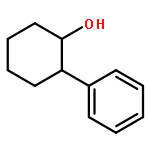
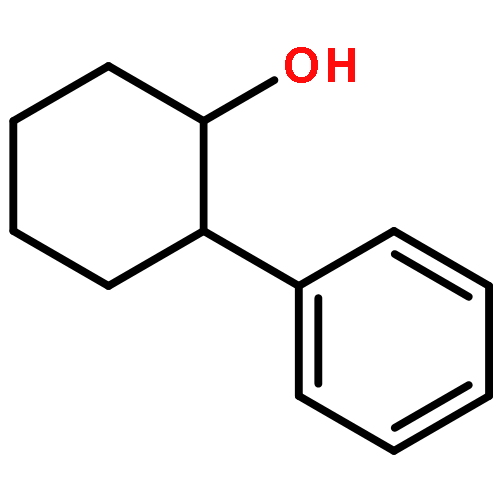
![Silane, (1,1-dimethylethyl)[(2E)-2-penten-4-ynyloxy]diphenyl-](http://img.cochemist.com/ccimg/81700/81677-37-4.png)
![Silane, (1,1-dimethylethyl)[(2E)-2-penten-4-ynyloxy]diphenyl-](http://img.cochemist.com/ccimg/81700/81677-37-4_b.png)
![Spiro[bicyclo[3.3.1]nonane-3,2'-[1,3]dioxolan]-7-one](http://img.cochemist.com/ccimg/66100/66077-97-2.png)
![Spiro[bicyclo[3.3.1]nonane-3,2'-[1,3]dioxolan]-7-one](http://img.cochemist.com/ccimg/66100/66077-97-2_b.png)
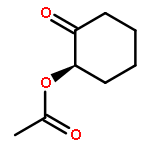
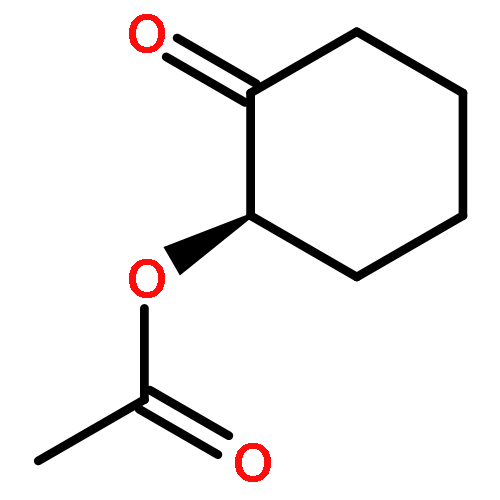





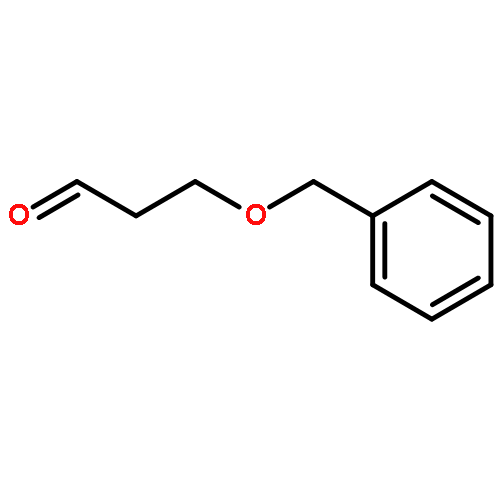
![7-Methylenebicyclo[3.3.1]nonan-3-one](http://img.cochemist.com/ccimg/18000/17933-29-8.png)
![7-Methylenebicyclo[3.3.1]nonan-3-one](http://img.cochemist.com/ccimg/18000/17933-29-8_b.png)


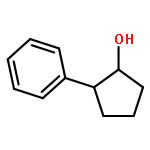
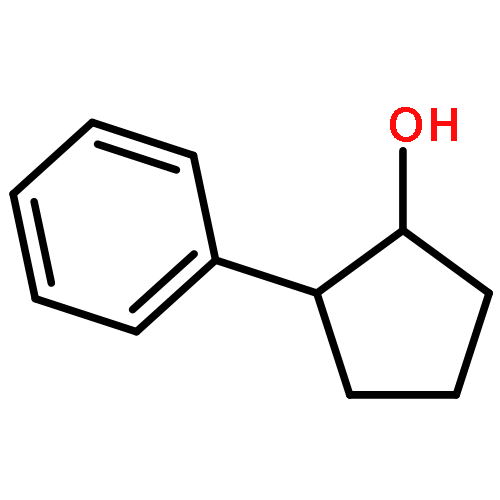
![8,17-Dioxabicyclo[14.1.0]heptadeca-4,10,12-trien-9-one,7-[(1S,3E,6S,7R,8R,9S,10S)-6,8-dihydroxy-10-methoxy-1,7,9-trimethyl-3-undecen-1-yl]-2,15-dihydroxy-10,12,14-trimethyl-,(1S,2R,4E,7S,10E,12E,14R,15S,16S)-](http://img.cochemist.com/ccimg/142400/142383-53-7.png)
![8,17-Dioxabicyclo[14.1.0]heptadeca-4,10,12-trien-9-one,7-[(1S,3E,6S,7R,8R,9S,10S)-6,8-dihydroxy-10-methoxy-1,7,9-trimethyl-3-undecen-1-yl]-2,15-dihydroxy-10,12,14-trimethyl-,(1S,2R,4E,7S,10E,12E,14R,15S,16S)-](http://img.cochemist.com/ccimg/142400/142383-53-7_b.png)


![2-Azatricyclo[3.3.1.13,7]dec-2-yloxy, 1-methyl-](http://img.cochemist.com/ccimg/872600/872598-44-2.png)
![2-Azatricyclo[3.3.1.13,7]dec-2-yloxy, 1-methyl-](http://img.cochemist.com/ccimg/872600/872598-44-2_b.png)
![2-Azatricyclo[3.3.1.1<sup>3,7</sup>]dec-2-yloxidanyl](http://img.cochemist.com/ccimg/57700/57625-08-8.png)
![2-Azatricyclo[3.3.1.1<sup>3,7</sup>]dec-2-yloxidanyl](http://img.cochemist.com/ccimg/57700/57625-08-8_b.png)