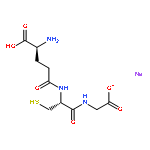
Co-reporter:Sven H. C. Askes, G. Upendar Reddy, Ralf Wyrwa, Sylvestre Bonnet, and Alexander Schiller
Journal of the American Chemical Society November 1, 2017 Volume 139(Issue 43) pp:15292-15292
Publication Date(Web):October 3, 2017
DOI:10.1021/jacs.7b07427
Applicability of phototherapeutic CO-releasing molecules (photoCORMs) is limited because they are activated by harmful and poorly tissue-penetrating near-ultraviolet light. Here, a strategy is demonstrated to activate classical photoCORM Mn2(CO)10 using red light (635 nm). By mixing in solution a triplet photosensitizer (PS) with the photoCORM and shining red light, energy transfer occurs from triplet excited-state 3PS* to a photolabile triplet state of Mn2(CO)10, which, like under near-UV irradiation, led to complete release of carbonyls. Crucially, such “triplet-sensitized CO-release” occurred in solid-state materials: when PS and Mn2(CO)10 were embedded in electrospun nonwoven fabrics, CO was liberated upon irradiation with low-intensity red light (≤36 mW 635 nm).
Co-reporter:Jordi-Amat Cuello-Garibo, Elena Pérez-Gallent, Lennard van der Boon, Maxime A. Siegler, and Sylvestre Bonnet
Inorganic Chemistry May 1, 2017 Volume 56(Issue 9) pp:4818-4818
Publication Date(Web):April 13, 2017
DOI:10.1021/acs.inorgchem.6b02794
Ruthenium polypyridyl complexes are good candidates for photoactivated chemotherapy (PACT) provided that they are stable in the dark but efficiently photosubstitute one of their ligands. Here the use of the natural amino acid l-proline as a protecting ligand for ruthenium-based PACT compounds is investigated in the series of complexes Λ-[Ru(bpy)2(l-prol)]PF6 ([1a]PF6; bpy = 2,2′-bipyridine and l-prol = l-proline), Λ-[Ru(bpy)(dmbpy)(l-prol)]PF6 ([2a]PF6 and [2b]PF6; dmbpy = 6,6′-dimethyl-2,2′-bipyridine), and Λ-[Ru(dmbpy)2(l-prol)]PF6 ([3a]PF6). The synthesis of the tris-heteroleptic complex bearing the dissymmetric proline ligand yielded only two of the four possible regioisomers, called [2a]PF6 and [2b]PF6. Both isomers were isolated and characterized by a combination of spectroscopy and density functional theory calculations. The photoreactivity of all four complexes [1a]PF6, [2a]PF6, [2b]PF6, and [3a]PF6 was studied in water (H2O) and acetonitrile (MeCN) using UV–vis spectroscopy, circular dichroism spectroscopy, mass spectrometry, and 1H NMR spectroscopy. In H2O, upon visible-light irradiation in the presence of oxygen, no photosubstitution took place, but the amine of complex [1a]PF6 was photooxidized to an imine. Contrary to expectations, enhancing the steric strain by the addition of two ([2b]PF6) or four ([3a]PF6) methyl substituents did not lead, in phosphate-buffered saline (PBS), to ligand photosubstitution. However, it prevented photoxidation, probably as a consequence of the electron-donating effect of the methyl substituents. In addition, whereas [2b]PF6 was photostable in PBS, [2a]PF6 quantitatively isomerized to [2b]PF6 upon light irradiation. In pure MeCN, [2a]PF6 and [3a]PF6 showed non-selective photosubstitution of both the l-proline and dmbpy ligands, whereas the non-strained complex [1a]PF6 was photostable. Finally, in H2O–MeCN mixtures, [3a]PF6 showed selective photosubstitution of l-proline, thus demonstrating the active role played by the solvent on the photoreactivity of this series of complexes. The role of the solvent polarity and coordination properties on the photochemical properties of polypyridyl complexes is discussed.
