Co-reporter:Abhinan Shyamsunder;Dr. Witali Beichel;Petra Klose;Quan Pang;Dr. Harald Scherer;Dr. Anke Hoffmann; Graham K. Murphy; Ingo Krossing; Linda F. Nazar
Angewandte Chemie 2017 Volume 129(Issue 22) pp:6288-6293
Publication Date(Web):2017/05/22
DOI:10.1002/ange.201701026
AbstractThe step-change in gravimetric energy density needed for electrochemical energy storage devices to power unmanned autonomous vehicles, electric vehicles, and enable low-cost clean grid storage is unlikely to be provided by conventional lithium ion batteries. Lithium–sulfur batteries comprising lightweight elements provide a promising alternative, but the associated polysulfide shuttle in typical ether-based electrolytes generates loss in capacity and low coulombic efficiency. The first new electrolyte based on a unique combination of a relatively hydrophobic sulfonamide solvent and a low ion-pairing salt, which inhibits the polysulfide shuttle, is presented. This system behaves as a sparingly solvating electrolyte at slightly elevated temperatures, where it sustains reversible capacities as high as 1200–1500 mAh g−1 over a wide range of current density (2C–C/5, respectively) when paired with a lithium metal anode, with a coulombic efficiency of >99.7 % in the absence of LiNO3 additive.
Co-reporter:Mario Schleep;Clarissa Hettich;Daniel Kratzert;Harald Scherer
Chemical Communications 2017 vol. 53(Issue 79) pp:10914-10917
Publication Date(Web):2017/10/03
DOI:10.1039/C7CC06201J
Examples of tin dications without closer contacts to the anion are rare, as are straightforward routes to weakly coordinated tin(II) dication salts. Here we report on the synthesis of [Sn(MeCN)6][Al(ORF)4]2 (RF = C(CF3)3) via NO+-oxidation of tin metal. Subsequently, [Sn(MeCN)6][Al(ORF)4]2 was used to prepare the mixed coordinated [Sn(pyr)2(MeCN)4][Al(ORF)4]2 and [Sn(PPh3)2-(MeCN)5][Al(ORF)4]2·MeCN. Additionally, [Sn(dmap)4][Al(ORF)4]2 was prepared via a reaction of dmap with [SnCp][Al(ORF)4]. The generality of the formation of tin(II) dications by reacting [SnCp]+ and L to give [SnLx]2+ and SnCp2 was investigated using DFT calculations. Extensions to [ECp*]+ cations (E = Si, Ge, Sn, and Pb) are also suggested to be useful for the preparation of E(II) dications.
Co-reporter:Eno Paenurk;Karl Kaupmees;Daniel Himmel;Agnes Kütt;Ivari Kaljurand;Ilmar A. Koppel;Ivo Leito
Chemical Science (2010-Present) 2017 vol. 8(Issue 10) pp:6964-6973
Publication Date(Web):2017/09/25
DOI:10.1039/C7SC01424D
The most comprehensive solvent acidity scale spanning 28 orders of magnitude of acidity was measured in the low-polarity solvent 1,2-dichloroethane (DCE). Its experimental core is linked to the unified acidity scale (pHabs) in an unprecedented and generalized approach only based on experimental values. This enables future measurements of acid strengths and acidity adjustments in low polarity solvents. The scale was cross-validated computationally. The purely experimental and computational data agree very well. The DCE scale includes 87 buffer systems with values between −13.0 and +15.4, i.e. similar to water at hypothetical and extreme pH values of −13.0 to +15.4. Unusually, such high acidities in DCE are not realized via solvated protons, but rather through strongly acidic molecules able to directly donate their proton, even to weak bases dissolved in the solution. Thus, in all examined cases, not a single solvated proton is present in one liter of DCE.
Co-reporter:Daniel Himmel;Robin J. White;Eberhard Jacob
Sustainable Energy & Fuels (2017-Present) 2017 vol. 1(Issue 5) pp:1177-1183
Publication Date(Web):2017/06/27
DOI:10.1039/C7SE00053G
Oxymethylene dimethyl ethers, of the structure CH3(OCH2)nOCH3, denoted as OMEn are receiving increasing interest (where n = 2–5) in a range of important applications including as sustainable fuels and solvents (e.g. as derived from green methanol). However, limited thermodynamic information from computational studies exists in the literature regarding their formation in the gas and liquid phases. In this context, this report describes the principal thermodynamic functions of gaseous and liquid phase OME formation derived from B3LYP-D3(BJ)/def2-TZVPP optimised structures and a series of CCSD(T) and MP2 calculations. The generated total energies are almost of CCSD(T)/A′VQZ quality, the “gold standard” of computational chemistry. Thermal corrections to enthalpy and entropy were included on the basis of analytical BP86-D3(BJ)/def-TZVP frequencies and empirical corrections for low anharmonic C–O–C–O torsional vibrations/hindered rotations and due to the neglect of other conformers/enantiomers. This yielded corrected values for the standard entropy S° of gaseous OMEn (n = 2–7). With the well-established experimental formation enthalpies of dimethyl ether (i.e. OME0) and OME1, the formation enthalpies of OME2–7 were obtained from those and the isodesmic reaction enthalpy of nOME1 → OMEn + (n − 1)OME0. Overall, an error bar on those gas phase values of <1 kJ mol−1 is assigned. From the known and extra- or interpolated phase change thermodynamics, the standard formation enthalpy H°, and the standard entropy S°, as well as the heat capacity cp were established for the liquid mixture of OME2–7. The internal consistency of these data was assessed based on the plots of H°/S° vs. n, presenting linear regressions and correlation coefficients very close to unity. Data quality was also evaluated against published combustion energies, suggesting our values are currently the most reliable, internally consistent dataset that should be used in future investigations for the design of sustainable ether-based fuels and chemicals.
Co-reporter:Tobias A. Engesser, Martin R. Lichtenthaler, Mario Schleep and Ingo Krossing
Chemical Society Reviews 2016 vol. 45(Issue 4) pp:789-899
Publication Date(Web):27 Nov 2015
DOI:10.1039/C5CS00672D
The chemistry of the p-block elements is a huge playground for fundamental and applied work. With their bonding from electron deficient to hypercoordinate and formally hypervalent, the p-block elements represent an area to find terra incognita. Often, the formation of cations that contain p-block elements as central ingredient is desired, for example to make a compound more Lewis acidic for an application or simply to prove an idea. This review has collected the reactive p-block cations (rPBC) with a comprehensive focus on those that have been published since the year 2000, but including the milestones and key citations of earlier work. We include an overview on the weakly coordinating anions (WCAs) used to stabilize the rPBC and give an overview to WCA selection, ionization strategies for rPBC-formation and finally list the rPBC ordered in their respective group from 13 to 18. However, typical, often more organic ion classes that constitute for example ionic liquids (imidazolium, ammonium, etc.) were omitted, as were those that do not fulfill the – naturally subjective – “reactive”-criterion of the rPBC. As a rule, we only included rPBC with crystal structure and only rarely refer to important cations published without crystal structure. This collection is intended for those who are simply interested what has been done or what is possible, as well as those who seek advice on preparative issues, up to people having a certain application in mind, where the knowledge on the existence of a rPBC that might play a role as an intermediate or active center may be useful.
Co-reporter:Maria Kaliner;Dr. Alexer Rupp;Dr. Ingo Krossing;Dr. Thomas Strassner
Chemistry - A European Journal 2016 Volume 22( Issue 29) pp:10044-10049
Publication Date(Web):
DOI:10.1002/chem.201601063
Abstract
Weakly coordinating borate or aluminate anions have recently been shown to yield interesting properties of the resulting ionic liquids (ILs). The same is true for large phenyl-substituted imidazolium cations, which can be tuned by the choice, position, or number of substituents on the aromatic ring. We were therefore interested to combine these aryl alkyl imidazolium cations with the weakly coordinating tetrakis((1,1,1,3,3,3-hexafluoropropan-2-yl)oxy)borate [B(hfip)4]− anions to study the physical properties and viscosities of these ionic liquids. Despite the large size and high molecular weight of these readily available ILs, they are liquid at room temperature and show remarkably low glass transition points and relatively high decomposition temperatures.
Co-reporter:Mario Schleep;Stefanie Reininger;Dr. Philipp Eiden;Petra Klose;Dr. Christoph Schulz;Dr. Harald Scherer;Stephan Laule;Simon Bodendorfer;Dr. Michael Schmidt;Dr. Arnd Garsuch;Dr. Ingo Krossing
ChemElectroChem 2016 Volume 3( Issue 5) pp:774-782
Publication Date(Web):
DOI:10.1002/celc.201500560
Abstract
The synthesis and characterization of Li[O2P{OCH(CF3)2}2] (1) was investigated together with a new synthesis for Li[O2P(OCH2CF3)2] (2). The electrochemical properties of both lithium bis(fluoroalkyl)phosphates were investigated. Compound 2 was prepared by deprotonation of the conjugate acid HO(O)P(OCH2CF3)2 (3), of which the crystal structure was solved. Lithium bis(fluoroalkyl)phosphates 1 and 2 were used to prepare gel electrolytes on the basis of a coordination network held together solely by ionic interactions between the lithium ions and the anions. The preparation of the gel electrolytes produced with 2 and their properties were explored.
Co-reporter:Dr. Przemys&x142;aw J. Malinowski;Dr. Daniel Himmel;Dr. Ingo Krossing
Angewandte Chemie International Edition 2016 Volume 55( Issue 32) pp:9262-9266
Publication Date(Web):
DOI:10.1002/anie.201603913
Abstract
The synergistic Ag+/X2 system (X=Cl, Br, I) is a very strong, but ill-defined oxidant—more powerful than X2 or Ag+ alone. Intermediates for its action may include [Agm(X2)n]m+ complexes. Here, we report on an unexpectedly variable coordination chemistry of diiodine towards this direction: (A)Ag-I2-Ag(A), [Ag2(I2)4]2+(A−)2 and [Ag2(I2)6]2+(A−)2⋅(I2)x≈0.65 form by reaction of Ag(A) (A=Al(ORF)4; RF=C(CF3)3) with diiodine (single crystal/powder XRD, Raman spectra and quantum-mechanical calculations). The molecular (A)Ag-I2-Ag(A) is ideally set up to act as a 2 e− oxidant with stoichiometric formation of 2 AgI and 2 A−. Preliminary reactivity tests proved this (A)Ag-I2-Ag(A) starting material to oxidize n-C5H12, C3H8, CH2Cl2, P4 or S8 at room temperature. A rough estimate of its electron affinity places it amongst very strong oxidizers like MF6 (M=4d metals). This suggests that (A)Ag-I2-Ag(A) will serve as an easily in bulk accessible, well-defined, and very potent oxidant with multiple applications.
Co-reporter:Dr. Przemys&x142;aw J. Malinowski;Dr. Daniel Himmel;Dr. Ingo Krossing
Angewandte Chemie International Edition 2016 Volume 55( Issue 32) pp:9259-9261
Publication Date(Web):
DOI:10.1002/anie.201603741
Abstract
The perfluorohexane-soluble and donor-free silver compound Ag(A) (A=Al(ORF)4; RF=C(CF3)3) prepared using a facile novel route has unprecedented capabilities to form unusual and weakly bound complexes. Here, we report on the three dihalogen–silver complexes Ag(Cl2)A, Ag(Br2)A, and Ag(I2)A derived from the soluble silver compound Ag(A) (characterized by single-crystal/powder XRD, Raman spectra, and quantum-mechanical calculations).
Co-reporter:Dr. Przemys&x142;aw J. Malinowski;Dr. Daniel Himmel;Dr. Ingo Krossing
Angewandte Chemie 2016 Volume 128( Issue 32) pp:9408-9412
Publication Date(Web):
DOI:10.1002/ange.201603913
Abstract
The synergistic Ag+/X2 system (X=Cl, Br, I) is a very strong, but ill-defined oxidant—more powerful than X2 or Ag+ alone. Intermediates for its action may include [Agm(X2)n]m+ complexes. Here, we report on an unexpectedly variable coordination chemistry of diiodine towards this direction: (A)Ag-I2-Ag(A), [Ag2(I2)4]2+(A−)2 and [Ag2(I2)6]2+(A−)2⋅(I2)x≈0.65 form by reaction of Ag(A) (A=Al(ORF)4; RF=C(CF3)3) with diiodine (single crystal/powder XRD, Raman spectra and quantum-mechanical calculations). The molecular (A)Ag-I2-Ag(A) is ideally set up to act as a 2 e− oxidant with stoichiometric formation of 2 AgI and 2 A−. Preliminary reactivity tests proved this (A)Ag-I2-Ag(A) starting material to oxidize n-C5H12, C3H8, CH2Cl2, P4 or S8 at room temperature. A rough estimate of its electron affinity places it amongst very strong oxidizers like MF6 (M=4d metals). This suggests that (A)Ag-I2-Ag(A) will serve as an easily in bulk accessible, well-defined, and very potent oxidant with multiple applications.
Co-reporter:Dr. Przemys&x142;aw J. Malinowski;Dr. Daniel Himmel;Dr. Ingo Krossing
Angewandte Chemie 2016 Volume 128( Issue 32) pp:9405-9407
Publication Date(Web):
DOI:10.1002/ange.201603741
Abstract
The perfluorohexane-soluble and donor-free silver compound Ag(A) (A=Al(ORF)4; RF=C(CF3)3) prepared using a facile novel route has unprecedented capabilities to form unusual and weakly bound complexes. Here, we report on the three dihalogen–silver complexes Ag(Cl2)A, Ag(Br2)A, and Ag(I2)A derived from the soluble silver compound Ag(A) (characterized by single-crystal/powder XRD, Raman spectra, and quantum-mechanical calculations).
Co-reporter:Hannes Böhrer, Nils Trapp, Daniel Himmel, Mario Schleep and Ingo Krossing
Dalton Transactions 2015 vol. 44(Issue 16) pp:7489-7499
Publication Date(Web):13 Mar 2015
DOI:10.1039/C4DT02822H
The possibility of obtaining frustrated Lewis pairs (FLPs) suitable for H2-activation based on the Lewis acid B(Ohfip)31 (Ohfip = OC(H)(CF3)2) was investigated. In this context, the crystal structure of 1 as well as the crystal structure of the very weak adduct 1·NCMe was determined. When reacting solutions of 1 with H2 (1 bar) and selected phosphanes, amines, pyridines and N-heterocyclic carbenes, dihydrogen activation was never observed. Without H2, adduct formation with 1 was observed to be an equilibrium process, regardless of the Lewis base adduct. Thus, the thermodynamics of H2 activation of 1 in comparison with the well-known B(C6F5)3 was analyzed using DFT calculations in the gas phase and different solvents (CH2Cl2, ortho-difluorobenzene and acetonitrile). These investigations indicated that FLP chemistry based on 1 is considerably less favored than that with B(C6F5)3. This is in agreement with control NMR experiments indicating hydride transfer from [H–B(Ohfip)3]− upon reaction with B(C6F5)3, giving [H–B(C6F5)3]− and B(Ohfip)3 in toluene and also MeCN. Induced by these unsuccessful reactions, the Lewis acidity towards HSAB hard and soft ions was investigated for gaining a deeper insight. A unified reference system based on the trimethylsilyl compounds Me3Si–Y (Y = F, Cl, H, Me) and their respective ions Me3Si+/Y− calculated at the G3 level was chosen as the anchor point. The individual ion affinities were then assessed based on subsequent isodesmic reactions calculated at a much less expensive level (RI-)BP86/SV(P). This method was validated by systematic calculations of smaller reference systems at the frozen core CCSD(T) level with correlation effects extrapolated to a full quadruple-ζ basis. Overall, 33 common and frequently used Lewis acids were ranked with respect to their FIA, CIA, HIA and MIA (fluoride/chloride/hydride/methyl ion affinity).
Co-reporter:Christoph Schulz, Philipp Eiden, Petra Klose, Andreas Ermantraut, Michael Schmidt, Arnd Garsuch and Ingo Krossing
Dalton Transactions 2015 vol. 44(Issue 15) pp:7048-7057
Publication Date(Web):18 Mar 2015
DOI:10.1039/C5DT00469A
Weakly coordinating anions (WCAs) with the difluorophosphato ligand (O2PF2) were the target of this study. Initial experiments were conducted towards the preparation of homoleptic aluminates of the well-studied [Al(OR)4]−-type. The preparation of the initial target structure Li[Al(O2PF2)4] failed due to the remaining Lewis acidic character of the central aluminum atom. Instead, the formation of Li3[Al(O2PF2)6] and Al(O2PF2)3 was observed with hexacoordinate aluminum atoms and verified by NMR, IR and X-ray crystallography. A possible mechanism towards these compounds was postulated in the solvent induced dismutation of the tetracoordinate Li[Al(O2PF2)4]. A singly charged WCA was realized by the exchange of the central aluminum atom for boron. The [B(O2PF2)4]− anion was prepared starting from BH3·S(CH3)2 and boron tribromide leading to the protic room temperature Ionic Liquid (IL) [H(S(CH3)2)][B(O2PF2)4] and the neat liquid Brønsted acid H[B(O2PF2)4], respectively, representing a significantly improved synthesis with regard to the first experiments of Dove et al. The basicity of the [B(O2PF2)4]− anion and its WCA quality were investigated on the basis of the IR-spectroscopic NH-scale and the salt [H(N(Oct)3)][B(O2PF2)4] that places it better than all oxyanions and close to the carboranate based WCAs. A pathway to the solvent free pure Li[B(O2PF2)4] salt was established on a multi-gram scale with excellent purities enabling electrochemical applications (verified by NMR, IR, X-ray crystallography and cyclovoltammetry).
Co-reporter:Hatem Abushammala, Ingo Krossing, Marie-Pierre Laborie
Carbohydrate Polymers 2015 Volume 134() pp:609-616
Publication Date(Web):10 December 2015
DOI:10.1016/j.carbpol.2015.07.079
•Cellulose nanocrystals can be directly extracted from wood using a low temperature pulping process with [EMIM][OAc].•The CNCs extracted from wood maintain cellulose I microstructure (CrI of 70–75%), display large aspect ratios (22–65) and are surface acetylated.•Wood polymers acetylation and the heterogeneous conditions for wood cellulose appear critical to the efficiency of the ionic liquid assisted extraction of CNCs directly from wood.We report for the first time the direct extraction of cellulose nanocrystals (CNCs) from wood by means of a 1-ethyl-3-methylimidazolium acetate ([EMIM][OAc]) treatment. A native cellulosic product could be recovered in 44% yield with respect to wood cellulose content. The product was analyzed for morphological (TEM, AFM, XRD), chemical (FTIR, 13C CP/MAS NMR), thermal (DSC, TGA) and surface properties (Zeta potential, contact angle). These analyses evidenced the presence of partially acetylated (surface DS = 0.28) nanocrystals of native cellulose I microstructure, with a crystallinity index of about 75% and aspect ratio of 65. Direct production of CNCs from wood is ascribed to the simultaneous capability of [EMIM][OAc] to (1) dissolve lignin in situ while only swelling cellulose, (2) decrease intermolecular cohesion in wood via acetylation, and (3) to catalyze cellulose hydrolysis.
Co-reporter:Franziska Scholz;Wiebke Unkrig;Philipp Eiden;Michael A. Schmidt;Arnd Garsuch
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 19) pp:3128-3138
Publication Date(Web):
DOI:10.1002/ejic.201500254
Abstract
Lithium chloro- and bromoaluminates were prepared, fully characterized, and investigated as conducting salts for battery applications. In addition, thermodynamic investigations of their lattice enthalpies and other quantities were carried out. The Li[AlX4] salts also show very high conductivities in solvents of very low polarity, which makes them suitable candidates for conducting salts and battery systems working with voltages below ca. 3.5 V (X = Br) or ca. 4.4 V (X = Cl), such as lithium-sulfur batteries (LSBs). In particular, the bromoaluminates, with their Pearson-soft character, which prevents ion-pair formation with the Pearson-hard lithium ion, are promising for this purpose. An interesting alternative solvent for LSBs might be o-difluorobenzene, in which Li[AlBr4] shows very high solubility and conductivity. The carbonate solvents typically used for LIBs are not suitable for the bromoaluminates, and Li[AlCl4] also shows corrosive behavior towards aluminum at potentials higher than 2.5 V. This can be prevented by, for example, addition of Li[PF6].
Co-reporter:Dipl.-Chem. Miriam M. Schwab;Dr. Daniel Himmel;Dr. Sylwia Kacprzak;Dr. Daniel Kratzert;Dr. Valentin Radtke;Philippe Weis;Dr. Kallol Ray;Dr. Ernst-Wilhelm Scheidt;Dr. Wolfgang Scherer;Dr. Bas deBruin;Dr. Stefan Weber;Dr. Ingo Krossing
Angewandte Chemie 2015 Volume 127( Issue 49) pp:14919-14922
Publication Date(Web):
DOI:10.1002/ange.201506475
Abstract
The straightforward synthesis of the cationic, purely organometallic NiI salt [Ni(cod)2]+[Al(ORF)4]− was realized through a reaction between [Ni(cod)2] and Ag[Al(ORF)4] (cod=1,5-cyclooctadiene). Crystal-structure analysis and EPR, XANES, and cyclic voltammetry studies confirmed the presence of a homoleptic NiI olefin complex. Weak interactions between the metal center, the ligands, and the anion provide a good starting material for further cationic NiI complexes.
Co-reporter:Dipl.-Chem. Miriam M. Schwab;Dr. Daniel Himmel;Dr. Sylwia Kacprzak;Dr. Daniel Kratzert;Dr. Valentin Radtke;Philippe Weis;Dr. Kallol Ray;Dr. Ernst-Wilhelm Scheidt;Dr. Wolfgang Scherer;Dr. Bas deBruin;Dr. Stefan Weber;Dr. Ingo Krossing
Angewandte Chemie International Edition 2015 Volume 54( Issue 49) pp:14706-14709
Publication Date(Web):
DOI:10.1002/anie.201506475
Abstract
The straightforward synthesis of the cationic, purely organometallic NiI salt [Ni(cod)2]+[Al(ORF)4]− was realized through a reaction between [Ni(cod)2] and Ag[Al(ORF)4] (cod=1,5-cyclooctadiene). Crystal-structure analysis and EPR, XANES, and cyclic voltammetry studies confirmed the presence of a homoleptic NiI olefin complex. Weak interactions between the metal center, the ligands, and the anion provide a good starting material for further cationic NiI complexes.
Co-reporter:Martin R. Lichtenthaler;Steffen Maurer;Robert J. Mangan;Florian Stahl;Florian Mönkemeyer;Julian Hamann;Dr. Ingo Krossing
Chemistry - A European Journal 2015 Volume 21( Issue 1) pp:157-165
Publication Date(Web):
DOI:10.1002/chem.201404833
Abstract
Using [Ga(C6H5F)2]+[Al(ORF)4]−(1) (RF=C(CF3)3) as starting material, we isolated bis- and tris-η6-coordinated gallium(I) arene complex salts of p-xylene (1,4-Me2C6H4), hexamethylbenzene (C6Me6), diphenylethane (PhC2H4Ph), and m-terphenyl (1,3-Ph2C6H4): [Ga(1,4-Me2C6H4)2.5]+ (2+), [Ga(C6Me6)2]+ (3+), [Ga(PhC2H4Ph)]+ (4+) and [(C6H5F)Ga(μ-1,3-Ph2C6H4)2Ga(C6H5F)]2+ (52+). 4+ is the first structurally characterized ansa-like bent sandwich chelate of univalent gallium and 52+ the first binuclear gallium(I) complex without a GaGa bond. Beyond confirming the structural findings by multinuclear NMR spectroscopic investigations and density functional calculations (RI-BP86/SV(P) level), [Ga(PhC2H4Ph)]+[Al(ORF)4]−(4) and [(C6H5F)Ga(μ-1,3-Ph2C6H4)2Ga(C6H5F)]2+{[Al(ORF)4] −}2 (5), featuring ansa-arene ligands, were tested as catalysts for the synthesis of highly reactive polyisobutylene (HR-PIB). In comparison to the recently published 1 and the [Ga(1,3,5-Me3C6H3)2]+[Al(ORF)4]− salt (6) (1,3,5-Me3C6H3=mesitylene), 4 and 5 gave slightly reduced reactivities. This allowed for favorably increased polymerization temperatures of up to +15 °C, while yielding HR-PIB with high contents of terminal olefinic double bonds (α-contents=84–93 %), low molecular weights (Mn=1000–3000 g mol−1) and good monomer conversions (up to 83 % in two hours). While the chelate complexes delivered more favorable results than 1 and 6, the reaction kinetics resembled and thus concurred with the recently proposed coordinative polymerization mechanism.
Co-reporter:Martin R. Lichtenthaler;Steffen Maurer;Robert J. Mangan;Florian Stahl;Florian Mönkemeyer;Julian Hamann;Dr. Ingo Krossing
Chemistry - A European Journal 2015 Volume 21( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/chem.201490221
Co-reporter:Dr. Franziska Scholz;Dr. Daniel Himmel;Lea Eisele;Wiebke Unkrig;Arthur Martens;Peter Schlüter ;Dr. Ingo Krossing
Chemistry - A European Journal 2015 Volume 21( Issue 20) pp:7489-7502
Publication Date(Web):
DOI:10.1002/chem.201405952
Abstract
Bulk protonated mesitylene, toluene, and benzene bromoaluminate salts were stabilized and characterized in the superacidic system HBr/n AlBr3 with NMR spectroscopy and X-ray analysis of [HC6H3(CH3)3]+[AlBr4]− (1), [HC6H5(CH3)]+[AlBr4]− (2), and [C6H7]+[Al2Br7]−⋅C6H6 (3). Protonation attempts in bromoaluminate ILs led to a complete protonation of mesitylene, and a protonation degree of up to 15 % for toluene in the IL BMP+[Al2Br7]−. Benzene could only be protonated in the more acidic IL BMP+[Al3Br10]−, with a degree of 25 %. Protonation attempts on aromatics provide evidence that the bromoaluminate ILs tolerate superacidic environments. On the basis of the absolute Brønsted acidity scale, quantum chemical calculations confirmed the superacidic properties, and rank the acidities in ILs down to a pHabs value of 164 with an error of less than one pH unit compared with experimental findings. The neat AlBr3/HBr system even may reach acidities down to pHabs 163.
Co-reporter:Dr. Michael Rohde;Dr. Philipp Eiden;Verena Leppert;Dr. Michael Schmidt;Dr. Arnd Garsuch;Dr. Guenter Semrau; Dr. Ingo Krossing
ChemPhysChem 2015 Volume 16( Issue 3) pp:666-675
Publication Date(Web):
DOI:10.1002/cphc.201402680
Abstract
A new Li salt with views to success in electrolytes is synthesized in excellent yields from lithium borohydride with excess 2,2,2-trifluorethanol (HOTfe) in toluene and at least two equivalents of 1,2-dimethoxyethane (DME). The salt Li[B(OTfe)4] is obtained in multigram scale without impurities, as long as DME is present during the reaction. It is characterized by heteronuclear magnetic resonance and vibrational spectroscopy (IR and Raman), has high thermal stability (Tdecomposition>271 °C, DSC) and shows long-term stability in water. The concentration-dependent electrical conductivity of Li[B(OTfe)4] is measured in water, acetone, EC/DMC, EC/DMC/DME, ethyl acetate and THF at RT In DME (0.8 mol L−1) it is 3.9 mS cm−1, which is satisfactory for the use in lithium-sulfur batteries (LiSB). Cyclic voltammetry confirms the electrochemical stability of Li[B(OTfe)4] in a potential range of 0 to 4.8 V vs. Li/Li+. The performance of Li[B(OTfe)4] as conducting salt in a 0.2 mol L−1 solution in 1:1 wt % DME/DOL is investigated in LiSB test cells. After the 40th cycle, 86 % of the capacity remains, with a coulombic efficiency of around 97 % for each cycle. This indicates a considerable performance improvement for LiSB, if compared to the standard Li[NTf2]/DOL/DME electrolyte system.
Co-reporter:Dr. Daniel Himmel;Dr. Sascha K. Goll;Dr. Franziska Scholz;Dr. Valentin Radtke; Ivo Leito; Dr. Ingo Krossing
ChemPhysChem 2015 Volume 16( Issue 7) pp:1428-1439
Publication Date(Web):
DOI:10.1002/cphc.201402906
Abstract
Although receiving large interest over the last years, some fundamental aspects of Brønsted acidity in ionic liquids (ILs) have up to now been insufficiently highlighted. In this work, standard states, activity, and activity coefficient definitions for IL solvent systems were developed from general thermodynamic considerations and then extended to a general mixed solvent standard state. By using the bromide/bromoaluminate systems as representative ILs, formulae for thermodynamically consistent pH scales for ILs with simple (Br−) and complex ([AlnBr3n+1]−) anions were derived on the basis of the chemical potential of the proton. Supported by quantum chemical [ccsd(t)/MP2/DFT/COSMO-RS] calculations, Gibbs solvation energies of the proton were calculated, which allowed the ILs to be ranked in absolute acidity, that is, pHabs or μabs(H+, IL), and additionally allowed their acidity to be compared with molecular Brønsted acid systems. It was shown that bromoaluminate ILs are suited for reaching superacidic conditions. The complexity of autoprotolysis processes in C6MIM+[AlBr4]− (C6MIM=1-hexyl-3-methylimidazolium) with or without the addition of basic (i.e. Br−) or acidic (AlBr3 and/or HBr) solutes was examined in detail by model calculations, and they indicated a large thermodynamic influence of small deviations from the exact stoichiometric composition.
Co-reporter:Dr. Alexer B. A. Rupp;Sabrina Welle;Petra Klose;Dr. Harald Scherer; Dr. Ingo Krossing
ChemPhysChem 2015 Volume 16( Issue 9) pp:1940-1947
Publication Date(Web):
DOI:10.1002/cphc.201500069
Abstract
Several ionic liquids (ILs) comprising [B(hfip)4]− [hfip=OCH(CF3)2] or [Al(hfip)4]− anions and imidazolium or ammonium cations were prepared and mixed with up to 270 mol % of dimethyl carbonate (DMC). The viscosities, conductivities, and self-diffusion constants of these mixtures and, where possible, of the neat ILs were measured and compared with common [NTf2]− based ILs and their mixtures with DMC. A tremendous decrease of the viscosities and a likewise increase of the conductivities and diffusion constants can be achieved for all classes of ILs. However, the order of the conductivities is partially reversed in the diffusion data. This is probably due to the low dielectric constant of DMC and the, thus, favored ion pairing, as evidenced, for example, by the calculated ionicities. Altogether, our data show that the chemically robust, but high-melting and more viscous [B(hfip)4]− ILs might be candidates for electrolytes when mixed with suitable molecular solvents.
Co-reporter:Martin R. Lichtenthaler;Florian Stahl;Daniel Kratzert;Boumahdi Benkmil;Hermann A. Wegner
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 26) pp:4335-4341
Publication Date(Web):
DOI:10.1002/ejic.201402360
Abstract
To answer the question as to whether gallium in its oxidation state +1 favors a σ- or a π-coordination of aromatic nitrogen bases, we reacted [Ga(C6H5F)2]+[Al(ORF)4]– {RF = C(CF3)} with pyrazine and 2,6-di-tert-butyl-4-methylpyridine (DTBMP). In doing so, we obtained the first tricoordinate, nonchelated, homoleptic N-donor complex of gallium(I): [Ga(pyrazine)3]+[Al(ORF)4]–, in which each gallium(I) cation is coordinated in a trigonal-pyramidal fashion by three η1-donating pyrazine ligands. Hence, the gallium(I) cations favor σ- over π-coordination. Depending on the reaction conditions, and due to the bifunctionality of pyrazine, 1D coordination polymers of {[Ga(μ-pyrazine)2(η1-pyrazine)]+[Al(ORF)4]–}∞ were also obtained. With the sterically demanding DTBMP, which is conventionally used as a proton scavenger, the mixed complex [Ga(C6H5F)2(DTBMP)]+[Al(ORF)4]– was isolated, thus proving incorrect the perception of DTBMP being “non-nucleophilic”. The structural findings were confirmed by multinuclear NMR investigations and density functional performed at the RI-BP86/SV(P) level.
Co-reporter:Philip J. W. Elder;Tracey L. Roemmele;Mona Taghavikish;Tobias A. Engesser;Harald Scherer;René T. Boeré;Tristram Chivers
Heteroatom Chemistry 2014 Volume 25( Issue 6) pp:501-513
Publication Date(Web):
DOI:10.1002/hc.21193
ABSTRACT
Density functional theory (DFT) calculations for the six-membered ring 1,4-(CH2)2(PtBu)4 (1), the dimer (PtBu2)2 (2) and the 2,5-chalcogenated derivatives of 1, 3a (E = S) and 3b (E = Se), and the corresponding cation radicals and dications predict significant structural changes upon oxidation. The formation of a transannular P–P single bond (ca. 2.25 Å) in the three cyclic dications 12+, 3a2+, and 3b2+ is indicated by geometry and consideration of the frontier orbitals. The calculations also indicate a weak transannular interaction in the cyclic cation radicals. The nature of these transannular P–P bonding interactions is analyzed through a consideration of the molecular orbitals involved. Cyclic Voltammetry studies of 1 and 2 reveal two well-separated oxidation processes. Both processes are irreversible for 1 at normal scan rates, whereas for 2 the first process is quasi-reversible. The cation radical 1+• could not be detected by in situ electron paramagnetic resonance studies of the first electrochemical oxidation, but a spectrum for the radical cation 2+• could be observed. The difference in the redox behavior of 1 and 2 is considered with respect to the structural parameters and DFT calculations. Chemical oxidation of 1 with NO+[Al(ORF)4]− (RF = C(CF3)3) in CH2Cl2 led to a complex mixture; the protonated cation H1+ (1,4-(CH2)2(PtBu)3(HPtBu)+) was identified as one of the major products on the basis of multinuclear NMR spectra.
Co-reporter:Elias Frei;Dr. Achim Schaadt;Dr. Thilo Ludwig;Dr. Harald Hillebrecht;Dr. Ingo Krossing
ChemCatChem 2014 Volume 6( Issue 6) pp:1721-1730
Publication Date(Web):
DOI:10.1002/cctc.201300665
Abstract
For heterogeneous catalysts, the constitution of the precursor is an important parameter to adjust the properties of the active catalyst. Therefore, we examined the influence of the temperature during the precipitation process and during the ageing time in the mother liquor for a Cu/ZnO/ZrO2 catalyst system obtained through a coprecipitation route. The variation of the temperature affects the ratio and crystallinity of the precursor phases zincian malachite and aurichalcite, as detected by powder XRD (phase and line width) and FTIR spectroscopy (characteristic asymmetric CO stretching modes of the carbonate anions at =1600–1100 cm−1). Therefore, the precatalyst surface area (As,BET) and pore distribution are adjustable (i.e., As,BET of 190 m2 g−1 was reached). The influence of the synthesis conditions on the catalysts activity for methanol production was analyzed and discussed up to the level of productivity/activity testing at 413/513 K and 40 bar total H2/CO2 pressure. The best catalyst showed a methanol productivity of 9.16 mmol gcat−1 h−1 (513 K, 40 bar, and gas hourly space velocity=8000) and is better than an industrial catalyst tested under the same conditions (8.34 mmol gcat−1 h−1). However, despite considerable differences in the precursor and precatalyst structure and morphology, their influence on the methanol productivity is only small. This demonstrates that the active catalyst is formed under reaction conditions.
Co-reporter:Dr. Witali Beichel;Dr. Nils Trapp;Dipl.-Phys. Christoph Hauf;Dipl.-Ing. Oliver Kohler;Dr. Georg Eickerling;Dr. Wolfgang Scherer;Dr. Ingo Krossing
Angewandte Chemie International Edition 2014 Volume 53( Issue 12) pp:3143-3146
Publication Date(Web):
DOI:10.1002/anie.201308760
Abstract
The charge scaling effect in ionic liquids was explored on the basis of experimental and theoretical charge-density analyses of [C1MIM][C1SO4] employing the quantum theory of atoms in molecules (QTAIM) approach. Integrated QTAIM charges of the experimental (calculated) charge density of the cation and anion resulted in non-integer values of ±0.90 (±0.87) e. Efficient charge transfer along the bond paths of the hydrogen bonds between the imidazolium ring and the anion was considered as the origin of these reduced charges. In addition, a detailed QTAIM analysis of the bonding situation in the [C1SO4]− anion revealed the presence of negative πOσ*S-O hyperconjugation.
Co-reporter:Dr. Chul-Woong Cho;Dr. Stefan Stolte;Dr. Johannes Ranke;Dr. Ulrich Preiss; Ingo Krossing; Jorg Thöming
ChemPhysChem 2014 Volume 15( Issue 11) pp:2351-2358
Publication Date(Web):
DOI:10.1002/cphc.201402092
Abstract
The molecular interaction potentials, including S (dipolarity/polarizability), A (hydrogen bonding acidity), and B (hydrogen bonding basicity), of anions are experimentally determined using multi-functionalized stationary phases in high-performance liquid chromatography (HPLC) systems. We employ three different multi-functionalized stationary phase columns (Obelisc R, Obelisc N, and Acclaim Trinity-P1) combined with two ingredients, namely, acetonitrile (ACN) and methanol (MeOH). These conditions can cause neutral, cationic, and anionic compounds to be retained. By using the retention characteristics of calibration compounds, including cations, anions, and neutral compounds, system parameters including the ionic interaction terms (zcZc, zaZa) are evaluated using multiple linear regression, resulting in a standard deviation (SD) of 0.090–0.158 log units. Based on the system parameters and retention characteristics of the anions of interest, their molecular interaction potentials are characterized on the same scale for neutral and cationic molecules. Furthermore, to verify the determined molecular interaction potentials, we predict anion hydrophobicity. The results show that the determined S, A, and B, together with the computable descriptors E (excess molar refraction) and V (McGowan volume), can predict anion hydrophobicity with R2=0.982 and SD=0.167 (dimensionless).
Co-reporter:Dipl.-Chem. Michael Rohde;Dr. Lutz O. Müller;Dr. Daniel Himmel;Dr. Harald Scherer ;Dr. Ingo Krossing
Chemistry - A European Journal 2014 Volume 20( Issue 5) pp:1218-1222
Publication Date(Web):
DOI:10.1002/chem.201303671
Abstract
Upon reaction of gaseous Me3SiF with the in situ prepared Lewis acid Al(ORF)3, the stable ion-like silylium compound Me3Si-F-Al(ORF)3 1 forms. The Janus-headed 1 is a readily available smart Lewis acid that differentiates between hard and soft nucleophiles, but also polymerizes isobutene effectively. Thus, in reactions of 1 with soft nucleophiles (Nu), such as phosphanes, the silylium side interacts in an orbital-controlled manner, with formation of [Me3SiNu]+ and the weakly coordinating [FAl(ORF)3]– or [(FRO)3Al-F-Al(ORF)3]– anions. If exchanged for hard nucleophiles, such as primary alcohols, the aluminum side reacts in a charge-controlled manner, with release of FSiMe3 gas and formation of the adduct R(H)OAl(ORF)3. Compound 1 very effectively initiates polymerization of 8 to 21 mL of liquid C4H8 in 50 mL of CH2Cl2 already at temperatures between −57 and −30 °C with initiator loads as low as 10 mg in a few seconds with 100 % yield but broad polydispersities.
Co-reporter:Dipl.-Chem. Franziska Scholz;Dr. Daniel Himmel;B.Sc. Lea Eisele;B.Sc. Wiebke Unkrig ;Dr. Ingo Krossing
Angewandte Chemie 2014 Volume 126( Issue 6) pp:1715-1718
Publication Date(Web):
DOI:10.1002/ange.201308120
Abstract
Die Reaktion von festem AlBr3, Benzol und HBr-Gas führte zur ausgeordneten Kristallstruktur des protonierten Benzols der Zusammensetzung [C6H7]+[Al2Br7]−⋅(C6H6) (1). Die Verbindung konnte mittels Röntgenstrukturanalyse, NMR- und Raman-Spektroskopie zweifelsfrei nachgewiesen werden. Dieser unerwartet einfache und leicht zugängliche Syntheseweg zeigt, dass HBr/AlBr3 eine Supersäure ist, deren Potential bisher unterschätzt wurde und noch weiter untersucht werden sollte.
Co-reporter:Dr. Witali Beichel;Dipl.-Chem. Johann M. U. Panzer;B.Sc. Julian Hätty;Dipl.-Chem. Xiaowei Ye;Dr. Daniel Himmel;Dr. Ingo Krossing
Angewandte Chemie International Edition 2014 Volume 53( Issue 26) pp:6637-6640
Publication Date(Web):
DOI:10.1002/anie.201402577
Abstract
The easily accessible hexafluoroisopropoxysulfuric acid (1, hfipOSO3H; hfip=C(H)(CF3)2) was synthesized by the reaction of hexafluoroisopropanol and chlorosulfonic acid on the kilogram scale and isolated in 98 % yield. The calculated gas-phase acidity (GA) value of 1 is 58 kJ mol−1 lower in ΔG° than that of sulfuric acid (GA value determined by a CCSD(T)-MP2 compound method). Considering the gas-phase dissociation constant as a measure for the intrinsic molecular acid strength, a hfipOSO3H molecule is more than ten orders of magnitude more acidic than a H2SO4 molecule. The acid is a liquid at room temperature, distillable at reduced pressure, stable for more than one year in a closed vessel, reactive towards common solvents, and decomposes above 180 °C. It is a versatile compound for further applications, such as the synthesis of ammonium- and imidazolium-based air- and moisture-stable protic ionic liquids (pILs). Among the six synthesized ionic compounds, five are pILs with melting points below 100 °C and three of them are liquids at nearly room temperature. The conductivities and viscosities of two representative ILs were investigated in terms of Walden plots, and the pILs were found to be little associated ILs, comparable to conventional aprotic ILs.
Co-reporter:Dipl.-Chem. Franziska Scholz;Dr. Daniel Himmel;B.Sc. Lea Eisele;B.Sc. Wiebke Unkrig ;Dr. Ingo Krossing
Angewandte Chemie International Edition 2014 Volume 53( Issue 6) pp:1689-1692
Publication Date(Web):
DOI:10.1002/anie.201308120
Abstract
Crystalline and properly ordered protonated benzene as the [C6H7]+[Al2Br7]−⋅(C6H6) salt 1 are obtained by the combination of solid AlBr3, benzene, and HBr gas. Compound 1 was characterized and verified by NMR, Raman and X-Ray spectroscopy. This unexpected simple and straight forward access shows that HBr/AlBr3 is an underestimated superacid that should be used more frequently.
Co-reporter:Dr. Daniel Himmel;Dr. Ingo Krossing;Dr. Andreas Schnepf
Angewandte Chemie 2014 Volume 126( Issue 24) pp:6159-6160
Publication Date(Web):
DOI:10.1002/ange.201403078
Co-reporter:Dr. Witali Beichel;Dipl.-Chem. Johann M. U. Panzer;B.Sc. Julian Hätty;Dipl.-Chem. Xiaowei Ye;Dr. Daniel Himmel;Dr. Ingo Krossing