Co-reporter:Sven H. C. Askes, Vincent C. Leeuwenburgh, Wim Pomp, Hadi Arjmandi-Tash, Stefania Tanase, Thomas Schmidt, and Sylvestre Bonnet
ACS Biomaterials Science & Engineering March 13, 2017 Volume 3(Issue 3) pp:322-322
Publication Date(Web):January 17, 2017
DOI:10.1021/acsbiomaterials.6b00678
Light upconversion by triplet–triplet annihilation (TTA-UC) in nanoparticles has received considerable attention for bioimaging and light activation of prodrugs. However, the mechanism of TTA-UC is inherently sensitive for quenching by molecular oxygen. A potential oxygen protection strategy is the coating of TTA-UC nanoparticles with a layer of oxygen-impermeable material. In this work, we explore if (organo)silica can fulfill this protecting role. Three synthesis routes are described for preparing water-dispersible (organo)silica-coated red-to-blue upconverting liposomes. Their upconversion properties are investigated in solution and in A549 lung carcinoma cells. Although it was found that the silica offered no protection from oxygen in solution and after uptake in A549 cancer cells, upon drying of the silica-coated liposome dispersion in an excess of (organo)silica precursor, interesting liposome–silica nanocomposite materials were obtained that were capable of generating blue light upon red light excitation in air.Keywords: light upconversion; liposomes; nanoparticles; oxygen quenching; photonic materials; silica coating;
Co-reporter:Hyo Jin Jang;Samantha L. Hopkins;Maxime A. Siegler
Dalton Transactions 2017 vol. 46(Issue 30) pp:9969-9980
Publication Date(Web):2017/08/01
DOI:10.1039/C7DT01540B
The synthesis and characterization of [Ru(tpy)(R2bpy)(L)](X)n complexes (tpy = 2,2′:6′,2′′-terpyridine, R2bpy = 4,4′-dimethyl-2,2′-bipyridine (dmbpy), or 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (tfmbpy), X = Cl− or PF6−, and n = 1 or 2) are described. The dmbpy and tfmbpy bidentate ligands allow for investigating the effects of electron-donating and electron-withdrawing ligands, respectively, on the frontier orbital energetics as well as the photoreactivity of these ruthenium polypyridyl complexes for five prototypical monodentate ligands L = Cl−, H2O, CH3CN, 2-(methylthio)ethanol (Hmte), or pyridine. According to spectroscopic and electrochemical studies, the dmbpy analogues displayed a singlet metal-to-ligand charge transfer (1MLCT) transition at higher energy than the tfmbpy analogues. The shift of the 1MLCT to higher energy results from the lowest unoccupied molecular orbital (LUMO) for the dmbpy analogues being tpy-based, whereas for the tfmbpy analogues orbital inversion occurs resulting in a tfmbpy-based LUMO. The energy level of the highest occupied molecular orbital (HOMO) was considerably affected by the nature of the monodentate ligand. Visible light irradiation of the complexes demonstrated that the tfmbpy analogue increased the rate and quantum yields of photosubstitution reactions, compared to the dmbpy analogue, suggesting that the electron-withdrawing substituents allowed better thermal accessibility of the triplet metal-centered (3MC) state from the photochemically generated triplet metal-to-ligand charge transfer (3MLCT) excited state. A correlation between the photolability of the monodentate ligands and the electrochemical reversibility of the metal-based oxidation is also reported.
Co-reporter:B. Siewert;M. Langerman;Y. Hontani;J. T. M. Kennis;V. H. S. van Rixel;B. Limburg;M. A. Siegler;V. Talens Saez;R. E. Kieltyka;S. Bonnet
Chemical Communications 2017 vol. 53(Issue 81) pp:11126-11129
Publication Date(Web):2017/10/10
DOI:10.1039/C7CC02989F
Coupling the notoriously non-emissive complex [Ru(tpy)(bpy)Cl]Cl (tpy = 2,2′:6′,2′′-terpyridine, bpy = 2,2′-bipyridine) to a C12 alkyl chain via an amide linker on the 4′ position of the terpyridine yielded a new amphiphilic ruthenium complex showing red emission and chloride-dependent aggregation properties. This emissive complex is highly cytotoxic in A549 non-small lung cancer cells where it can be followed by confocal microscopy. Uptake occurs within minutes, first by insertion into the cellular membrane, and then by migration to the peri-nuclear region.