Angewandte Chemie 2014 Volume 126( Issue 26) pp:6755-6758
Publication Date(Web):
DOI:10.1002/ange.201402577
Abstract
The easily accessible hexafluoroisopropoxysulfuric acid (1, hfipOSO3H; hfip=C(H)(CF3)2) was synthesized by the reaction of hexafluoroisopropanol and chlorosulfonic acid on the kilogram scale and isolated in 98 % yield. The calculated gas-phase acidity (GA) value of 1 is 58 kJ mol−1 lower in ΔG° than that of sulfuric acid (GA value determined by a CCSD(T)-MP2 compound method). Considering the gas-phase dissociation constant as a measure for the intrinsic molecular acid strength, a hfipOSO3H molecule is more than ten orders of magnitude more acidic than a H2SO4 molecule. The acid is a liquid at room temperature, distillable at reduced pressure, stable for more than one year in a closed vessel, reactive towards common solvents, and decomposes above 180 °C. It is a versatile compound for further applications, such as the synthesis of ammonium- and imidazolium-based air- and moisture-stable protic ionic liquids (pILs). Among the six synthesized ionic compounds, five are pILs with melting points below 100 °C and three of them are liquids at nearly room temperature. The conductivities and viscosities of two representative ILs were investigated in terms of Walden plots, and the pILs were found to be little associated ILs, comparable to conventional aprotic ILs.
Co-reporter:Tobias A. Engesser, Ingo Krossing
Coordination Chemistry Reviews 2013 Volume 257(5–6) pp:946-955
Publication Date(Web):March 2013
DOI:10.1016/j.ccr.2012.07.025
The electronegative non metallic elements have high electron affinities and ionization potentials and thus a strong tendency to form anions. To work against the naturally negative charge of the non-metals, inspired and continues to inspire chemists around the world. Nevertheless, it still is one of the tough challenges in inorganic chemistry to selectively synthesize pure homopolyatomic cations of the non metals. Over the last hundred years the characterization of cations of most of those elements were described in the literature. Even the very recent years brought new and exciting examples, e.g. cations of N, P, Sb and others. Herein the advances towards the syntheses of homopolyatomic cations of the non metallic elements over the last fifteen years are reviewed.Highlights► We review the recent advances in the syntheses of cations of non metallic elements. ► New examples of the homopolyatomic cations of the group 14–18 elements are included. ► We discuss the role of the oxidants, weakly coordinating anions and reaction media. ► An outlook of applications of the already and other new cations is given.
Co-reporter:P. v. R. Schleyer;F. W. Heinemann;K. Meyer;D. Himmel;F. Scholz;I. Krossing
Science 2013 Volume 341(Issue 6141) pp:62-64
Publication Date(Web):05 Jul 2013
DOI:10.1126/science.1238849
A Nonclassical Conclusion
The concept of valence, which underlies the Periodic Table, originated in studies of reactivity rather than structure. Nonetheless, when studies in the mid-20th century suggested that the transient norbornyl cation (C7H11+) reacted as though a carbon center had adopted a formally pentacoordinate motif, this nonclassical structural hypothesis engendered tremendous controversy. Scholz et al. (p. 62) have now succeeded in characterizing the norbornyl cation by x-ray crystallography and confirm the symmetrical fivefold motif.
Co-reporter:Dipl.Chem. Franziska Scholz;Dr. Daniel Himmel;Dr. Harald Scherer ;Dr. Ingo Krossing
Chemistry - A European Journal 2013 Volume 19( Issue 1) pp:109-116
Publication Date(Web):
DOI:10.1002/chem.201203260
Abstract
The room-temperature ionic liquid (RT-IL) [C(CH3)3]+ [Al2Br7]− (m.p. 2 °C) was generated by bromide abstraction from tert-butyl bromide with the Lewis acid aluminum bromide in the absence of solvent. The crystal structure of the tert-butyl cation salt was determined by X-ray diffraction. NMR, IR, and Raman spectroscopy, as well as quantum-chemical and thermodynamic calculations, confirm the composition of this RT-IL. Thus, one may consider this RT-IL to be a readily accessible (and on a large scale) cationic Brønsted acid (protonated isobutene) with the potential for further reactivity. Based on the new absolute Brønsted acidity scale, we calculated an absolute pHabs value of 171 for liquid bulk [C(CH3)3]+ [Al2Br7]−. This value is about as acidic as 100 % sulfuric acid (pHabs=171) and, thus, on the edge of superacidity.
Co-reporter:Alexer Higelin;Sarah Keller;Christian Göhringer; Cameron Jones;Dr. Ingo Krossing
Angewandte Chemie International Edition 2013 Volume 52( Issue 18) pp:4941-4944
Publication Date(Web):
DOI:10.1002/anie.201209757
Co-reporter:Mathias Hill;Patrick Baron;Dr. Keith Cobry;Sascha K. Goll;Dr. Philipp Lang; Carsten Knapp;Dr. Harald Scherer; Peter Woias;Pengcheng Zhang; Ingo Krossing
ChemPlusChem 2013 Volume 78( Issue 4) pp:292-301
Publication Date(Web):
DOI:10.1002/cplu.201200267
Abstract
A novel minireactor for direct fluorination of organic and inorganic substances was tested. The reactor consists of nickel-coated copper blocks with mechanically machined 1 mm channels and is equipped with an active cooling system. The direct fluorination of ethylene carbonate and propylene carbonate is described. For the fluorinated propylene carbonate, the NMR data of various fluorinated isomers were determined. The Gibbs reaction energies for the direct fluorination of ethylene and propylene carbonate were calculated at the reliable G3 level of theory. The excellent decomposition stability of the cyclic carbonates against high fluorine and HF concentrations also qualifies them as good solvents for direct fluorination processes, especially for ionic substrates. In this respect, the direct fluorination of the inorganic salt closo-K2[B12H12] in cyclic carbonates is presented.
Co-reporter:Mathias Hill;Patrick Baron;Dr. Keith Cobry;Sascha K. Goll;Dr. Philipp Lang; Carsten Knapp;Dr. Harald Scherer; Peter Woias;Pengcheng Zhang; Ingo Krossing
ChemPlusChem 2013 Volume 78( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/cplu.201300055
Abstract
Invited for this month’s cover are the groups of Prof. Ingo Krossing and Prof. Peter Woias based at Albert-Ludwigs-Universität Freiburg as well as Prof. Carsten Knapp who is now at Bergische Universität Wuppertal. The background of the cover picture shows the Rhine Falls (Schaffhausen, Switzerland), which is the largest waterfall in Europe and only one hour away from the laboratories in Freiburg. The waterfall symbolizes flow chemistry and the wild nature of direct fluorination reactions.
Co-reporter:Mathias Hill;Patrick Baron;Dr. Keith Cobry;Sascha K. Goll;Dr. Philipp Lang; Carsten Knapp;Dr. Harald Scherer; Peter Woias;Pengcheng Zhang; Ingo Krossing
ChemPlusChem 2013 Volume 78( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/cplu.201390014
Co-reporter:Martin R. Lichtenthaler, Alexander Higelin, Anne Kraft, Sarah Hughes, Alberto Steffani, Dietmar A. Plattner, John M. Slattery, and Ingo Krossing
Organometallics 2013 Volume 32(Issue 22) pp:6725-6735
Publication Date(Web):October 31, 2013
DOI:10.1021/om4005516
The scope of the univalent gallium salts [Ga(C6H5F)2]+[Al(ORF)4]− and the new completely characterized [Ga(1,3,5-Me3C6H3)2]+[Al(ORF)4]− (RF = C(CF3)3) was investigated in terms of initiating or catalyzing the synthesis of highly reactive poly(2-methylpropylene)—highly reactive polyisobutylene (HR-PIB)—in several solvents. A series of polymerization reactions proved the high efficiency and quality of the univalent gallium salts for the polymerization of isobutylene. The best results were obtained using very low concentrations of [Ga(C6H5F)2]+[Al(ORF)4]− (down to 0.007 mol%) while working at reaction temperatures of up to ±0 °C and in the noncarcinogenic and non-water hazardous solvent toluene. Under these conditions, HR-PIB with an α-content of terminal olefinic double bonds up to 91 mol% and a molecular weight of 1000–2000 was obtained in good yields. Upon changing [Ga(C6H5F)2]+[Al(ORF)4]− for the electron richer [Ga(1,3,5-Me3C6H3)2]+[Al(ORF)4]−, polymerization temperatures could be increased to +10 °C. The reactivity of the gallium(I) cations therefore seems to be tunable through ligand exchange reactions. Experimental results, density functional theory calculations, and mass spectrometric investigations point toward a coordinative polymerization mechanism.
Co-reporter:Alexander Higelin, Christoph Haber, Stefan Meier and Ingo Krossing
Dalton Transactions 2012 vol. 41(Issue 39) pp:12011-12015
Publication Date(Web):31 May 2012
DOI:10.1039/C2DT30379E
The recently reported homologous low-valent indium and gallium salts M+[Al(ORF)4]− (M = Ga, In; RF = C(CF3)3) were used to extend the coordination chemistry of GaI and InI to the isolated [18]crown-6 complexes [M([18]crown-6)(PhF)2]+[Al(ORF)4]− in fluorobenzene solution (PhF = C6H5F). In contrast to known ion-paired compounds for M = In, our complexes are undisturbed and in the solid state free of contacts to the anion. A peculiar combination of very weak η1- and η6-coordination to the PhF-solvent was observed that allows speculation about the presence of a stereochemically active lone pair at MI. Structure and energetics of these novel salts were rationalized on the basis of DFT calculations.
Co-reporter:Dr. Tobias Köchner;Dipl.-Chem. Tobias A. Engesser;Dr. Harald Scherer;Dr. Dietmar A. Plattner;Dr. Alberto Steffani;Dr. Ingo Krossing
Angewandte Chemie 2012 Volume 124( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/ange.201203991
Co-reporter:Dipl.-Chem. Tobias A. Engesser;Dr. Peter Hrobárik;Dr. Nils Trapp;Dr. Philipp Eiden;Dr. Harald Scherer;Dr. Martin Kaupp;Dr. Ingo Krossing
ChemPlusChem 2012 Volume 77( Issue 8) pp:643-651
Publication Date(Web):
DOI:10.1002/cplu.201200025
Abstract
TeX3[Al(ORF)4] (X=Cl, Br, I; RF=C(CF3)3) were synthesized by the reaction of Ag[Al(ORF)4] and TeX4 or the reaction of AuX, Ag[Al(ORF)4], and elemental tellurium in liquid SO2. The compounds were characterized by 125Te NMR in solution and by X-ray diffraction, Raman, and IR spectroscopy in the solid state. The vibrational spectra and the crystal structure show very weak secondary interactions, indicating “pseudo gas phase conditions” in the condensed phase. The observed trend of the 125Te NMR chemical shifts along the [TeX3]+ series follows neither the monotonous decrease known as “normal halogen dependence” nor the increase known as “inverse halogen dependence”. By relativistic two-component calculations based on the ZORA approach, we find that this “abnormal halogen dependence” results from an interplay of relativistic and solvent effects, where non-negligible scalar relativistic effects and intermediate-sized spin-orbit effects compensate to some extent. The reasons for these trends are evaluated in the context of the Te s-orbital character of the TeX bonds and compared with the halogen dependence(s) within the isoelectronic [SeX3]+ and PX3 series and related trihalomethyl [CX3]+ cations.
Co-reporter:Dipl.-Chem. Julia Schaefer;Dipl.-Chem. Alberto Steffani;Dr. Dietmar A. Plattner;Dr. Ingo Krossing
Angewandte Chemie International Edition 2012 Volume 51( Issue 24) pp:6009-6012
Publication Date(Web):
DOI:10.1002/anie.201201642
Co-reporter:Dr. Tobias Köchner;Dipl.-Chem. Tobias A. Engesser;Dr. Harald Scherer;Dr. Dietmar A. Plattner;M.Sc. Alberto Steffani;Dr. Ingo Krossing
Angewandte Chemie International Edition 2012 Volume 51( Issue 26) pp:6529-6531
Publication Date(Web):
DOI:10.1002/anie.201201262
Co-reporter:Dr. Tobias Köchner;Dipl.-Chem. Tobias A. Engesser;Dr. Harald Scherer;Dr. Dietmar A. Plattner;M.Sc. Alberto Steffani;Dr. Ingo Krossing
Angewandte Chemie International Edition 2012 Volume 51( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/anie.201203991
Co-reporter:Dipl.-Chem. Mara Bürchner;Anna M. T. Erle;Dr. Harald Scherer;Dr. Ingo Krossing
Chemistry - A European Journal 2012 Volume 18( Issue 8) pp:2254-2262
Publication Date(Web):
DOI:10.1002/chem.201102460
Abstract
Straightforward access to hydridoborate-based ionic liquids (BILs) is provided. They fall into a barely developed area of research and are of interest as, for example, reagents for organic synthesis. A series of pure [BH4]− ILs with 1-butyl-2,3-dimethylimidazolium (BMMIM), 1-ethyl-3-methylimidazolium (EMMIM), 1-propyl-1-methylpiperidinium (PropMPip), and1-butyl-1-methylpyrrolidinium (BMP) cations were prepared. All synthesized ILs are well soluble in CH2Cl2. We developed a procedure that gives clean products with correct elemental analyses. In contrast to earlier reports, which when conducted by us yielded only mixtures of the boranate anion with major halide contamination (maximum [BH4]− content: 77.5 %). These materials can be viewed as the starting material for the (hypothetical) hydrogen-storage redox shuttling sequence between [BH4]− and [B12H12]2−, in which the triboranate anion [B3H8]− is a formal intermediate. Here we also developed a facile route to [B3H8]− ILs with [BMMIM]+, [EMMIM]+, [PropMPip]+, and [NBu4]+, in which Na[BH4] reacts in situ (enhanced by ultrasound) with the solvent CH2Cl2 as the oxidizing agent to give the triboranate IL in high yield and purity according to the equation: 3 [BH4]−+2 CH2Cl2+[Cat]+[B3H8]−[Cat]++H2+2 CH3Cl+2 Cl−. We further investigated this reaction path by additional NMR spectroscopic experiments, powder-XRD analysis, and quantum chemical DFT calculations.
Co-reporter:Dr. Daniel Himmel;Dipl.-Chem. Sascha K. Goll;Dr. Ivo Leito;Dr. Ingo Krossing
Chemistry - A European Journal 2012 Volume 18( Issue 30) pp:9333-9340
Publication Date(Web):
DOI:10.1002/chem.201104025
Abstract
The capability of a gaseous Brønsted acid HB to deliver protons to a base is usually described by the gas-phase acidity (GA) value of the acid. However, GA values are standard Gibbs energy differences and refer to individual gas pressures of 1 bar for acid HB, base B−, and proton H+. We show that the GA value is not suited to describe the bulk acidity of a gaseous acid. Here the pressure dependence of the activities of HB, H(HB)n+, and B(HB)m− that result from gaseous autoprotolysis have to be considered. In this work, the pressure-dependent absolute chemical potential of the proton in the representative gaseous proton acids CH4, NH3, H2O, HF, and HCl was worked out and the general theory to describe bulk gas phase acidity—that can directly be compared with solution acidity—was developed.
Co-reporter:Alexer Higelin;Ulf Sachs;Sarah Keller ;Dr. Ingo Krossing
Chemistry - A European Journal 2012 Volume 18( Issue 32) pp:10029-10034
Publication Date(Web):
DOI:10.1002/chem.201104040
Abstract
In a new oxidative route, Ag+[Al(ORF)4]− (RF=C(CF3)3) and metallic indium were sonicated in aromatic solvents, such as fluorobenzene (PhF), to give a precipitate of silver metal and highly soluble [In(PhF)n]+ salts (n=2, 3) with the weakly coordinating [Al(ORF)4]− anion in quantitative yield. The In+ salt and the known analogous Ga+[Al(ORF)4]− were used to synthesize a series of homoleptic PR3 phosphane complexes [M(PR3)n]+, that is, the weakly PPh3-bridged [(Ph3P)3In–(PPh3)–In(PPh3)3]2+ that essentially contains two independent [In(PPh3)3]+ cations or, with increasing bulk of the phosphane, the carbene-analogous [M(PtBu3)2]+ (M=Ga, In) cations. The MIP distances are 27 to 29 pm longer for indium, and thus considerably longer than the difference between their tabulated radii (18 pm). The structure, formation, and frontier orbitals of these complexes were investigated by calculations at the BP86/SV(P), B3LYP/def2-TZVPP, MP2/def2-TZVPP, and SCS-MP2/def2-TZVPP levels.
Co-reporter:Chul-Woong Cho;Dr. Christian Jungnickel;Dr. Stefan Stolte;Dr. Ulrich Preiss;Dr. Jürgen Arning;Dr. Johannes Ranke; Ingo Krossing; Jorg Thöming
ChemPhysChem 2012 Volume 13( Issue 3) pp:780-787
Publication Date(Web):
DOI:10.1002/cphc.201100872
Abstract
In order to understand molecular interaction potentials of 30 cations of ionic liquids (ILs), the well-known linear free energy relationship concept (LFER) was applied. The LFER descriptors for the excess molar refractivity and the molar volume were calculated in silico and for hydrogen-bonding acidity and basicity, and the polarizability/dipolarity of IL cations were experimentally determined through high performance liquid chromatography (HPLC) measurements. For the study, three different columns (RP-select B, Cyan, and Diol) and buffered mobile phases, based on two organic solvents acetonitrile (ACN) and methanol (MeOH), were selectively combined to the HPLC separation systems RP-select B-ACN, RP-select B-MeOH, Cyan-MeOH, Diol-ACN, and Diol-MeOH. By measuring the retention factors of 45 neutral calibration compounds and calculating LFER descriptors of three cations in the HPLC systems, the system parameters, including an ionic z coefficient, were determined. Conversely, the LFER descriptors of 30 ionic liquid cations were determined, based on the parameters of five systems and their retention factors in the HPLC systems. The results showed that the type of head group, alkyl chain length and further substituents of the cation have a significant influence on the dipolarity/polarizability and the hydrogen-bonding acidity, and functionalized groups (hydroxyl, ether, and dimethylamino) lead to hydrogen-bonding basicity of the cation. The characterization of cationic LFER descriptors opens up the chance for a more quantitative understanding of molecular interaction potentials and physicochemical properties of ILs.
Co-reporter:Dr. Safak Bulut;M. A. Ab Rani; Dr. Tom Welton; Dr. Paul D. Lickiss; Dr. Ingo Krossing
ChemPhysChem 2012 Volume 13( Issue 7) pp:1802-1805
Publication Date(Web):
DOI:10.1002/cphc.201200028
Abstract
Two new ionic liquids (ILs) with siloxane-functionalized cations and the weakly coordinating tetraalkoxyaluminate [Al(hfip)4]− (hfip=hexafluoroisopropoxy) are prepared and characterized by nuclear magnetic resonance (NMR), infrared (IR) and Raman spectroscopy. With melting points below 0 °C they qualify as room temperature ILs (RTILs). Their temperature-dependent viscosities and conductivities, together with those of two [Tf2N]− ILs with the same cations and a further siloxane-functionalized [Tf2N]− IL, are measured between 0 and 80 °C, and all are described by the Vogel–Fulcher–Tammann (VFT) equations. We note that the [Al(hfip)4]− ILs have lower viscosities than their [Tf2N]− analogues at all measured temperatures and higher conductivities at room temperature.
Co-reporter:Dr. Tobias Köchner;Dipl.-Chem. Tobias A. Engesser;Dr. Harald Scherer;Dr. Dietmar A. Plattner;Dr. Alberto Steffani;Dr. Ingo Krossing
Angewandte Chemie 2012 Volume 124( Issue 26) pp:6635-6637
Publication Date(Web):
DOI:10.1002/ange.201201262
Co-reporter:Nils Trapp, Harald Scherer, Stuart A. Hayes, Raphael J. F. Berger, Agnes Kütt, Norbert W. Mitzel, Jaan Saame and Ingo Krossing
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 13) pp:6184-6191
Publication Date(Web):25 Feb 2011
DOI:10.1039/C0CP02596H
The syntheses of the perfluorinated alcohols (F5C6)(F3C)2COH (1) and (F5C6)(C5F10)COH (2) are described. Both compounds were prepared in reasonable yields (1: 65%, 2: 85%) by reacting the corresponding ketone with C6F5MgBr, followed by acidic work-up. The alcohols were characterized by NMR, vibrational spectroscopy, single-crystal X-ray diffraction, acidity measurements and gas-phase electron diffraction. A combination of appropriate 2D NMR experiments allowed the unambiguous assignment of all signals in the 19F spin systems, of which that of 2 was especially complex. High acidity of the alcohols is indicated by acidity measurements as well as the calculated gas phase acidities. It is also supported by the crystal structure of 2, which exhibits only a single weak intermolecular hydrogen bridge with an O⋯O distance of 301 pm. This shows the low donor strength of the oxygen atom in the compound, which is partly compensated through formation of two intramolecular CF⋯H contacts of 220 and 232 pm length to the proton not involved in the hydrogen bridge. The pKa values in acetonitrile are 22.2 for 1 and 22.0 for 2; their calculated gas phase acidities are 1367 and 1343 kJ mol−1 (MP2/TZVPP level).
Co-reporter:Safak Bulut, Petra Klose and Ingo Krossing
Dalton Transactions 2011 vol. 40(Issue 32) pp:8114-8124
Publication Date(Web):18 Jul 2011
DOI:10.1039/C1DT10722D
The fast, high yield synthesis and full characterization of Na[B(hfip)4] (hfip: OC(H)(CF3)2) from NaBH4 and hexafluoroisopropanol (hfipH) is presented. By anion metathesis, five [B(hfip)4]− salts with classical/functionalized ionic liquid (IL) cations with melting points between 0 ([C6MIM]+[B(hfip)4]−) and 113 °C ([C4MMorph]+[B(hfip)4]−) were prepared. Four of these qualify as ILs and one as room temperature IL (RTIL). The properties of the borate anion [B(hfip)4]− and its aluminum analogue [Al(hfip)4]− were compared based on the available structural information from XRD. Viscosities (10.3 (90 °C) to 855 (0 °C) mPa s−1) and conductivities (0.603 (30 °C) to 4.844 (90 °C) mS cm−1) were measured between 0 and 90 °C, and described by the Vogel–Fulcher–Tammann (VFT) equations. The properties of the [B(hfip)4]− ILs were analyzed in the context of the anion-dependent molecular volume Vm-viscosity-/conductivity-correlations, also in comparison to ILs with [BF4]−/[PF6]−, [N(CN)2]−, [Tf2N]− and [Al(hfip)4]− counterions. The viscosities and conductivities of [B(hfip)4]− ILs are slightly inferior to [Al(hfip)4]− ILs, similar to/better than all other anions given above. According to the Walden plots, the ionicity of the [B(hfip)4]− ILs may at least be classified as “good”. By sharp contrast to the [Al(hfip)4]− ILs, the [B(hfip)4]− ILs have good stability against humidity/water. Thus, handling of [B(hfip)4]− ILs in an open laboratory atmosphere over hours and days is allowed and further facilitates the use of this new IL class.
Co-reporter:Anna J. Lehner, Nils Trapp, Harald Scherer and Ingo Krossing
Dalton Transactions 2011 vol. 40(Issue 7) pp:1448-1452
Publication Date(Web):03 Dec 2010
DOI:10.1039/C0DT01076F
The CX3+ salts [CCl3]+[Al(ORF)4]−1, [CCl3]+[(RFO)3Al–F–Al(ORF)3]−2, [CBr3]+[Al(ORF)4]−3, [CBr3]+[(RFO)3Al–F–Al(ORF)3]−4 (RF = C(CF3)3) were prepared in 56 to 85% yield from CX4 (X = Cl, Br) and the corresponding silver salts (weight balance, NMR, IR, X-ray structure of 1). The most convenient solvent for the preparation of 1 and 2 is SO2ClF but for 3 and 4 it is SO2. The reactions are complete after about three days stirring at −30 to −40 °C. The salts are stable for weeks in solution at −40 °C and stable for a few hours at RT in the solid state. In SO2ClF (1, 2) or SO2 (3, 4) solution they decompose slowly at −20 °C and within several hours at RT; in general the CBr3+ salts are more stable than the CCl3+ homologues. The decomposition products were assigned as CCl3F and primarily CBr2F2 (which likely forms as a Lewis acid induced disproportionation product of the initial CBr3F). The C-X vibrations of the salts were found in the expected range and the assignments were made based on experimental and calculated data. The IR spectrum of a CBr3+ salt is for the first time reported here.
Co-reporter:Chul-Woong Cho, Ulrich Preiss, Christian Jungnickel, Stefan Stolte, Jürgen Arning, Johannes Ranke, Andreas Klamt, Ingo Krossing, and Jorg Thöming
The Journal of Physical Chemistry B 2011 Volume 115(Issue 19) pp:6040-6050
Publication Date(Web):April 19, 2011
DOI:10.1021/jp200042f
In this article, we present evolutionary models to predict the octanol–water partition coefficients (log P), water solubilities, and critical micelle concentrations (CMCs) of ionic liquids (ILs), as well as the anionic activity coefficients and hydrophobicities in pure water and octanol–water. They are based on a polyparameter linear free energy relationship (LFER) using measured and/or DFT-calculated LFER parameters: hydrogen-bonding acidity (A), hydrogen-bonding basicity (B), polarizability/dipolarity (S), excess molar refraction (E), and McGowan volume (V) of IL ions. With both calculated or experimental LFER descriptors of IL ions, the physicochemical parameters were predicted with an errors of 0.182–0.217 for the octanol–water partition coefficient and 0.131–0.166 logarithmic units for the water solubility. Because experimentally determined solute parameters of anions are not currently available, the CMC, anionic activity coefficient, and hydrophobicity were predicted with quantum-chemical methods with R2 values of at least 0.99, as well as errors below 0.168 logarithmic units. These new approaches will facilitate the assessment of the technical applicability and environmental fate of ionic compounds even before their synthesis.
Co-reporter:Dr. Ulrich Preiss;Dr. Sergey P. Verevkin;Dr. Thorsten Koslowski;Dr. Ingo Krossing
Chemistry - A European Journal 2011 Volume 17( Issue 23) pp:6508-6517
Publication Date(Web):
DOI:10.1002/chem.201003150
Abstract
We present the full enthalpic phase transition cycle for ionic liquids (ILs) as examples of non-classical salts. The cycle was closed for the lattice, solvation, dissociation, and vaporization enthalpies of 30 different ILs, relying on as much experimental data as was available. High-quality dissociation enthalpies were calculated at the G3 MP2 level. From the cycle, we could establish, for the first time, the lattice and solvation enthalpies of ILs with imidazolium ions. For vaporization, lattice, and dissociation enthalpies, we also developed new prediction methods in the course of our investigations. Here, as only single-ion values need to be calculated and the tedious optimization of an ion pair can be circumvented, the computational time is short. For the vaporization enthalpy, a very simple approach was found, using a surface term and the calculated enthalpic correction to the total gas-phase energy. For the lattice enthalpy, the most important constituent proved to be the calculated conductor-like screening model (COSMO) solvation enthalpy in the ideal electric conductor. A similar model was developed for the dissociation enthalpy. According to our assessment, the typical error of the lattice enthalpy would be 9.4 kJ mol−1, which is less than half the deviation we get when using the (optimized) Kapustinskii equation or the recent volume-based thermodynamics (VBT) theory. In contrast, the non-optimized VBT formula gives lattice enthalpies 20 to 140 kJ mol−1 lower than the ones we assessed in the cycle, because of the insufficient description of dispersive interactions. Our findings show that quantum-chemical calculations can greatly improve the VBT approaches, which were parameterized for simple, inorganic salts with ideally point-shaped charges. In conclusion, we suggest the term “augmented VBT”, or “aVBT”, to describe this kind of theoretical approach.
Co-reporter:Philipp Eiden, Safak Bulut, Tobias Köchner, Christian Friedrich, Thomas Schubert, and Ingo Krossing
The Journal of Physical Chemistry B 2011 Volume 115(Issue 2) pp:300-309
Publication Date(Web):December 7, 2010
DOI:10.1021/jp108059x
The viscosity (η) and electrical conductivity (κ) of ionic liquids are, next to the melting point, the two key properties of general interest. The knowledge of temperature-dependent η and κ data before their first synthesis would permit a much more target-oriented development of ionic liquids. We present in this work a novel approach to predict the viscosity and electrical conductivity of an ionic liquid without further input of experimental data. For the viscosity, only some basic physical observables like the Gibbs solvation energy (ΔGsolv*,∞), which was calculated at the affordable DFT-level (RI-)BP86/TZVP/COSMO, the molecular radius, calculated from the molecular volume Vm of the ion volumes, and the symmetry number (σ), according to group theory, are necessary as input. The temperature dependency (253−373 K) of the viscosity (4−19000 mPa s) was modeled by an Arrhenius approach. An alternative way, which avoids the deficits of the Arrhenius relation by a series expansion in the exponential term, is also presented. On the basis of their close connection, the same set of parameters is suitable to describe the electrical conductivity as well (238−468 K, 0.003−193 mS/cm). Nevertheless, more elegant alternatives like the usage of the Stokes−Einstein/Nernst−Einstein relation or the Walden rule are highlighted in this work. During this investigation, we additionally found an approach to predict the dielectric constant ε* of an ionic liquid at 298 K by using Vm and ΔGsolv*,∞ between ε* = 9 and 43.
Co-reporter:Dr. Tobias Köchner;Dr. Nils Trapp;Dipl.-Chem. Tobias A. Engesser;Dipl.-Chem. Anna J. Lehner;Dr.-Ing. Caroline Röhr;Dr. Sebastian Riedel;Dr. Carsten Knapp;Dr. Harald Scherer ;Dr. Ingo Krossing
Angewandte Chemie International Edition 2011 Volume 50( Issue 47) pp:11253-11256
Publication Date(Web):
DOI:10.1002/anie.201104666
Co-reporter:Dr. Safak Bulut;Dr. Philipp Eiden;Witali Beichel;Dr. John M. Slattery;Dr. Tom F. Beyersdorff;Dr. Thomas J. S. Schubert; Dr. Ingo Krossing
ChemPhysChem 2011 Volume 12( Issue 12) pp:2296-2310
Publication Date(Web):
DOI:10.1002/cphc.201100214
Abstract
A series of bis(trifluoromethylsulfonyl)imide ionic liquids (ILs) with classical as well as mildly functionalized cations was prepared and their viscosities and conductivities were determined as a function of the temperature. Both were analyzed with respect to Arrhenius, Litovitz and Vogel–Fulcher–Tammann (VFT) behaviors, as well as in the context of their molecular volume (Vm). Their viscosity and conductivity are highly correlated with Vm/T or related expressions (R2≥0.94). With the knowledge of Vm of new cations, these correlations allow the temperature-dependent prediction of the viscosity and conductivity of hitherto unknown, non- or mildly functionalized ILs with low error bars (0.05 and 0.04 log units, respectively). The influence of the cation structure and mild functionalization on the physical properties was studied with systematically altered cations, in which Vm remained similar. The To parameter obtained from the VFT fits was compared to the experimental glass temperature (Tg) and the Tg/To ratio for each IL was calculated using both experimental values and Angell’s relationship. With Walden plots we investigated the IL ionicity and interpreted it in relation to the cation effects on the physical IL properties. We checked the validity of these Vm/T relations by also including the recently published variable temperature viscosity and conductivity data of the [Al(ORF)4]− ILs with RF=C(H)(CF3)2 (error bars for the prediction: 0.09 and 0.10 log units, respectively).
Co-reporter:Dr. Ulrich P. Preiss;Witali Beichel;Anna M. T. Erle;Dr. Yauheni U. Paulechka; Dr. Ingo Krossing
ChemPhysChem 2011 Volume 12( Issue 16) pp:2959-2972
Publication Date(Web):
DOI:10.1002/cphc.201100522
Abstract
An investigation of the melting points of 520 organic 1:1 salts is presented with the aim of developing a universal, simple, physically well-founded prediction scheme. The general reliability and reproducibility of the recorded experimental data are discussed with respect to purity, phase behavior, disorder and thermal history of a given substance. Additionally, mistakes, systematic errors, or lack of conventions can lead to considerable differences in the experimental measurements. A rough error bar for the reproducibility of the melting points of organic salts of ±5 to ±15 °C can be assigned. With this restraint, we developed two simple, semiempirical, five- and nine-parameter schemes with easy-to-calculate quantities. With these, we could predict the melting temperature of a given organic salt in the temperature range of −25 to +300 °C with an average error of 33.5 °C and a relative error of 9.3 %. All calculated quantities are assessed with the help of conventional DFT, COSMO and COSMO-RS calculations, and are currently implemented into the IL-Prop module of the upcoming version of COSMOtherm. These prediction schemes are suitable for high-throughput computational screening of substances in the context of “computer-aided synthesis”. Therefore, they are valuable tools to find a compound with a suitable melting point before its first synthesis.
Co-reporter:Dr. Ulrich P. Preiss;Witali Beichel;Anna M. T. Erle;Dr. Yauheni U. Paulechka; Dr. Ingo Krossing
ChemPhysChem 2011 Volume 12( Issue 16) pp:
Publication Date(Web):
DOI:10.1002/cphc.201190080
Co-reporter:Dr. Daniel Himmel;Dipl.-Chem. Sascha K. Goll;Dr. Ivo Leito;Dr. Ingo Krossing
Chemistry - A European Journal 2011 Volume 17( Issue 21) pp:5808-5826
Publication Date(Web):
DOI:10.1002/chem.201003164
Abstract
The COSMO cluster-continuum (CCC) solvation model is introduced for the calculation of standard Gibbs solvation energies of protons. The solvation sphere of the proton is divided into an inner proton–solvent cluster with covalent interactions and an outer solvation sphere that interacts electrostatically with the cluster. Thus, the solvation of the proton is divided into two steps that are calculated separately: 1) The interaction of the proton with one or more solvent molecules is calculated in the gas phase with high-level quantum-chemical methods (modified G3 method). 2) The Gibbs solvation energy of the proton–solvent cluster is calculated by using the conductor-like screening model (COSMO). For every solvent, the solvation of the proton in at least two (and up to 11) proton–solvent clusters was calculated. The resulting Gibbs solvation energies of the proton were weighted by using Boltzmann statistics. The model was evaluated for the calculation of Gibbs solvation energies by using experimental data of water, MeCN, and DMSO as a reference. Allowing structural relaxation of the proton–solvent clusters and the use of structurally relaxed Gibbs solvation energies improved the accordance with experimental data especially for larger clusters. This variation is denoted as the relaxed COSMO cluster-continuum (rCCC) model, for which we estimate a 1σ error bar of 10 kJ mol−1. Gibbs solvation energies of protons in the following representative solvents were calculated: Water, acetonitrile, sulfur dioxide, dimethyl sulfoxide, benzene, diethyl ether, methylene chloride, 1,2-dichloroethane, sulfuric acid, fluorosulfonic acid, and hydrogen fluoride. The obtained values are absolute chemical standard potentials of the proton (pH=0 in this solvent). They are used to anchor the individual solvent specific acidity (pH) scales to our recently introduced absolute acidity scale.
Co-reporter:Dr. Daniel Himmel;Dipl.-Chem. Sascha K. Goll;Dr. Ivo Leito;Dr. Ingo Krossing
Chemistry - A European Journal 2011 Volume 17( Issue 21) pp:
Publication Date(Web):
DOI:10.1002/chem.201190104
Co-reporter:Dipl.-Chem. Anne Kraft;Jennifer Beck ;Dr. Ingo Krossing
Chemistry - A European Journal 2011 Volume 17( Issue 46) pp:12975-12980
Publication Date(Web):
DOI:10.1002/chem.201102077
Abstract
The pnictocenium salts [Cp*PCl]+[μCl]− (1 a), [Cp*PCl]+[ClAl(ORF)3]− (1 b), [Cp*AsCl]+[ClAl(ORF)3]− (2), and [(Cp*)2P]+[μCl]− (3), in which Cp*=Me5C5, μCl=(FRO)3AlClAl(ORF)3, and ORF=OC(CF3)3, were prepared by halide abstraction from the respective halopnictines with the Lewis superacid PhFAl(ORF)3.1 The X-ray crystal structures of 1 a, 2, and 3 established that in the half as well as in the sandwich cations the Cp* rings are attached in an η2-fashion. By using one or two equivalents of the Lewis acid, the two new weakly coordinating anions [μCl]− and [ClAl(ORF)3]− resulted. They also stabilize the highly reactive cations in PhF or 1,2-F2C6H4 solution at room temperature. The chloride ion affinities (CIAs) of a range of classical strong Lewis acids were also investigated. The calculations are based on a set of isodesmic BP86/SV(P) reactions and a non-isodesmic reference reaction assessed at the G3MP2 level.
Co-reporter:Dr. Tobias Köchner;Dr. Nils Trapp;Dipl.-Chem. Tobias A. Engesser;Dipl.-Chem. Anna J. Lehner;Dr.-Ing. Caroline Röhr;Dr. Sebastian Riedel;Dr. Carsten Knapp;Dr. Harald Scherer ;Dr. Ingo Krossing
Angewandte Chemie 2011 Volume 123( Issue 47) pp:11449-11452
Publication Date(Web):
DOI:10.1002/ange.201104666
Co-reporter:Michael P. Stewart, Lacey Marina Paradee, Ines Raabe, Nils Trapp, John S. Slattery, Ingo Krossing, William E. Geiger
Journal of Fluorine Chemistry 2010 Volume 131(Issue 11) pp:1091-1095
Publication Date(Web):November 2010
DOI:10.1016/j.jfluchem.2010.03.001
Anodic voltammetry and electrolysis of the metallocenes ferrocene, ruthenocene, and nickelocene have been studied in dichloromethane containing two different fluorine-containing anions in the supporting electrolyte. The perfluoroalkoxyaluminate anion [Al(OC(CF3)3)4]− has very low nucleophilicity, as shown by its inertness towards the strong electrophile [RuCp2]+ and by computation of its electrostatic potential in comparison to other frequently used electrolyte anions. The low ion-pairing ability of this anion was shown by the large spread in E1/2 potentials (ΔE1/2 = 769 mV) for the two one-electron oxidations of bis(fulvalene)dinickel. The hexafluoroarsenate anion [AsF6]−, on the other hand, reacts rapidly with the ruthenocenium ion and is much more strongly ion-pairing towards oxidized bis(fulvalene)dinickel (ΔE1/2 = 492 mV). In terms of applications of these two anions to the anodic oxidation of organometallic sandwich complexes, the behavior of [Al(OC(CF3)3)4]− is similar to that of other weakly-coordinating anions such as [B(C6F5)4]−, whereas that of [AsF6]− is similar to the more traditional electrolyte anions such as [PF6]− and [BF4]−. Additionally, the synthesis and crystal structure of [Cp2Fe][Al(OC(CF3)3)4] are reported.The anodic electrochemical behavior of several monometallic and dimetallic sandwich complexes was studied in dichloromethane containing either [Al(OC(CF3)3)4]− or [AsF6]− as the supporting electrolyte anion. The former behaves as a weakly-coordinating, non-nucleophilic anion towards reactive organometallic cation radicals, whereas the latter behaves more like a traditional small anion.
Co-reporter:Dipl.-Chem. Tobias Köchner;Dr. Sebastian Riedel;Dipl.-Chem. Anna J. Lehner;Dr. Harald Scherer;Dr. Ines Raabe;Tobias A. Engesser;Dipl.-Chem. Franziska W. Scholz;Dipl.-Chem. Urs Gellrich;Dipl.-Chem. Philipp Eiden;Dipl.-Chem. Roberto A. PazSchmidt;Dr. Dietmar A. Plattner;Dr. Ingo Krossing
Angewandte Chemie 2010 Volume 122( Issue 44) pp:8316-8320
Publication Date(Web):
DOI:10.1002/ange.201003031
Co-reporter:Ulrich Preiss, Safak Bulut and Ingo Krossing
The Journal of Physical Chemistry B 2010 Volume 114(Issue 34) pp:11133-11140
Publication Date(Web):August 6, 2010
DOI:10.1021/jp104679m
The melting points (Tfus) of crystalline ionic liquids are calculated from the ratio of the fusion enthalpy and entropy at the melting point where solid and liquid phases are in chemical equilibrium (ΔGT = 0), and therefore, Tfus = ΔfusHT/ΔfusST (if T = Tfus). We specify two variants of this method that have no need for experimental input or tedious simulations but rely on simple calculations feasible with standard quantum chemical program codes and may further be augmented by COSMO-RS. Only single ions are used as input, making the demanding calculation of ion pairs superfluous. The fusion enthalpy is obtained by the principles of volume-based thermodynamics (ion volumes as the major contributor), which may additionally be augmented by COSMO-RS interaction enthalpies for increased accuracy. The calculation of the fusion entropy largely relies on a procedure originally developed for neutral organic molecules that was extended to molecular ionic compounds. Its contributors are the site symmetry σ and the number of torsion angles τ, which are both determined individually for the cation and the anion and are included as their geometric mean. The two methods were tested on several sets of ionic liquids (ILs) and a combination of all sets (67 ILs) that span an experimental melting temperature range of 337 °C. The average error of the simpler, volume-based model (only ion volumes, σ, and τ as input) is 36.4 °C and that of the augmented method (using ion volumes, σ, τ, and COSMO-RS output) is 24.5 °C.
Co-reporter:Dr. Ulrich Preiss;Dr. Vladimir N. Emel'yanenko; Dr. Sergey P. Verevkin;Dr. Daniel Himmel;Dr. Yauheni U. Paulechka; Dr. Ingo Krossing
ChemPhysChem 2010 Volume 11( Issue 16) pp:3425-3431
Publication Date(Web):
DOI:10.1002/cphc.201000614
Abstract