Co-reporter:Jordi-Amat Cuello-Garibo;Michael S. Meijer
Chemical Communications 2017 vol. 53(Issue 50) pp:6768-6771
Publication Date(Web):2017/06/20
DOI:10.1039/C7CC03469E
In metal-based photoactivated chemotherapy (PACT), two photoproducts are generated by light-triggered photosubstitution of a metal-bound ligand: the free ligand itself and an aquated metal complex. By analogy with cisplatin, the aquated metal complex is usually presented as the biologically active species, as it can typically bind to DNA. In this work, we show that this qualitative assumption is not necessarily valid by comparing the biological activity, log P, and cellular uptake of three ruthenium-based PACT complexes: [Ru(bpy)2(dmbpy)]2+, [Ru(bpy)2(mtmp)]2+, and [Ru(Ph2phen)2(mtmp)]2+. For the first complex, the photoreleased dmbpy ligand is responsible for the observed phototoxicity, whereas the second complex is not phototoxic, and for the third complex it is the ruthenium bis-aqua photoproduct that is the sole cytotoxic species.
Co-reporter:Lucien N. Lameijer;Daniël Ernst;Dr. Samantha L. Hopkins;Michael S. Meijer;Dr. Sven H. C. Askes;Dr. Sylvia E. Le Dévédec;Dr. Sylvestre Bonnet
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11707-11711
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201703890
AbstractWe describe two water-soluble ruthenium complexes, [1]Cl2 and [2]Cl2, that photodissociate to release a cytotoxic nicotinamide phosphoribosyltransferase (NAMPT) inhibitor with a low dose (21 J cm−2) of red light in an oxygen-independent manner. Using a specific NAMPT activity assay, up to an 18-fold increase in inhibition potency was measured upon red-light activation of [2]Cl2, while [1]Cl2 was thermally unstable. For the first time, the dark and red-light-induced cytotoxicity of these photocaged compounds could be tested under hypoxia (1 % O2). In skin (A431) and lung (A549) cancer cells, a 3- to 4-fold increase in cytotoxicity was found upon red-light irradiation for [2]Cl2, whether the cells were cultured and irradiated with 1 % or 21 % O2. These results demonstrate the potential of photoactivated chemotherapy for hypoxic cancer cells, in which classical photodynamic therapy, which relies on oxygen activation, is poorly efficient.
Co-reporter:Sven H. C. Askes, Philip Brodie, Gilles Bruylants, and Sylvestre Bonnet
The Journal of Physical Chemistry B 2017 Volume 121(Issue 4) pp:
Publication Date(Web):January 6, 2017
DOI:10.1021/acs.jpcb.6b10039
Understanding the temperature dependency of triplet–triplet annihilation upconversion (TTA-UC) is important for optimizing biological applications of upconversion. Here the temperature dependency of red-to-blue TTA-UC is reported in a variety of neutral PEGylated phospholipid liposomes. In these systems a delicate balance between lateral diffusion rate of the dyes, annihilator aggregation, and sensitizer self-quenching leads to a volcano plot, with the maximum upconversion intensity occurring near the main order–disorder transition temperature of the lipid membrane.
Co-reporter:B. Limburg, E. Bouwman, and S. Bonnet
ACS Catalysis 2016 Volume 6(Issue 8) pp:5273
Publication Date(Web):June 29, 2016
DOI:10.1021/acscatal.6b00107
The kinetics of homogeneous photocatalytic water oxidation is reported using [Ru(bpy)3]Cl2 as photosensitizer, Na2S2O8 as sacrificial electron acceptor, and three different water-oxidation catalysts: the ruthenium catalyst [Ru(bda)(isoq)2] ([1], H2bda = 2,2′-bipyridine-6,6′-dicarboxylic acid, isoq = isoquinoline), Co(NO3)2 ([2]), and [Ir(Cp*)(dmiz)(OH)2] ([3], Cp* = pentamethylcyclopentadienyl, dmiz =1,3-dimethylimidazol-2-ylidene). At pH 7.0, in a phosphate buffer and under blue light irradiation, the production of O2 at the catalyst is rate determining when [2] or [3] is used as a water-oxidation catalyst. However, when [1] is used as the catalyst under identical conditions the turnover at the water-oxidation catalyst is not the rate-limiting step of the photocatalysis. Instead, the step limiting dioxygen production is the transfer of electrons from the catalyst to the photooxidized photosensitizer [Ru(bpy)3]3+. Due to the instability of [Ru(bpy)3]3+ in neutral aqueous solutions, slow electron transfer results in significant photosensitizer decomposition, which limits the overall stability of the photocatalytic system. When the catalyst [1] is used, decomposition of both the photosensitizer and the catalyst [1] occurs in parallel. However, the photosensitizer and the catalyst also stabilize each other, i.e., the TON increases when more photosensitizer is added, while the photocatalytic turnover number PTON increases when more catalyst [1] is added. These data demonstrate not only that new and more stable water oxidation catalysts should be developed in the future but also that new and more stable photosensitizers are needed.Keywords: electron transfer; kinetics; photocatalysis; ruthenium tris(bipyridine); water oxidation
Co-reporter:V. H. S. van Rixel, B. Siewert, S. L. Hopkins, S. H. C. Askes, A. Busemann, M. A. Siegler and Sylvestre Bonnet