Modeling of the temperature-dependent liquid entropy of ionic liquids (ILs) with great accuracy using COSMO-RS is demonstrated. The minimum structures of eight IL ion pairs are investigated and the entropy, calculated from ion pairs, is found to differ on average only 2 % from the available experimental values (119 data points). For calculations with single ions, the average error amounts to 2.6 % and stronger-coordinating ions tend to give higher deviations. Additionally, the first parameterization of the standard liquid entropy for ILs is presented in the context of traditional volume-based thermodynamics (Sl0=1.585 kJ mol−1 K−1 nm−3⋅rm3+14.09 J mol−1 K−1), which sheds light on the statistical treatment of ionic interactions. The findings provide the first direct access to accurate predictions of liquid entropies of ILs, which are tedious and time-consuming to measure.
Co-reporter:Dipl.-Chem. Safak Bulut;Petra Klose;M.Sc. Mian-Mian Huang;Dr. Hermann Weingärtner;Dr. Paul J. Dyson;Dr. Gábor Laurenczy;Dr. Christian Friedrich;Jakob Menz;Dr. Klaus Kümmerer;Dr. Ingo Krossing
Chemistry - A European Journal 2010 Volume 16( Issue 44) pp:13139-13154
Publication Date(Web):
DOI:10.1002/chem.201000982
Abstract
A large series of ionic liquids (ILs) based on the weakly coordinating alkoxyaluminate [Al(hfip)4]− (hfip: hexafluoroisopropoxy) with classical as well as functionalized cations were prepared, and their principal physical properties determined. Melting points are between 0 ([C4MMIM][Al(hfip)4]) and 69 °C ([C3MPip][Al(hfip)4]); three qualify as room-temperature ILs (RTILs). Crystal structures for six ILs were determined; their structural parameters and anion–cation contacts are compared here with known ILs, with a special focus on their influence on physical properties. Moreover, the biodegradability of the compounds was investigated by using the closed-bottle and the manometric respirometry test. Temperature-dependent viscosities and conductivities were measured between 0 and 80 °C, and described by either the Vogel–Fulcher–Tammann (VFT) or the Arrhenius equations. Moreover, conductivities and viscosities were investigated in the context of the molecular volume, Vm. Physical property–Vm correlations were carried out for various temperatures, and the temperature dependence of the molecular volume was analyzed by using crystal structure data and DFT calculations. The IL ionicity was investigated by Walden plots; according to this analysis, [Al(hfip)4]− ILs may be classified as “very good to good ILs”; while [C2MIM][Al(hfip)4] is a better IL than [C2MIM][NTf2]. The dielectric constants of ten [Al(hfip)4]− ILs were determined, and are unexpectedly high (εr=11.5 to 16.8). This could be rationalized by considering additional calculated dipole moments of the structures frozen in the solid state by DFT. The determination of hydrogen gas solubility in [Al(hfip)4]− RTILs by high-pressure NMR spectroscopy revealed very high hydrogen solubilities at 25 °C and 1 atm. These results indicate the significant potential of this class of ILs in manifold applications.
Co-reporter:JohnM. Slattery Dr.;Alexer Higelin;Thomas Bayer Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 18) pp:3228-3231
Publication Date(Web):
DOI:10.1002/anie.201000156
Co-reporter:JohnM. Slattery Dr.;Alexer Higelin;Thomas Bayer Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 18) pp:
Publication Date(Web):
DOI:10.1002/anie.201001595
Co-reporter:Dr. Daniel Himmel;Dipl.-Chem. SaschaK. Goll;Dr. Ivo Leito;Dr. Ingo Krossing
Angewandte Chemie International Edition 2010 Volume 49( Issue 38) pp:6885-6888
Publication Date(Web):
DOI:10.1002/anie.201000252
Co-reporter:Dipl.-Chem. Tobias Köchner;Dr. Sebastian Riedel;Dipl.-Chem. Anna J. Lehner;Dr. Harald Scherer;Dr. Ines Raabe;Tobias A. Engesser;Dipl.-Chem. Franziska W. Scholz;Dipl.-Chem. Urs Gellrich;Dipl.-Chem. Philipp Eiden;Dipl.-Chem. Roberto A. PazSchmidt;Dr. Dietmar A. Plattner;Dr. Ingo Krossing
Angewandte Chemie International Edition 2010 Volume 49( Issue 44) pp:8139-8143
Publication Date(Web):
DOI:10.1002/anie.201003031
Co-reporter:Dr. Daniel Himmel;Dipl.-Chem. SaschaK. Goll;Dr. Ivo Leito;Dr. Ingo Krossing
Angewandte Chemie 2010 Volume 122( Issue 38) pp:7037-7040
Publication Date(Web):
DOI:10.1002/ange.201000252
Co-reporter:Ulrich P. R. M. Preiss, John M. Slattery and Ingo Krossing
Industrial & Engineering Chemistry Research 2009 Volume 48(Issue 4) pp:2290-2296
Publication Date(Web):January 5, 2009
DOI:10.1021/ie801268a
We present a fast and reliable general estimation of the heat capacity, Cp, and temperature-dependent density, ρ, of ionic liquids, based on a new in silico method to calculate the molecular volume, Vm. The knowledge of Vm allows the prediction of many physical properties of hitherto unknown ionic liquids as well as the prediction of lattice energies and entropies of salts.
Co-reporter:Uro&x161; Gro&x161;elj;Dieter Seebach;D.Michael Badine;W.Bernd Schweizer;AlbertK. Beck;Petra Klose;Yujiro Hayashi;Tadafumi Uchimaru
Helvetica Chimica Acta 2009 Volume 92( Issue 7) pp:1225-1259
Publication Date(Web):
DOI:10.1002/hlca.200900179
Abstract
Structures of the reactive intermediates (enamines and iminium ions) of organocatalysis with diarylprolinol derivatives have been determined. To this end, diarylprolinol methyl and silyl ethers, 1, and aldehydes, PhCH2CHO, tBuCH2CHO, PhCH=CHCHO, are condensed to the corresponding enamines, A and 3 (Scheme 2), and cinnamoylidene iminium salts, B and 4 (Scheme 3). These are isolated and fully characterized by melting/decomposition points, [α]D, elemental analysis, IR and NMR spectroscopy, and high-resolution mass spectrometry (HR-MS). Salts with BF4, PF6, SbF6, and the weakly coordinating Al[OC(CF3)3]4 anion were prepared. X-Ray crystal structures of an enamine and of six iminium salts have been obtained and are described herein (Figs. 2 and 4–8, and Tables 2 and 7) and in a previous preliminary communication (Helv. Chim. Acta2008, 91, 1999). According to the NMR spectra (in CDCl3, (D6)DMSO, (D6)acetone, or CD3OD; Table 1), the major isomers 4 of the iminium salts have (E)-configuration of the exocyclic NC(1′) bond, but there are up to 11% of the (Z)-isomer present in these solutions (Fig. 1). In all crystal structures, the iminium ions have (E)-configuration, and the conformation around the exocyclic N-CC-O bond is synclinal-exo (cf.C and L), with one of the phenyl groups over the pyrrolidine ring, and the RO group over the π-system. One of the meta-substituents (Me in 4b, CF3 in 4c and 4e) on a 3,5-disubstituted phenyl group is also located in the space above the π-system. DFT Calculations at various levels of theory (Tables 3–6) confirm that the experimentally determined structures (cf. Fig. 10) are by far (up to 8.3 kcal/mol) the most stable ones. Implications of the results with respect to the mechanism of organocatalysis by diarylprolinol derivatives are discussed.
Co-reporter:Andreas Reisinger Dr.;Nils Trapp Dr.;Carsten Knapp Dr.;Daniel Himmel Dr.;Frank Breher ;Heinz Rüegger Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 37) pp:9505-9520
Publication Date(Web):
DOI:10.1002/chem.200900100
Abstract
Compounds including the free or coordinated gas-phase cations [Ag(η2-C2H4)n]+ (n=1–3) were stabilized with very weakly coordinating anions [A]− (A=Al{OC(CH3)(CF3)2}4, n=1 (1); Al{OC(H)(CF3)2}4, n=2 (3); Al{OC(CF3)3}4, n=3 (5); {(F3C)3CO}3Al-F-Al{OC(CF3)3}3, n=3 (6)). They were prepared by reaction of the respective silver(I) salts with stoichiometric amounts of ethene in CH2Cl2 solution. As a reference we also prepared the isobutene complex [(Me2CCH2)Ag(Al{OC(CH3)(CF3)2}4)] (2). The compounds were characterized by multinuclear solution-NMR, solid-state MAS-NMR, IR and Raman spectroscopy as well as by their single crystal X-ray structures. MAS-NMR spectroscopy shows that the [Ag(η2-C2H4)3]+ cation in its [Al{OC(CF3)3}4]− salt exhibits time-averaged D3h-symmetry and freely rotates around its principal z-axis in the solid state. All routine X-ray structures (2θmax.<55°) converged within the 3σ limit at CC double bond lengths that were shorter or similar to that of free ethene. In contrast, the respective Raman active CC stretching modes indicated red-shifts of 38 to 45 cm−1, suggesting a slight CC bond elongation. This mismatch is owed to residual librational motion at 100 K, the temperature of the data collection, as well as the lack of high angular data owing to the anisotropic electron distribution in the ethene molecule. Therefore, a method for the extraction of the CC distance in [M(C2H4)] complexes from experimental Raman data was developed and meaningful CC distances were obtained. These spectroscopic CC distances compare well to newly collected X-ray data obtained at high resolution (2θmax.=100°) and low temperature (100 K). To complement the experimental data as well as to obtain further insight into bond formation, the complexes with up to three ligands were studied theoretically. The calculations were performed with DFT (BP86/TZVPP, PBE0/TZVPP), MP2/TZVPP and partly CCSD(T)/AUG-cc-pVTZ methods. In most cases several isomers were considered. Additionally, [M(C2H4)3] (M=Cu+, Ag+, Au+, Ni0, Pd0, Pt0, Na+) were investigated with AIM theory to substantiate the preference for a planar conformation and to estimate the importance of σ donation and π back donation. Comparing the group 10 and 11 analogues, we find that the lack of π back bonding in the group 11 cations is almost compensated by increased σ donation.
Co-reporter:Gustavo Santiso-Quiñones Dr.;Alexer Higelin;Julia Schaefer;Robin Brückner;Carsten Knapp Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 27) pp:6663-6677
Publication Date(Web):
DOI:10.1002/chem.200900244
Co-reporter:Philipp Eiden;Qunxian Liu Dr.;Sherif ZeinElAbedin Dr.;Frank Endres Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 14) pp:3426-3434
Publication Date(Web):
DOI:10.1002/chem.200801616
Co-reporter:Gustavo Santiso-Quiñones Dr.;Robin Brückner;Carsten Knapp Dr.;Isabelle Dionne Dr.;Jack Passmore Dr. Dr.
Angewandte Chemie 2009 Volume 121( Issue 6) pp:1153-1157
Publication Date(Web):
DOI:10.1002/ange.200804021
Co-reporter:Gustavo Santiso-Quiñones Dr.;Robin Brückner;Carsten Knapp Dr.;Isabelle Dionne Dr.;Jack Passmore Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 6) pp:1133-1137
Publication Date(Web):
DOI:10.1002/anie.200804021
Co-reporter:Ines Raabe Dr.;Katrin Wagner;Kristin Guttsche;Mingkui Wang Dr.;Michael Grätzel Dr.;Gustavo Santiso-Quiñones Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 8) pp:1966-1976
Publication Date(Web):
DOI:10.1002/chem.200800417
Co-reporter:U. Preiss;C. Jungnickel Dr.;J. Thöming Dr. Dr.;J. &x141;uczak;M. Diedenhofen Dr.;A. Klamt Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 35) pp:8880-8885
Publication Date(Web):
DOI:10.1002/chem.200900024
Abstract
Some ionic liquids (ILs) are structurally analogous to surfactants, especially those that consist of a combination of organic and inorganic ions. The critical micelle concentration (CMC) is a basic parameter of surface chemistry and colloid science. A significant amount of research has already been carried out to determine the CMCs of ILs. However, because of the many varied cation/anion combinations, it is a daunting task to measure the CMCs of all possible ILs. Herein we suggest a general rule for predicting the CMCs of ionic surfactants in water based on data from COSMO-RS calculations. In accordance with the Stauff–Klevens rule, the molecular volume (Vm) is sufficient to describe similar homologous series of cationic surfactants such as imidazolium- and ammonium-based ionic liquids with varying side-chain lengths. However, to also include anionic surfactants like Na[CnSO4] in a more general correlation, Vm has to be exchanged by the cubed molecular radius () and the molecular surface has to be used as an additional descriptor. Furthermore, to describe double amphiphilic compounds like [C4MIm][C8SO4], the enthalpies of mixtures calculated by COSMO-RS have to be taken into account. The resulting equation had allowed us to predict the CMCs of all of the 36 tested surfactants with an error similar to or smaller than the usual experimental errors (18 different cations, 10 different anions: root mean squared error (rmse)=0.191 logarithmic units; R2=0.994). We discuss the factors governing micelle formation on the basis of our calculations and show that the structure of our equation can be related to Gibbs’ theory of crystallization.
Co-reporter:Stephan Schulz ;Daniella Schuchmann ;Daniel Himmel Dr.;Dieter Bläser;Rol Boese
Angewandte Chemie International Edition 2009 Volume 48( Issue 31) pp:5748-5751
Publication Date(Web):
DOI:10.1002/anie.200902202
Co-reporter:Ines Raabe, Daniel Himmel, Sonja Müller, Nils Trapp, Martin Kaupp and Ingo Krossing
Dalton Transactions 2008 (Issue 7) pp:946-956
Publication Date(Web):18 Dec 2007
DOI:10.1039/B714271D
The room-temperature stable CI3+ salts [CI3]+[pftb]−1 and [CI3]+[al-f-al]−2 ([pftb]− = [Al(OC(CF3)3)4]−; [al-f-al]− = [((CF3)3CO)3Al-F-Al(OC(CF3)3)3]−) were prepared in quantitative yields from purified CI4 and the corresponding silver aluminates with total exclusion of light (NMR, IR, UV-VIS, X-ray diffraction). The isolated CI3+ cation is trigonal planar with a sum of ∠(I–C–I) = 360.0° (1) and 359.9° (2). Attempts to prepare CHI2+ and CH2I+ salts from CHI3 or CH2I2/Ag[pftb] mixtures remained unsuccessful; the reaction with CH2I2 leads to the formation of the adduct [Ag(CH2I2)3]+[pftb]−3, while for HCI3, dismutation with formation of 1 as well as 3 was observed. All particles were also calculated at the MP2/TZVPP level to predict the vibrational and electronic spectra as well as to calculate the Gibbs free energies of all reactions (ΔG°, gas phase and CH2Cl2 solution). Quantum chemical calculations were also used to investigate the stability of the [pftb]− anion against the electrophilic attack of the CX3+ and CHnX3–n+ cations (X = F–I, n = 1–3). The strength of the Lewis acidity of these cations and of the isoelectronic boron halides BX3 and BHnX3–n have been established on the basis of their fluoride ion affinities (FIAs). The FIAs of the carbon and the boron containing compounds show opposite trends, with fluorinated halomethyl cations being stronger acids than their heavier congeners but iodinated holoboranes being stronger acids than their lighter homologues.
Co-reporter:LutzO. Müller Dr.;Daniel Himmel Dr.;Julia Stauffer;Gunther Steinfeld Dr.;John Slattery Dr.;Gustavo Santiso-Quiñones Dr.;Volker Brecht Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 40) pp:7659-7663
Publication Date(Web):
DOI:10.1002/anie.200800783
Co-reporter:Daniel Himmel Dr.;Nils Trapp Dipl.-Chem. Dr.;Sra Altmannshofer Dipl.-Chem.;Verena Herz Dipl.-Phys.;Georg Eickerling Dr.;Wolfgang Scherer Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 41) pp:7798-7801
Publication Date(Web):
DOI:10.1002/anie.200802616
Co-reporter:LutzO. Müller;Daniel Himmel Dr.;Julia Stauffer;Gunther Steinfeld Dr.;John Slattery Dr.;Gustavo Santiso-Quiñones Dr.;Volker Brecht Dr.
Angewandte Chemie 2008 Volume 120( Issue 40) pp:7772-7776
Publication Date(Web):
DOI:10.1002/ange.200800783
Co-reporter:Daniel Himmel Dr.;Nils Trapp Dipl.-Chem. Dr.;Sra Altmannshofer Dipl.-Chem.;Verena Herz Dipl.-Phys.;Georg Eickerling Dr.;Wolfgang Scherer Dr.
Angewandte Chemie 2008 Volume 120( Issue 41) pp:7914-7917
Publication Date(Web):
DOI:10.1002/ange.200802616
Co-reporter:Gustavo Santiso-Quiñones, Andreas Reisinger, John Slattery and Ingo Krossing
Chemical Communications 2007 (Issue 47) pp:5046-5048
Publication Date(Web):27 Sep 2007
DOI:10.1039/B710899K
Stable salts of the first homoleptic Cu–phosphorus and Cu–ethene complexes, [Cu(η2-P4)2]+ and [Cu(η2-C2H4)3]+, isolated by the aid of the weakly coordinating anion (WCA) [Al(OC(CF3)3)4]−, were obtained.
Co-reporter:Ines Raabe, Caroline Röhr and Ingo Krossing
Dalton Transactions 2007 (Issue 46) pp:5376-5386
Publication Date(Web):11 Oct 2007
DOI:10.1039/B711639J
What is the preferred coordination site of CI3+? Recent computational work suggested the iodine atoms of the Lewis acid CI3+ to be more electrophilic than the classically expected carbon atom, e.g. the complex with water is of type I2C–I⋯OH2+ and not the classically expected I3C–OH2+. If this structure is correct, one may also anticipate reactions of CI3+ as an I+ donor. Thus, we were interested in investigating the chemistry of CI3+ in the room-temperature stable salt [CI3]+[pftb]− ([pftb]− = [Al(OC(CF3)3)4]−) with weak nucleophiles that i) mimic water (OEt2) or ii) are electronically deactivated weak nucleophiles (PX3, X = Cl–I; AsI3). One question was: is it possible to obtain iodine-coordinated Lewis acid–base adducts of the CI3+ cation? With Et2O as a base, the cation behaves as a strong Lewis acid and cleaves the ether to give I3C–OEt, C2H4 and [H(Et2O)2]+. By contrast PX3 and AsI3 coordinate to the CI3+ cations and the adducts have classical, carbon-bound ethane-like structures, as proven by X-ray single-crystal diffraction, IR, UV-Vis and NMR spectroscopy. From variable temperature 13C NMR studies, it followed for the I3C–AsI3+ salt that the equilibrium between CI3+ and AsI3 is reversible and temperature dependent in solution. The I3C–PI3+ salt decomposes at room temperature giving PI4+ and C2I4, likely through an iodine coordinated I2C–I⋯PI3+ intermediate. Thus CI3+ may also act as an I+ donor. All reactions are in agreement with ab initio quantum chemical calculations at the MP2/TZVPP level and assignments of experimental spectra were aided by quantum chemistry.
Co-reporter:Andreas Reisinger Dr.;Nils Trapp Dipl.-Chem. Dr.;Sra Altmannshofer Dipl.-Chem.;Verena Herz Dipl.-Phys.;Manuel Presnitz Dipl.-Phys.;Wolfgang Scherer Dr.
Angewandte Chemie 2007 Volume 119(Issue 43) pp:
Publication Date(Web):26 OCT 2007
DOI:10.1002/ange.200790216
Hoch droben im ewigen Eis würden sich die homoleptischen Silber(I)-Acetylen-Komplexe, die I. Krossing, W. Scherer und Mitarbeiter in ihrer Zuschrift auf S. 8445 ff. vorstellen, richtig wohl fühlen. Für die Synthese, Handhabung und weitere Charakterisierung der empfindlichen Salze musste eine permanente Kühlkette bereitgestellt werden, und dennoch gelang es, die Ladungsdichte des Modellkomplexes [Ag(C2H2)][Al(ORF)4] (RF = C(CH3)(CF3)2) bei 10 K experimentell zu bestimmen. Diese zeigt, dass die Bindungsverhältnisse teils elektrostatischer, teils schwach kovalenter Natur sind.
Co-reporter:Andreas Reisinger Dr.;Nils Trapp Dipl.-Chem. Dr.;Sra Altmannshofer Dipl.-Chem.;Verena Herz Dipl.-Phys.;Manuel Presnitz Dipl.-Phys.;Wolfgang Scherer Dr.
Angewandte Chemie 2007 Volume 119(Issue 43) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/ange.200702688
Starke Komplexe dank schwacher Anionen: Die Festkörperstrukturen der ersten homoleptischen Metall-HCCH-Komplexe [M(C2H2)x]n+ (M=beliebiges Metall; n, x=beliebige Zahl) in Form zweier [Ag(η2-C2H2)n]+-Salze (n=3, 4) werden beschrieben (siehe Bild). Zudem gelang es, die elektronische Struktur einer [Ag(η2-C2H2)]+-Modellverbindung durch experimentelle Ladungsdichtestudien zu untersuchen.
Co-reporter:John M. Slattery Dr.;Corinne Daguenet Dr.;Paul J. Dyson ;Thomas J. S. Schubert Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 28) pp:
Publication Date(Web):14 JUN 2007
DOI:10.1002/ange.200700941
Das MolekülvolumenVm (d. h. die Summe der Ionenvolumina Vion der Bausteine) einer ionischen Flüssigkeit (IL) und eine anionenabhängige empirische Beziehung genügen, um physikalische Eigenschaften wie Viskosität, Leitfähigkeit und Dichte von ILs mit [N(CN)2]−, [BF4]−, [PF6]− und [N(SO2CF3)2]− als Anionen vorherzusagen – auch von ILs, die bisher nur auf dem Papier existieren.
Co-reporter:Andreas Reisinger Dr.;Nils Trapp Dipl.-Chem. Dr.;Sra Altmannshofer Dipl.-Chem.;Verena Herz Dipl.-Phys.;Manuel Presnitz Dipl.-Phys.;Wolfgang Scherer Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 43) pp:
Publication Date(Web):26 OCT 2007
DOI:10.1002/anie.200790216
High up in permanent ice is where the homoleptic silver(I) acetylene complexes described by I. Krossing, W. Scherer, and co-workers in their Communication on page 8295 ff. would feel comfortable. Permanent cooling was required for synthesis, handling, and characterization. Nevertheless, even experimental charge-density studies could be carried out, which indicate a bonding situation that is intermediate between purely electrostatic and weakly covalent.
Co-reporter:Andreas Reisinger Dr.;Nils Trapp Dipl.-Chem. Dr.;Sra Altmannshofer Dipl.-Chem.;Verena Herz Dipl.-Phys.;Manuel Presnitz Dipl.-Phys.;Wolfgang Scherer Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 43) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/anie.200702688
Hanging on to acetylene: The solid-state structures of the first homoleptic metal HCCH complexes [M(C2H2)x]n (M=any metal; n, x=any number), salts of [Ag(η2-C2H2)n]+ (n=3,4), are described (see picture). The electronic structure of a [Ag(η2-C2H2)] model complex has been investigated by experimental charge-density studies.
Co-reporter:John M. Slattery Dr.;Corinne Daguenet Dr.;Paul J. Dyson ;Thomas J. S. Schubert Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 28) pp:
Publication Date(Web):14 JUN 2007
DOI:10.1002/anie.200700941
The molecular volumeVm (that is, the sum of the ionic volumes Vion of the constituent ions) of an ionic liquid (IL) in combination with an anion-dependent empirical relationship is all one needs to predict physical properties such as viscosity, conductivity, and density of [N(CN)2]−, [BF4]−, [PF6]−, and [N(SO2CF3)2]− ionic liquids, including those which may as yet only exist on paper.
Co-reporter:Ines Raabe Dipl.-Chem.;Sasa Antonijevic Dr. Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 26) pp:
Publication Date(Web):6 JUL 2007
DOI:10.1002/chem.200601885
In an earlier publication (J. Am. Chem. Soc.2002, 124, 7111) we showed that polymeric cationic [Ag(P4S3)n]+ complexes (n=1, 2) are accessible if partnered with a suitable weakly coordinating counterion of the type [Al(ORF)4]− (ORF: poly- or perfluorinated alkoxide). The present work addresses the following questions that could not be answered in the initial report: How many P4S3 cages can be bound to a Ag+ ion? Why are these complexes completely dynamic in solution in the 31P NMR experiments? Can these dynamics be frozen out in a low-temperature 31P MAS NMR experiment? What are the principal binding sites of the P4S3 cage towards the Ag+ ion? What are likely other isomers on the [Ag(P4S3)n]+ potential energy surface? Counterion influence: Reactions of P4S3 with Ag[Al{OC(CH3)(CF3)2}4] (Ag[hftb]) and Ag[{(CF3)3CO}3Al-F-Al{OC(CF3)3)}3] (Ag[al-f-al]) gave [(P4S3)Ag[hftb]]∞ (7) as a molecular species, whereas [Ag2(P4S3)6]2+[al-f-al]−2 (8) is an isolated 2:1 salt. We suggest that a maximum of three P4S3 cages may be bound on average to an Ag+ ion. Only isolated dimeric dications are formed with the largest cation, but polymeric species are obtained with all other smaller aluminates. Thermodynamic Born–Haber cycles, DFT calculations, as well as solution NMR and ESI mass spectrometry indicate that 8 exhibits an equilibrium between the dication [Ag2(P4S3)6]2+ (in the solid state) and two [Ag(P4S3)3]+ monocations (in the gas phase and in solution). Dynamics: 31P MAS NMR spectroscopy showed these solid adducts to be highly dynamic, to an extent that the 2JP,P coupling within the cages could be resolved (J-res experiment). This is supported by DFT calculations, which show that the extended PES of [Ag(P4S3)n]+ (n=1–3) and [Ag2(P4S3)2]+ is very flat. The structures of α- and γ-P4S3 were redetermined. Their variable-temperature 31P MAS NMR spectra are discussed jointly with those of all four currently known [Ag(P4S3)n]+ adducts with n=1, 2, and 3.
Co-reporter:Andreas Reisinger;Daniel Himmel Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 42) pp:
Publication Date(Web):22 SEP 2006
DOI:10.1002/anie.200600805
Over 30 years after the first successful syntheses of CuI alkoxides and silyl oxides, the first homologous AgI compounds were synthesized and characterized (see scheme). AgOC(CF3)3 was furthermore found to be a promising precursor for chemical vapor deposition processes and the deposition of AgF.
Co-reporter:Andreas Reisinger;Daniel Himmel Dr.
Angewandte Chemie 2006 Volume 118(Issue 42) pp:
Publication Date(Web):22 SEP 2006
DOI:10.1002/ange.200600805
Über dreißig Jahre nach der erfolgreichen Synthese von CuI-Alkoxiden und -Siloxiden wurden nun die ersten homologen AgI-Verbindungen AgOR hergestellt und charakterisiert (siehe Schema). Darüber hinaus erwies sich AgOC(CF3)3 als vielversprechende Ausgangsverbindung für Gasphasenabscheidungen und die Abscheidung von AgF.
Co-reporter:Marcin Gonsior Dr.;Lutz Müller Dipl.-Chem.
Chemistry - A European Journal 2006 Volume 12(Issue 22) pp:
Publication Date(Web):23 MAY 2006
DOI:10.1002/chem.200501188
While reinvestigating the published synthesis of OPI3, it became evident from the experiments that phosphoryl triodide may only be formed as an intermediate and that the end products of the reaction of OPCl3 with LiI are PV oxides, PI3, I2, and LiCl. This is also in agreement with MP2/TZVPP calculations, which assign ΔrH° (ΔrG°) [ΔrG° in CHCl3] for the disproportionation of OPI3 as −7 (−18) [−17 kJ mol−1] (assuming P4O10 as the PV oxide). The first products of this reaction visible in a low-temperature in situ 31P NMR experiment are P2I4 and PI3, as well as traces of a compound that may be OPCl2I. By contrast, it was possible to prepare and structurally characterize Lewis acid [A] stabilized [A]OPX3 adducts, where [A] is Al(ORF)3 for X=Br and Al(ORF)2(μ-F)Al(ORF)3 for X=I (RF=C(CF3)3). These adducts are formed on decomposition of PX4+[Al(ORF)4]−; high yields of Br3POAl(ORF)3 (δ(31P)=−65) were obtained, while I3POAl(ORF)3 (δ(31P)=−337) and I3POAl(ORF)2(μ-F)Al(ORF)3 (δ(31P)=−332) are only formed as by-products. The main product of the room-temperature decomposition of PI4+[Al(ORF)4]− is PI4+[(RFO)3Al(μ-F)Al(ORF)3]−, which was also characterized by X-ray crystallography and was independently prepared from Ag+[(RFO)3Al(μ-F)Al(ORF)3]−, PI3, and I2.
Co-reporter:Gustavo Santiso-Quiñones, Andreas Reisinger, John Slattery and Ingo Krossing
Chemical Communications 2007(Issue 47) pp:
Publication Date(Web):
DOI:10.1039/B710899K
Co-reporter:Hannes Böhrer, Nils Trapp, Daniel Himmel, Mario Schleep and Ingo Krossing
Dalton Transactions 2015 - vol. 44(Issue 16) pp:NaN7499-7499
Publication Date(Web):2015/03/13
DOI:10.1039/C4DT02822H
The possibility of obtaining frustrated Lewis pairs (FLPs) suitable for H2-activation based on the Lewis acid B(Ohfip)31 (Ohfip = OC(H)(CF3)2) was investigated. In this context, the crystal structure of 1 as well as the crystal structure of the very weak adduct 1·NCMe was determined. When reacting solutions of 1 with H2 (1 bar) and selected phosphanes, amines, pyridines and N-heterocyclic carbenes, dihydrogen activation was never observed. Without H2, adduct formation with 1 was observed to be an equilibrium process, regardless of the Lewis base adduct. Thus, the thermodynamics of H2 activation of 1 in comparison with the well-known B(C6F5)3 was analyzed using DFT calculations in the gas phase and different solvents (CH2Cl2, ortho-difluorobenzene and acetonitrile). These investigations indicated that FLP chemistry based on 1 is considerably less favored than that with B(C6F5)3. This is in agreement with control NMR experiments indicating hydride transfer from [H–B(Ohfip)3]− upon reaction with B(C6F5)3, giving [H–B(C6F5)3]− and B(Ohfip)3 in toluene and also MeCN. Induced by these unsuccessful reactions, the Lewis acidity towards HSAB hard and soft ions was investigated for gaining a deeper insight. A unified reference system based on the trimethylsilyl compounds Me3Si–Y (Y = F, Cl, H, Me) and their respective ions Me3Si+/Y− calculated at the G3 level was chosen as the anchor point. The individual ion affinities were then assessed based on subsequent isodesmic reactions calculated at a much less expensive level (RI-)BP86/SV(P). This method was validated by systematic calculations of smaller reference systems at the frozen core CCSD(T) level with correlation effects extrapolated to a full quadruple-ζ basis. Overall, 33 common and frequently used Lewis acids were ranked with respect to their FIA, CIA, HIA and MIA (fluoride/chloride/hydride/methyl ion affinity).
Co-reporter:Ines Raabe, Caroline Röhr and Ingo Krossing
Dalton Transactions 2007(Issue 46) pp:NaN5386-5386
Publication Date(Web):2007/10/11
DOI:10.1039/B711639J
What is the preferred coordination site of CI3+? Recent computational work suggested the iodine atoms of the Lewis acid CI3+ to be more electrophilic than the classically expected carbon atom, e.g. the complex with water is of type I2C–I⋯OH2+ and not the classically expected I3C–OH2+. If this structure is correct, one may also anticipate reactions of CI3+ as an I+ donor. Thus, we were interested in investigating the chemistry of CI3+ in the room-temperature stable salt [CI3]+[pftb]− ([pftb]− = [Al(OC(CF3)3)4]−) with weak nucleophiles that i) mimic water (OEt2) or ii) are electronically deactivated weak nucleophiles (PX3, X = Cl–I; AsI3). One question was: is it possible to obtain iodine-coordinated Lewis acid–base adducts of the CI3+ cation? With Et2O as a base, the cation behaves as a strong Lewis acid and cleaves the ether to give I3C–OEt, C2H4 and [H(Et2O)2]+. By contrast PX3 and AsI3 coordinate to the CI3+ cations and the adducts have classical, carbon-bound ethane-like structures, as proven by X-ray single-crystal diffraction, IR, UV-Vis and NMR spectroscopy. From variable temperature 13C NMR studies, it followed for the I3C–AsI3+ salt that the equilibrium between CI3+ and AsI3 is reversible and temperature dependent in solution. The I3C–PI3+ salt decomposes at room temperature giving PI4+ and C2I4, likely through an iodine coordinated I2C–I⋯PI3+ intermediate. Thus CI3+ may also act as an I+ donor. All reactions are in agreement with ab initio quantum chemical calculations at the MP2/TZVPP level and assignments of experimental spectra were aided by quantum chemistry.
Co-reporter:Ines Raabe, Daniel Himmel, Sonja Müller, Nils Trapp, Martin Kaupp and Ingo Krossing
Dalton Transactions 2008(Issue 7) pp:NaN956-956
Publication Date(Web):2007/12/18
DOI:10.1039/B714271D
The room-temperature stable CI3+ salts [CI3]+[pftb]−1 and [CI3]+[al-f-al]−2 ([pftb]− = [Al(OC(CF3)3)4]−; [al-f-al]− = [((CF3)3CO)3Al-F-Al(OC(CF3)3)3]−) were prepared in quantitative yields from purified CI4 and the corresponding silver aluminates with total exclusion of light (NMR, IR, UV-VIS, X-ray diffraction). The isolated CI3+ cation is trigonal planar with a sum of ∠(I–C–I) = 360.0° (1) and 359.9° (2). Attempts to prepare CHI2+ and CH2I+ salts from CHI3 or CH2I2/Ag[pftb] mixtures remained unsuccessful; the reaction with CH2I2 leads to the formation of the adduct [Ag(CH2I2)3]+[pftb]−3, while for HCI3, dismutation with formation of 1 as well as 3 was observed. All particles were also calculated at the MP2/TZVPP level to predict the vibrational and electronic spectra as well as to calculate the Gibbs free energies of all reactions (ΔG°, gas phase and CH2Cl2 solution). Quantum chemical calculations were also used to investigate the stability of the [pftb]− anion against the electrophilic attack of the CX3+ and CHnX3–n+ cations (X = F–I, n = 1–3). The strength of the Lewis acidity of these cations and of the isoelectronic boron halides BX3 and BHnX3–n have been established on the basis of their fluoride ion affinities (FIAs). The FIAs of the carbon and the boron containing compounds show opposite trends, with fluorinated halomethyl cations being stronger acids than their heavier congeners but iodinated holoboranes being stronger acids than their lighter homologues.
Co-reporter:Safak Bulut, Petra Klose and Ingo Krossing
Dalton Transactions 2011 - vol. 40(Issue 32) pp:NaN8124-8124
Publication Date(Web):2011/07/18
DOI:10.1039/C1DT10722D
The fast, high yield synthesis and full characterization of Na[B(hfip)4] (hfip: OC(H)(CF3)2) from NaBH4 and hexafluoroisopropanol (hfipH) is presented. By anion metathesis, five [B(hfip)4]− salts with classical/functionalized ionic liquid (IL) cations with melting points between 0 ([C6MIM]+[B(hfip)4]−) and 113 °C ([C4MMorph]+[B(hfip)4]−) were prepared. Four of these qualify as ILs and one as room temperature IL (RTIL). The properties of the borate anion [B(hfip)4]− and its aluminum analogue [Al(hfip)4]− were compared based on the available structural information from XRD. Viscosities (10.3 (90 °C) to 855 (0 °C) mPa s−1) and conductivities (0.603 (30 °C) to 4.844 (90 °C) mS cm−1) were measured between 0 and 90 °C, and described by the Vogel–Fulcher–Tammann (VFT) equations. The properties of the [B(hfip)4]− ILs were analyzed in the context of the anion-dependent molecular volume Vm-viscosity-/conductivity-correlations, also in comparison to ILs with [BF4]−/[PF6]−, [N(CN)2]−, [Tf2N]− and [Al(hfip)4]− counterions. The viscosities and conductivities of [B(hfip)4]− ILs are slightly inferior to [Al(hfip)4]− ILs, similar to/better than all other anions given above. According to the Walden plots, the ionicity of the [B(hfip)4]− ILs may at least be classified as “good”. By sharp contrast to the [Al(hfip)4]− ILs, the [B(hfip)4]− ILs have good stability against humidity/water. Thus, handling of [B(hfip)4]− ILs in an open laboratory atmosphere over hours and days is allowed and further facilitates the use of this new IL class.
Co-reporter:Anna J. Lehner, Nils Trapp, Harald Scherer and Ingo Krossing
Dalton Transactions 2011 - vol. 40(Issue 7) pp:NaN1452-1452
Publication Date(Web):2010/12/03
DOI:10.1039/C0DT01076F
The CX3+ salts [CCl3]+[Al(ORF)4]−1, [CCl3]+[(RFO)3Al–F–Al(ORF)3]−2, [CBr3]+[Al(ORF)4]−3, [CBr3]+[(RFO)3Al–F–Al(ORF)3]−4 (RF = C(CF3)3) were prepared in 56 to 85% yield from CX4 (X = Cl, Br) and the corresponding silver salts (weight balance, NMR, IR, X-ray structure of 1). The most convenient solvent for the preparation of 1 and 2 is SO2ClF but for 3 and 4 it is SO2. The reactions are complete after about three days stirring at −30 to −40 °C. The salts are stable for weeks in solution at −40 °C and stable for a few hours at RT in the solid state. In SO2ClF (1, 2) or SO2 (3, 4) solution they decompose slowly at −20 °C and within several hours at RT; in general the CBr3+ salts are more stable than the CCl3+ homologues. The decomposition products were assigned as CCl3F and primarily CBr2F2 (which likely forms as a Lewis acid induced disproportionation product of the initial CBr3F). The C-X vibrations of the salts were found in the expected range and the assignments were made based on experimental and calculated data. The IR spectrum of a CBr3+ salt is for the first time reported here.
Co-reporter:Alexander Higelin, Christoph Haber, Stefan Meier and Ingo Krossing
Dalton Transactions 2012 - vol. 41(Issue 39) pp:NaN12015-12015
Publication Date(Web):2012/05/31
DOI:10.1039/C2DT30379E
The recently reported homologous low-valent indium and gallium salts M+[Al(ORF)4]− (M = Ga, In; RF = C(CF3)3) were used to extend the coordination chemistry of GaI and InI to the isolated [18]crown-6 complexes [M([18]crown-6)(PhF)2]+[Al(ORF)4]− in fluorobenzene solution (PhF = C6H5F). In contrast to known ion-paired compounds for M = In, our complexes are undisturbed and in the solid state free of contacts to the anion. A peculiar combination of very weak η1- and η6-coordination to the PhF-solvent was observed that allows speculation about the presence of a stereochemically active lone pair at MI. Structure and energetics of these novel salts were rationalized on the basis of DFT calculations.
Co-reporter:Christoph Schulz, Philipp Eiden, Petra Klose, Andreas Ermantraut, Michael Schmidt, Arnd Garsuch and Ingo Krossing
Dalton Transactions 2015 - vol. 44(Issue 15) pp:NaN7057-7057
Publication Date(Web):2015/03/18
DOI:10.1039/C5DT00469A
Weakly coordinating anions (WCAs) with the difluorophosphato ligand (O2PF2) were the target of this study. Initial experiments were conducted towards the preparation of homoleptic aluminates of the well-studied [Al(OR)4]−-type. The preparation of the initial target structure Li[Al(O2PF2)4] failed due to the remaining Lewis acidic character of the central aluminum atom. Instead, the formation of Li3[Al(O2PF2)6] and Al(O2PF2)3 was observed with hexacoordinate aluminum atoms and verified by NMR, IR and X-ray crystallography. A possible mechanism towards these compounds was postulated in the solvent induced dismutation of the tetracoordinate Li[Al(O2PF2)4]. A singly charged WCA was realized by the exchange of the central aluminum atom for boron. The [B(O2PF2)4]− anion was prepared starting from BH3·S(CH3)2 and boron tribromide leading to the protic room temperature Ionic Liquid (IL) [H(S(CH3)2)][B(O2PF2)4] and the neat liquid Brønsted acid H[B(O2PF2)4], respectively, representing a significantly improved synthesis with regard to the first experiments of Dove et al. The basicity of the [B(O2PF2)4]− anion and its WCA quality were investigated on the basis of the IR-spectroscopic NH-scale and the salt [H(N(Oct)3)][B(O2PF2)4] that places it better than all oxyanions and close to the carboranate based WCAs. A pathway to the solvent free pure Li[B(O2PF2)4] salt was established on a multi-gram scale with excellent purities enabling electrochemical applications (verified by NMR, IR, X-ray crystallography and cyclovoltammetry).
Co-reporter:Tobias A. Engesser, Martin R. Lichtenthaler, Mario Schleep and Ingo Krossing
Chemical Society Reviews 2016 - vol. 45(Issue 4) pp:NaN899-899
Publication Date(Web):2015/11/27
DOI:10.1039/C5CS00672D
The chemistry of the p-block elements is a huge playground for fundamental and applied work. With their bonding from electron deficient to hypercoordinate and formally hypervalent, the p-block elements represent an area to find terra incognita. Often, the formation of cations that contain p-block elements as central ingredient is desired, for example to make a compound more Lewis acidic for an application or simply to prove an idea. This review has collected the reactive p-block cations (rPBC) with a comprehensive focus on those that have been published since the year 2000, but including the milestones and key citations of earlier work. We include an overview on the weakly coordinating anions (WCAs) used to stabilize the rPBC and give an overview to WCA selection, ionization strategies for rPBC-formation and finally list the rPBC ordered in their respective group from 13 to 18. However, typical, often more organic ion classes that constitute for example ionic liquids (imidazolium, ammonium, etc.) were omitted, as were those that do not fulfill the – naturally subjective – “reactive”-criterion of the rPBC. As a rule, we only included rPBC with crystal structure and only rarely refer to important cations published without crystal structure. This collection is intended for those who are simply interested what has been done or what is possible, as well as those who seek advice on preparative issues, up to people having a certain application in mind, where the knowledge on the existence of a rPBC that might play a role as an intermediate or active center may be useful.
Co-reporter:Nils Trapp, Harald Scherer, Stuart A. Hayes, Raphael J. F. Berger, Agnes Kütt, Norbert W. Mitzel, Jaan Saame and Ingo Krossing
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 13) pp:NaN6191-6191
Publication Date(Web):2011/02/25
DOI:10.1039/C0CP02596H
The syntheses of the perfluorinated alcohols (F5C6)(F3C)2COH (1) and (F5C6)(C5F10)COH (2) are described. Both compounds were prepared in reasonable yields (1: 65%, 2: 85%) by reacting the corresponding ketone with C6F5MgBr, followed by acidic work-up. The alcohols were characterized by NMR, vibrational spectroscopy, single-crystal X-ray diffraction, acidity measurements and gas-phase electron diffraction. A combination of appropriate 2D NMR experiments allowed the unambiguous assignment of all signals in the 19F spin systems, of which that of 2 was especially complex. High acidity of the alcohols is indicated by acidity measurements as well as the calculated gas phase acidities. It is also supported by the crystal structure of 2, which exhibits only a single weak intermolecular hydrogen bridge with an O⋯O distance of 301 pm. This shows the low donor strength of the oxygen atom in the compound, which is partly compensated through formation of two intramolecular CF⋯H contacts of 220 and 232 pm length to the proton not involved in the hydrogen bridge. The pKa values in acetonitrile are 22.2 for 1 and 22.0 for 2; their calculated gas phase acidities are 1367 and 1343 kJ mol−1 (MP2/TZVPP level).