Chemical Science 2016 vol. 7(Issue 8) pp:4922-4929
Publication Date(Web):25 Apr 2016
DOI:10.1039/C6SC00167J
In this work, two new photopharmacological ruthenium prodrugs are described that can be activated by green light. They are based on the tetrapyridyl biqbpy ligand (6,6′-bis[N-(isoquinolyl)-1-amino]-2,2′-bipyridine), which coordinates to the basal plane of the metal centre and leaves two trans coordination sites for the binding of monodentate sulphur ligands. Due to the distortion of the coordination sphere these trans ligands are photosubstituted by water upon green light irradiation. In vitro cytotoxicity data on A431 and A549 cancer cell lines shows an up to 22-fold increase in cytotoxicity after green light irradiation (520 nm, 75 J cm−2), compared to the dark control. Optical microscopy cell imaging and flow cytometry indicate that the cancer cells die via apoptosis. Meanwhile, very low singlet oxygen quantum yields (∼1–2%) and cell-free DNA binding studies conclude that light-induced cell death is not caused by a photodynamic effect, but instead by the changes induced in the coordination sphere of the metal by light, which modifies how the metal complexes bind to biomolecules.
Co-reporter:B. Limburg, J. Wermink, S. S. van Nielen, R. Kortlever, M. T. M. Koper, E. Bouwman, and S. Bonnet
ACS Catalysis 2016 Volume 6(Issue 9) pp:5968
Publication Date(Web):July 19, 2016
DOI:10.1021/acscatal.6b00151
A tris(bipyridine)ruthenium(II) photosensitizer (PS) was anchored to the lipid bilayer of liposomes together with Ru-, Co-, or Ir-based water-oxidation catalysts in order to study the effect of liposomes on photocatalytic water oxidation in the presence of Na2S2O8. The Ru-based and Co-based systems both showed O2 production upon light irradiation, whereas the Ir-based system did not. Membrane anchoring caused a large shift in the quantum yield of oxidative quenching of the photosensitizer excited state by peroxodisulfate, which decreased from 180% in homogeneous solution to 7.3% at the surface of liposomes. For the Ru-based system the electron-transfer rate between the photosensitizer PS+ and the water-oxidation catalyst was increased relative to oxidative quenching. Consequently, the rate-limiting step of the photocatalytic water oxidation reaction at liposomes was oxidative quenching, whereas previous work showed that in homogeneous solution it is the reduction of the oxidized photosensitizer PS+ by the catalyst that limits O2 production. Overall, a lower dioxygen production rate was observed when photocatalytic water oxidation occurred at liposomes, but the stability of the liposomal system increased in comparison to that of the homogeneous system. Such stabilization is caused by the decreased concentration of the unstable PS+ species at liposomes, whereas this species accumulates in homogeneous solution, leading to faster degradation. Overall, liposomal water oxidation was found to be more tolerant to changes in light intensity and electron acceptor concentration, which is an interesting property for the building of solar fuel production devices.Keywords: artificial photosynthesis; kinetics; liposomes; photocatalysis; stability; water oxidation
Co-reporter:S. L. Hopkins, B. Siewert, S. H. C. Askes, P. Veldhuizen, R. Zwier, Michal Heger and Sylvestre Bonnet
Photochemical & Photobiological Sciences 2016 vol. 15(Issue 5) pp:644-653
Publication Date(Web):31 Mar 2016
DOI:10.1039/C5PP00424A
Traditionally, ultraviolet light (100–400 nm) is considered an exogenous carcinogen while visible light (400–780 nm) is deemed harmless. In this work, a LED irradiation system for in vitro photocytotoxicity testing is described. The LED irradiation system was developed for testing photopharmaceutical drugs, but was used here to determine the basal level response of human cancer cell lines to visible light of different wavelengths, without any photo(chemo)therapeutic. The effects of blue (455 nm, 10.5 mW cm−2), green (520 nm, 20.9 mW cm−2), and red light (630 nm, 34.4 mW cm−2) irradiation was measured for A375 (human malignant melanoma), A431 (human epidermoid carcinoma), A549 (human lung carcinoma), MCF7 (human mammary gland adenocarcinoma), MDA-MB-231 (human mammary gland adenocarcinoma), and U-87 MG (human glioblastoma-grade IV) cell lines. In response to a blue light dose of 19 J cm−2, three cell lines exhibited a minimal (20%, MDA-MB-231) to moderate (30%, A549 and 60%, A375) reduction in cell viability, compared to dark controls. The other cell lines were not affected. Effective blue light doses that produce a therapeutic response in 50% of the cell population (ED50) compared to dark conditions were found to be 10.9 and 30.5 J cm−2 for A375 and A549 cells, respectively. No adverse effects were observed in any of the six cell lines irradiated with a 19 J cm−2 dose of 520 nm (green) or 630 nm (red) light. The results demonstrate that blue light irradiation can have an effect on the viability of certain human cancer cell types and controls should be used in photopharmaceutical testing, which uses high-energy (blue or violet) visible light activation.