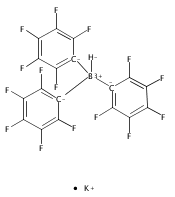
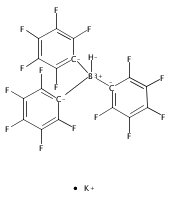











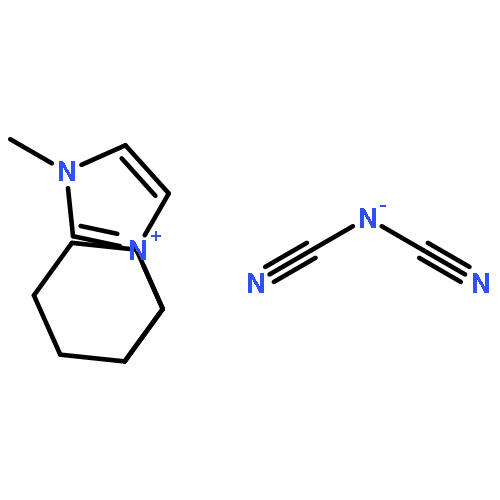





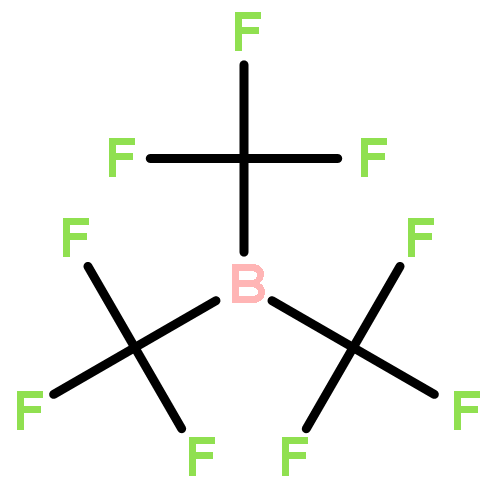


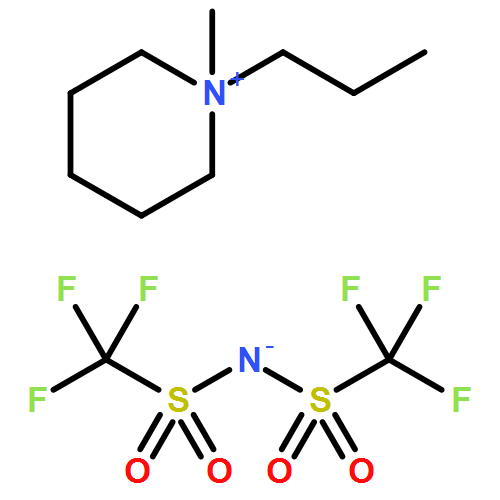














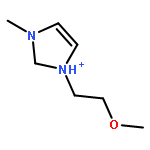
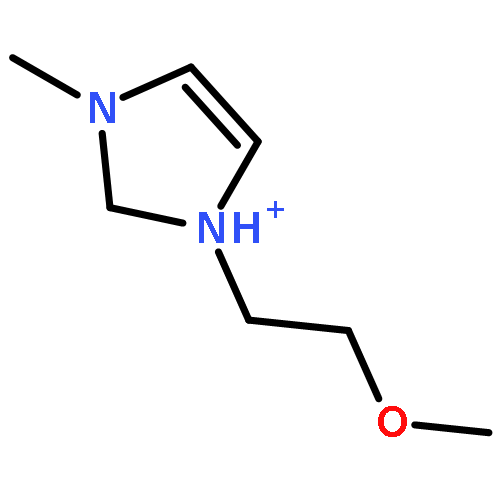
![Borane, tris[3,5-bis(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/169200/169116-84-1.png)
![Borane, tris[3,5-bis(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/169200/169116-84-1_b.png)




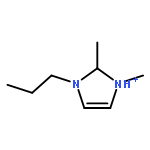


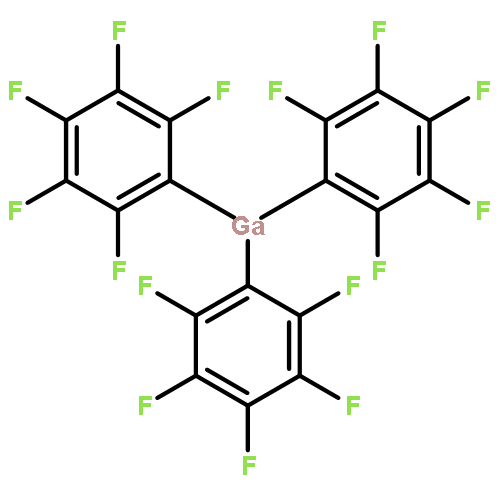



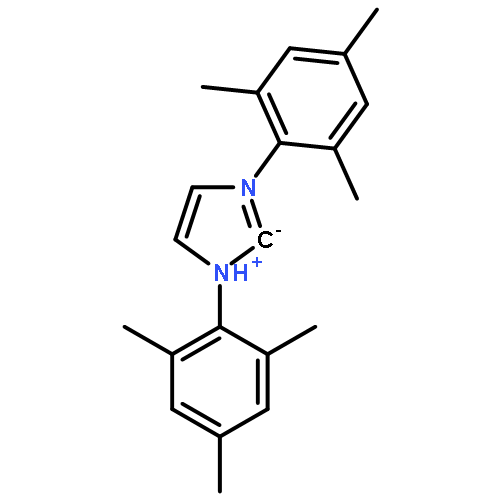



![D-Valine, N-[N-[(phenylmethoxy)carbonyl]-D-valyl]-, methyl ester](http://img.cochemist.com/ccimg/106600/106561-19-7.png)
![D-Valine, N-[N-[(phenylmethoxy)carbonyl]-D-valyl]-, methyl ester](http://img.cochemist.com/ccimg/106600/106561-19-7_b.png)
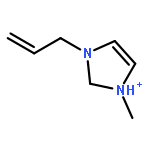
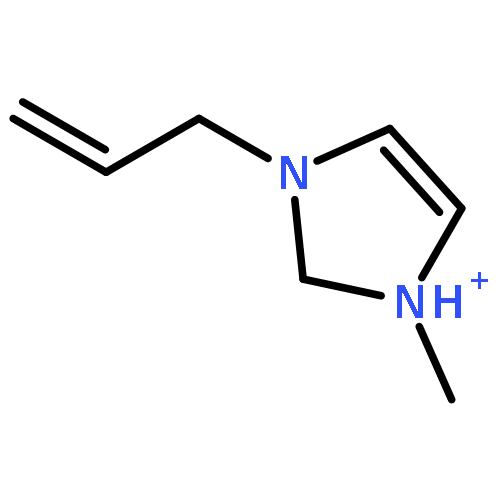
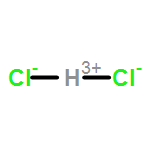
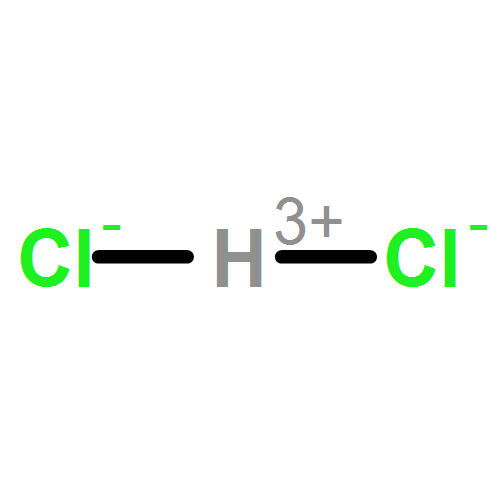

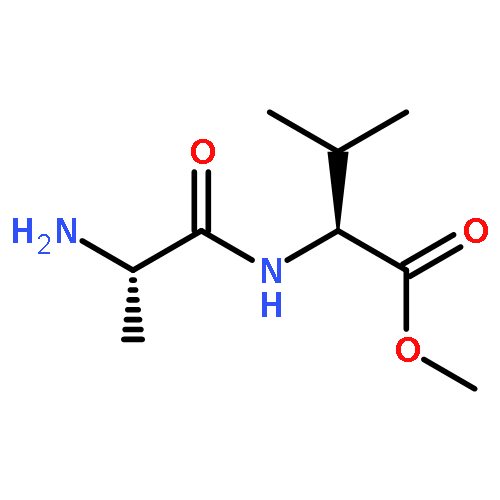











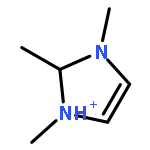
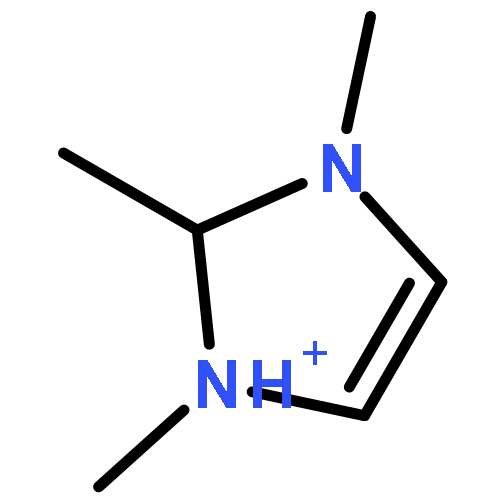
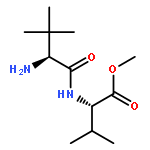




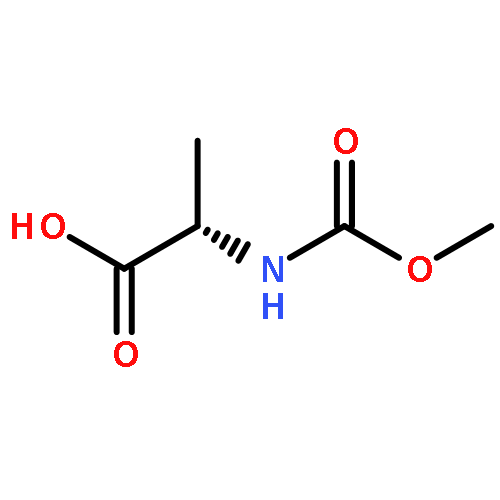

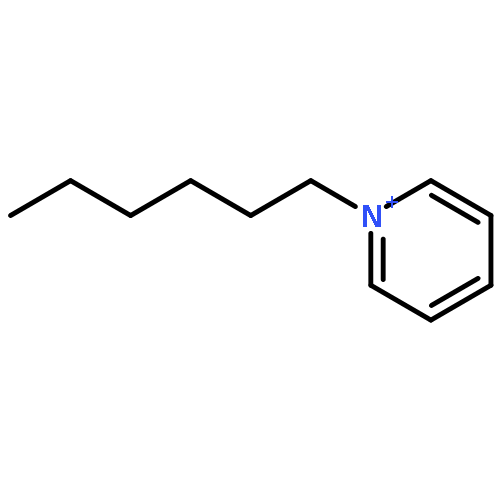
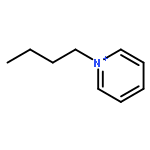
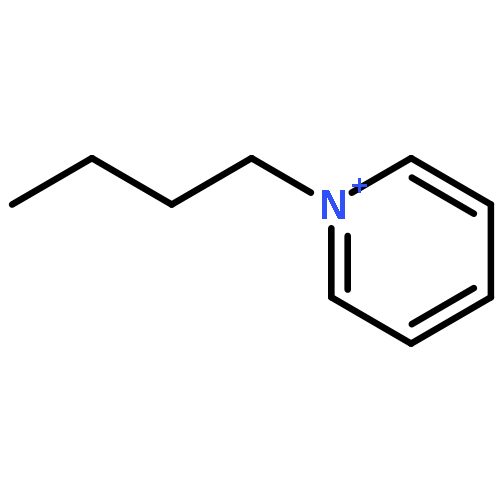
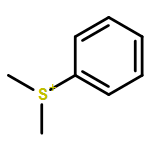
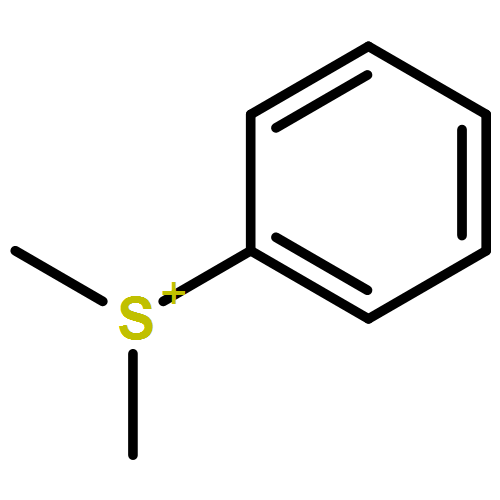










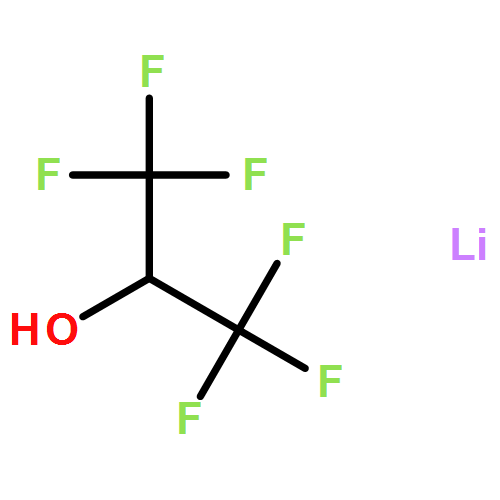



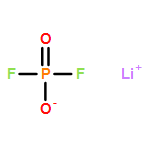
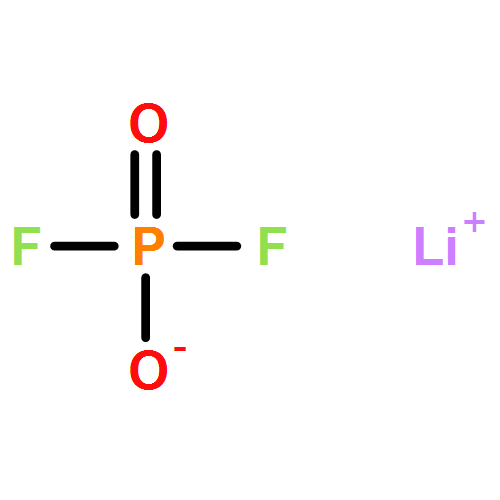
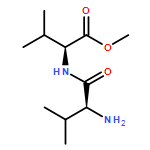


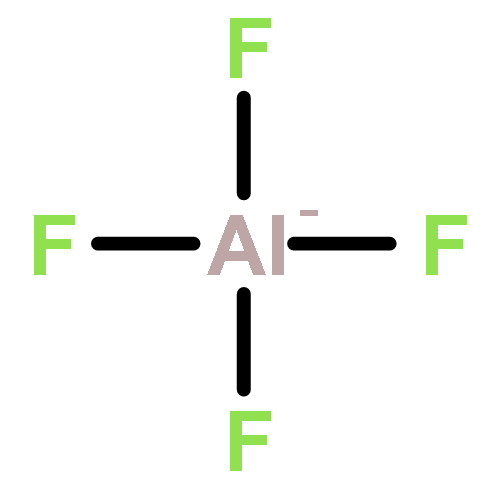
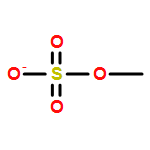
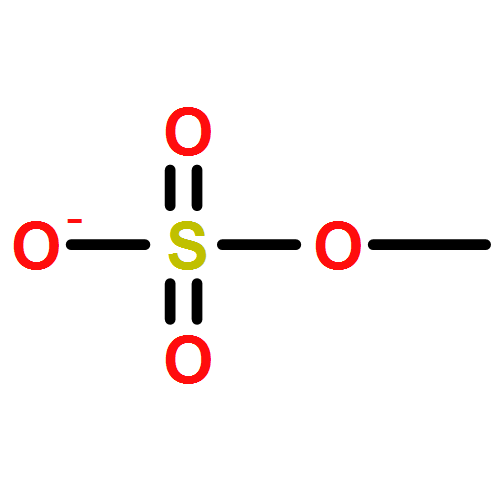





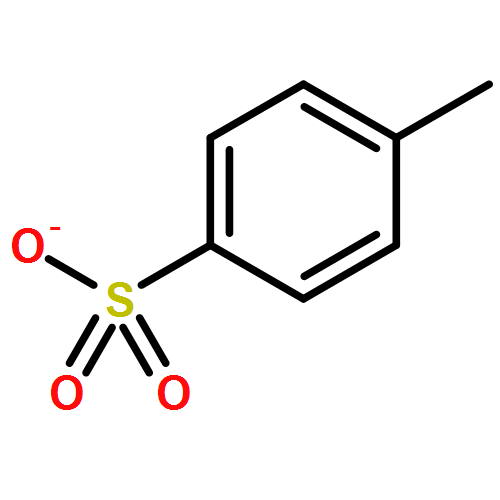













![N~2~-[(4-fluorophenyl)sulfonyl]-N~2~-(4-methylphenyl)-N-(1-phenylethyl)glycinamide](http://img.cochemist.com/ccimg/5800/5731-85-1.png)
![N~2~-[(4-fluorophenyl)sulfonyl]-N~2~-(4-methylphenyl)-N-(1-phenylethyl)glycinamide](http://img.cochemist.com/ccimg/5800/5731-85-1_b.png)
![L-Valine,N-[(phenylmethoxy)carbonyl]-L-alanyl-, methyl ester](http://img.cochemist.com/ccimg/4900/4864-38-4.png)
![L-Valine,N-[(phenylmethoxy)carbonyl]-L-alanyl-, methyl ester](http://img.cochemist.com/ccimg/4900/4864-38-4_b.png)








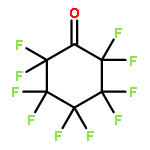
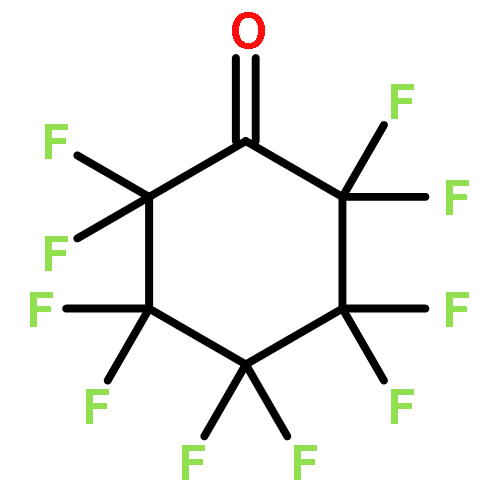




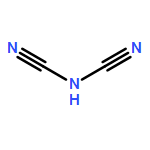



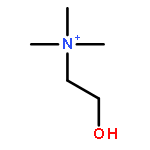




![Propanamide, N-[6-(diphenylphosphino)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/851600/851542-12-6.png)
![Propanamide, N-[6-(diphenylphosphino)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/851600/851542-12-6_b.png)
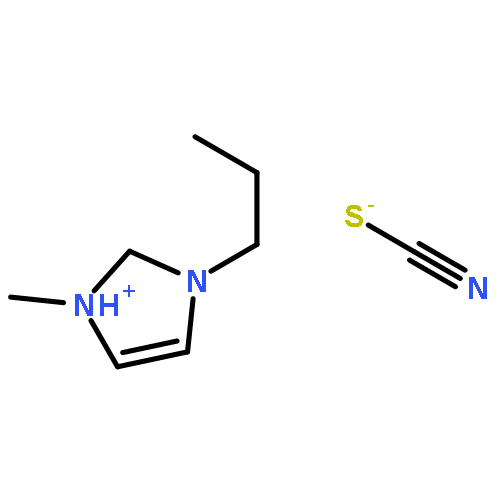








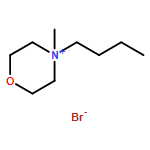
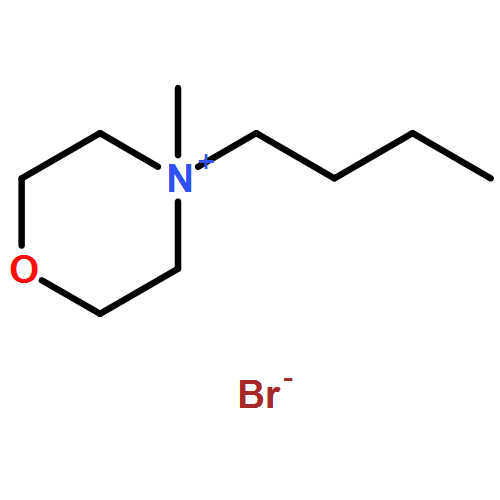
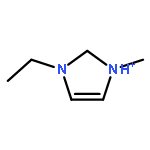

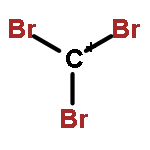
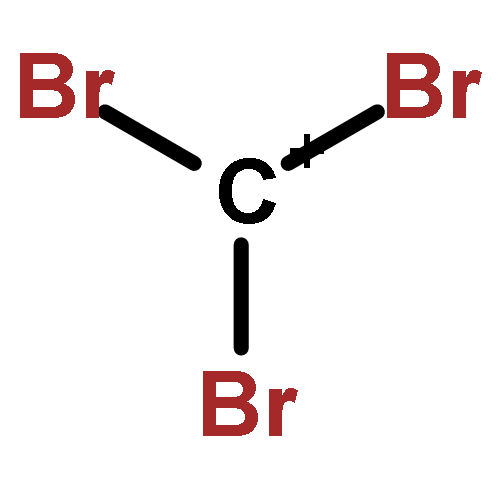



![L-Alanine,N-[(phenylmethoxy)carbonyl]-L-valyl-, methyl ester](http://img.cochemist.com/ccimg/4900/4817-92-9.png)
![L-Alanine,N-[(phenylmethoxy)carbonyl]-L-valyl-, methyl ester](http://img.cochemist.com/ccimg/4900/4817-92-9_b.png)








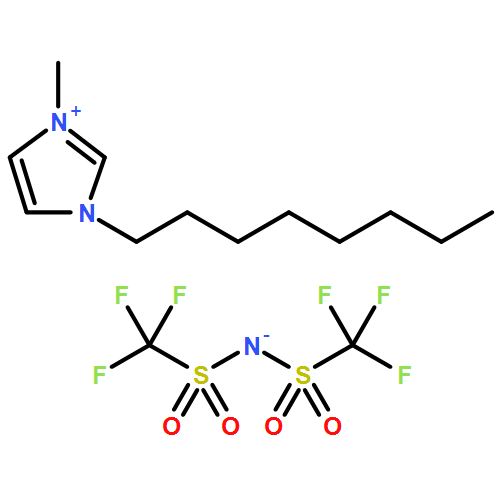





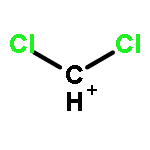
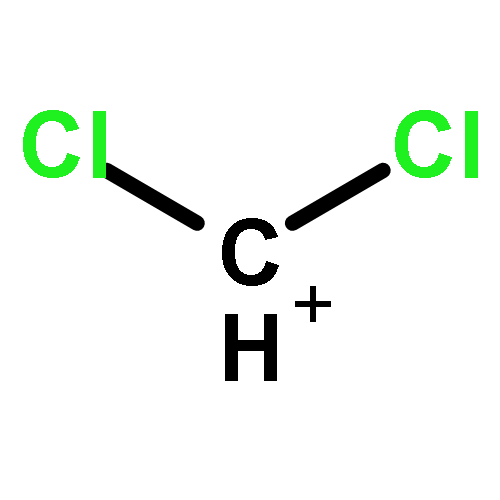
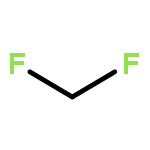
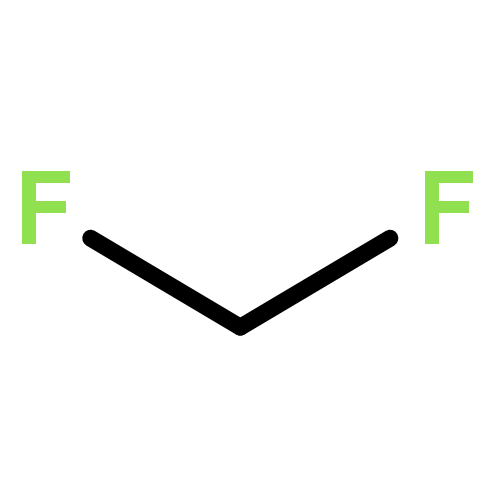




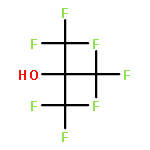

![Aluminate(1-),tetrakis[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)-2-propanolato-kO]-, lithium (1:1), (T-4)-](http://img.cochemist.com/ccimg/275000/274933-96-9.png)
![Aluminate(1-),tetrakis[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)-2-propanolato-kO]-, lithium (1:1), (T-4)-](http://img.cochemist.com/ccimg/275000/274933-96-9_b.png)