Co-reporter:Dr. Bianka Siewert;Vincent H. S. vanRixel;Eva J. vanRooden;Dr. Samantha L. Hopkins;Miriam J. B. Moester;Freek Ariese;Dr. Maxime A. Siegler;Dr. Sylvestre Bonnet
Chemistry - A European Journal 2016 Volume 22( Issue 31) pp:10960-10968
Publication Date(Web):
DOI:10.1002/chem.201600927
Abstract
The crystal structure and in vitro cytotoxicity of the amphiphilic ruthenium complex [3](PF6)2 are reported. Complex [3](PF6)2 contains a Ru−S bond that is stable in the dark in cell-growing medium, but is photosensitive. Upon blue-light irradiation, complex [3](PF6)2 releases the cholesterol–thioether ligand 2 and an aqua ruthenium complex [1](PF6)2. Although ligand 2 and complex [1](PF6)2 are by themselves not cytotoxic, complex [3](PF6)2 was unexpectedly found to be as cytotoxic as cisplatin in the dark, that is, with micromolar effective concentrations (EC50), against six human cancer cell lines (A375, A431, A549, MCF-7, MDA-MB-231, and U87MG). Blue-light irradiation (λ=450 nm, 6.3 J cm−2) had little influence on the cytotoxicity of [3](PF6)2 after 6 h of incubation time, but it increased the cytotoxicity of the complex by a factor 2 after longer (24 h) incubation. Exploring the unexpected biological activity of [3](PF6)2 in the dark elucidated an as-yet unknown bifaceted mode of action that depended on concentration, and thus, on the aggregation state of the compound. At low concentration, it acts as a monomer, inserts into the membrane, and can deliver [1]2+ inside the cell upon blue-light activation. At higher concentrations (>3–5 μm), complex [3](PF6)2 forms supramolecular aggregates that induce non-apoptotic cell death by permeabilizing cell membranes and extracting lipids and membrane proteins.
Co-reporter:Dr. Sipeng Zheng;Niels R. M. Reintjens;Dr. Maxime A. Siegler;Dr. Olivier Roubeau; Elisabeth Bouwman;Andrii Rudavskyi; Remco W. A. Havenith;Dr. Sylvestre Bonnet
Chemistry - A European Journal 2016 Volume 22( Issue 1) pp:331-339
Publication Date(Web):
DOI:10.1002/chem.201503119
Abstract
The tetrapyridyl ligand bbpya (bbpya=N,N-bis(2,2′-bipyrid-6-yl)amine) and its mononuclear coordination compound [Fe(bbpya)(NCS)2] (1) were prepared. According to magnetic susceptibility, differential scanning calorimetry fitted to Sorai’s domain model, and powder X-ray diffraction measurements, 1 is low-spin at room temperature, and it exhibits spin crossover (SCO) at an exceptionally high transition temperature of T1/2=418 K. Although the SCO of compound 1 spans a temperature range of more than 150 K, it is characterized by a wide (21 K) and dissymmetric hysteresis cycle, which suggests cooperativity. The crystal structure of the LS phase of compound 1 shows strong NH⋅⋅⋅S intermolecular H-bonding interactions that explain, at least in part, the cooperative SCO behavior observed for complex 1. DFT and CASPT2 calculations under vacuum demonstrate that the bbpya ligand generates a stronger ligand field around the iron(II) core than its analogue bapbpy (N,N′-di(pyrid-2-yl)-2,2′-bipyridine-6,6′-diamine); this stabilizes the LS state and destabilizes the HS state in 1 compared with [Fe(bapbpy)(NCS)2] (2). Periodic DFT calculations suggest that crystal-packing effects are significant for compound 2, in which they destabilize the HS state by about 1500 cm−1. The much lower transition temperature found for the SCO of 2 compared to 1 appears to be due to the combined effects of the different ligand field strengths and crystal packing.
Co-reporter:B. Limburg, M. Hilbers, A. M. Brouwer, E. BouwmanS. Bonnet
The Journal of Physical Chemistry B 2016 Volume 120(Issue 50) pp:12850-12862
Publication Date(Web):November 28, 2016
DOI:10.1021/acs.jpcb.6b09635
Recently, the addition of negatively charged liposomes was shown to increase the quantum yield of the photocatalytic reduction of 5,5′-dithio(2-nitrobenzoic acid) (H2DTNB) to 2-nitro-5-thiobenzoic acid (H2NTB) by triethanolamine using meso-tetra(4-(N-methylpyridinium)porphyrinato zinc chloride as photosensitizer. In this work, we investigate in detail the kinetics of this photocatalytic reaction both in homogeneous solution and at the surface of negatively charged liposomes, to unravel the effects of liposomes on the mechanism of the photoreaction. In homogeneous solution, the reaction is initiated by oxidative quenching. Both static (singlet) and dynamic (triplet) quenching of the photosensitizer contribute to the formation of the photoproduct. In these conditions, the reaction is limited by the low efficiency of reductive regeneration of the photosensitizer, compared to charge recombination. Upon adsorption of the positively charged photosensitizer to the negative surface of the liposomes, however, both static and dynamic oxidative quenching become ineffective due to electrostatic repulsion of the dianionic DTNB2– from the negatively charged membrane. In such conditions, photoreduction occurs via reductive quenching, showing that the addition of liposomes can truly modify the mechanism of photocatalyzed redox reactions.
Co-reporter:B. Limburg, E. Bouwman, and S. Bonnet
The Journal of Physical Chemistry B 2016 Volume 120(Issue 28) pp:6969-6975
Publication Date(Web):June 20, 2016
DOI:10.1021/acs.jpcb.6b03947
Liposomes are interesting scaffolds for photocatalysis. In particular, charged liposomes were shown to increase the quantum efficiency of photocatalytic reactions involving charged porphyrin photosensitizers and charged electron acceptors. In this work, the effects of adding positively charged liposomes (DMPC/eDMPCCl 1:1) on the mechanism of the photocatalytic reduction of methyl viologen (MV2+) by cysteine in the presence of sodium meso-tetra-(4-sulfonato)porphyrinatozinc (Na41) were probed by modeling UV–vis spectroscopy data using a steady-state approximation. By varying the concentration of methyl viologen, we found that the liposomes not only prevent the formation of a 1:1 complex between ground-state photosensitizer 14– and MV2+ but also that they increase the cage-escape yield in the excited state. Furthermore, the electrostatic repulsion between the liposomes and MV2+ diminishes by 1 order of magnitude the rate of oxidative quenching of the photosensitizer triplet excited state (T14–) by MV2+. By varying the amount of sacrificial electron donor (cysteine), the effect of liposome addition on the charge recombination reactions could also be studied. Because of the positive charge borne by the photoproduct MV•+, it was also repelled from the membrane, which significantly slows charge recombination at the surface of the liposome. Overall, compared to a liposome-free solution, the rates of most elementary steps of the photocatalytic reduction of MV2+ by cysteine are strongly modified when the negative photosensitizer is adsorbed on a positively charged liposome surface. These results not only explain the much higher efficiency of the liposome-containing system but also illustrate the power of supramolecular chemistry for the tuning of photocatalysis.
Co-reporter:B. Limburg, E. Bouwman and S. Bonnet
Chemical Communications 2015 vol. 51(Issue 96) pp:17128-17131
Publication Date(Web):07 Oct 2015
DOI:10.1039/C5CC07745A
Unidirectional photocatalytic electron transfer from a hydrophilic electron donor encapsulated in the interior of a liposome, to a hydrophilic electron acceptor on the other side of the membrane, has been achieved using the simple membrane-soluble electron relay 1-methoxy-N-methylphenazinium (MMP+). The total amount of photoproduct (>140 nmol) exceeds the number of moles of MMP+ present (125 nmol), thus showing that the transport of electrons is catalytic.
Co-reporter:Sven H. C. Askes, Néstor López Mora, Rolf Harkes, Roman I. Koning, Bram Koster, Thomas Schmidt, Alexander Kros and Sylvestre Bonnet
Chemical Communications 2015 vol. 51(Issue 44) pp:9137-9140
Publication Date(Web):27 Apr 2015
DOI:10.1039/C5CC02197A
Red-to-blue triplet–triplet annihilation upconversion was obtained in giant unilamellar vesicles. The upconverted light was homogeneously distributed across the membrane and could be utilized for the imaging of individual giant vesicles in three dimensions. These results show the great potential of TTA-UC for imaging applications under anoxic conditions.
Co-reporter:Sven H. C. Askes, Miroslav Kloz, Gilles Bruylants, John T. M. Kennis and Sylvestre Bonnet
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 41) pp:27380-27390
Publication Date(Web):22 Sep 2015
DOI:10.1039/C5CP04352B
Upconversion is a promising way to trigger high-energy photochemistry with low-energy photons. However, combining upconversion schemes with non-radiative energy transfer is challenging because bringing several photochemically active components in close proximity results in complex multi-component systems where quenching processes may deactivate the whole assembly. In this work, PEGylated liposomes were prepared that contained three photoactive components: a porphyrin dye absorbing red light, a perylene moiety emitting in the blue, and a light-activatable ruthenium prodrug sensitive to blue light. Time-dependent spectroscopic studies demonstrate that singlet perylene excited states are non-radiatively transferred to the nearby ruthenium complex by Förster resonance energy transfer (FRET). Under red-light irradiation of the three-component membranes, triplet–triplet annihilation upconversion (TTA-UC) occurs followed by FRET, which results in a more efficient activation of the ruthenium prodrug compared to a physical mixture of two-component upconverting liposomes and liposomes containing only the ruthenium complex. This work represents a rare example where TTA-UC and Förster resonance energy transfer are combined to achieve prodrug activation in the phototherapeutic window.
Co-reporter:Sipeng Zheng, Maxime A. Siegler, Olivier Roubeau, and Sylvestre Bonnet
Inorganic Chemistry 2014 Volume 53(Issue 24) pp:13162-13173
Publication Date(Web):November 20, 2014
DOI:10.1021/ic502381m
Coordination of the ligand bapbpy (1, bapbpy = N,N′-di(pyrid-2-yl)-2,2′-bipyridine-6,6′-diamine), of one of its four dimethyl-substituted analogues 2–5 (R2bapbpy = N,N′-di(methylpyrid-2-yl)-2,2′-bipyridine-6,6′-diamine), or of one of its three bis(iso)quinoline analogues 6–8 (R2bapbpy= N,N′-di(quinolyl)-2,2′-bipyridine-6,6′-diamine), to Fe(NCSe)2, afforded eight new iron(II) compounds of the type [Fe(R2bapbpy)(NCSe)2] (9–16). Three of these compounds (11, 13, and 16) were structurally characterized by single crystal X-ray diffraction, which showed similar molecular geometry and packing compared to their thiocyanate analogues. Magnetic susceptibility measurements were carried out for all iron compounds and revealed thermal spin-crossover (SCO) behavior for compounds 9, 11, 13, 15, and 16. Compounds 11, 13, 15, and 16 show an increased transition temperature compared to the thiocyanate analogues. [Fe(bapbpy)(NCSe)2] (9) shows a gradual, one-step SCO, whereas its thiocyanate analogue [Fe(bapbpy)(NCS)2] is known for its cooperative two-step SCO. To discuss the influence of S-to-Se substitution on the cooperativity of the SCO, heat capacity measurements were carried out for compounds 9, 11, 13, 15, and 16, and fitted to the Sorai domain model. The number n of like-spin SCO centers per interacting domain, which is a quantitative measure of the cooperativity of the spin transition, was found to be high for compounds 11 and 15, and low for compounds 9, 11, and 13. Compound 15 is one of the few known SCO compounds that is more cooperative than its thiocyanate analogue. Altogether, X-ray diffraction, calorimetry, and magnetic data give a consistent structure–property relationship for this family of compounds: hydrogen-bonding networks made of intermolecular N–H···Se interactions are of paramount importance for the cooperativity of the SCO.
Co-reporter:Azadeh Bahreman, Jordi-Amat Cuello-Garibo and Sylvestre Bonnet
Dalton Transactions 2014 vol. 43(Issue 11) pp:4494-4505
Publication Date(Web):05 Dec 2013
DOI:10.1039/C3DT52643G
The ruthenium complex [Ru(terpy)(bpy)(Hmte)]2+ ([1]2+), where terpy is 2,2′;6′,2′′-terpyridine, bpy is 2,2′-bipyridine, and Hmte is 2-methylthioethan-1-ol, poorly absorbs yellow light, and although its quantum yield for the photosubstitution of Hmte by water is comparable at 570 nm and at 452 nm (0.011(4) vs. 0.016(4) at 298 K at neutral pH), the photoreaction using yellow photons is very slow. Complex [1]2+ was thus functionalized with rhodamine B, an organic dye known for its high extinction coefficient for yellow light. Complex [Ru(Rterpy)(bpy)(Hmte)]3+ ([2]3+) was synthesized, where Rterpy is a terpyridine ligand covalently bound to rhodamine B via a short saturated linker. [2]Cl3 shows a very high extinction coefficient at 570 nm (44000 M−1 cm−1), but its luminescence upon irradiation at 570 nm is completely quenched in aqueous solution. The quantum yield for the photosubstitution of Hmte by water in [2]3+ was comparable to that in [1]2+ at 570 nm (0.0085(6) vs. 0.011(4), respectively), which, in combination with the much better photon collection, resulted in a higher photosubstitution rate constant for [2]3+ than for [1]2+. The energy of yellow photons is thus transferred efficiently from the rhodamine antenna to the ruthenium center, leading to efficient photosubstitution of Hmte. These results bring new opportunities for extending the photoactivation of polypyridyl ruthenium complexes towards longer wavelengths.
Co-reporter:Sven H. C. Askes;Azadeh Bahreman ;Dr. Sylvestre Bonnet
Angewandte Chemie International Edition 2014 Volume 53( Issue 4) pp:1029-1033
Publication Date(Web):
DOI:10.1002/anie.201309389
Abstract
Liposomes capable of generating photons of blue light in situ by triplet–triplet annihilation upconversion of either green or red light, were prepared. The red-to-blue upconverting liposomes were capable of triggering the photodissociation of ruthenium polypyridyl complexes from PEGylated liposomes using a clinical grade photodynamic therapy laser source (630 nm).
Co-reporter:Sven H. C. Askes;Azadeh Bahreman ;Dr. Sylvestre Bonnet
Angewandte Chemie 2014 Volume 126( Issue 4) pp:1047-1051
Publication Date(Web):
DOI:10.1002/ange.201309389
Abstract
Liposomes capable of generating photons of blue light in situ by triplet–triplet annihilation upconversion of either green or red light, were prepared. The red-to-blue upconverting liposomes were capable of triggering the photodissociation of ruthenium polypyridyl complexes from PEGylated liposomes using a clinical grade photodynamic therapy laser source (630 nm).
Co-reporter:Bart Limburg;Guillaume Laisné;Elisabeth Bouwman
Chemistry - A European Journal 2014 Volume 20( Issue 29) pp:8965-8972
Publication Date(Web):
DOI:10.1002/chem.201402712
Abstract
Photocatalytic systems often suffer from poor quantum yields due to fast charge recombination: The energy-wasting annihilation of the photochemically created charge-separated state. In this report, we show that the efficiency of photoinduced electron transfer from a sacrificial electron donor to positively charged methyl viologen, or to negatively charged 5,5′-dithiobis(2-nitrobenzoate), increases dramatically upon addition of charged phospholipid vesicles if the charge of the lipids is of the same sign as that of the electron acceptor. Centrifugation and UV/Vis titration experiments showed that the charged photosensitizers adsorb at the liposome surface, that is, where the photocatalytic reaction takes place. The increased photoelectron transfer efficiency in the presence of charged liposomes has been ascribed to preferential electrostatic interactions between the photosensitizer and the membrane, which prevents the formation of photosensitizer–electron-acceptor complexes that are inactive towards photoreduction. Furthermore, it is shown that the addition of liposomes results in a decrease in photoproduct inhibition, which is caused by repulsion of the reduced electron acceptor by the photocatalytic site. Thus, liposomes can be used as a support to perform efficient photocatalysis; the charged photoproducts are pushed away from the liposomes and represent “soluble electrons” that can be physically separated from the place where they were generated.
Co-reporter:Dr. Azadeh Bahreman;Martin Rabe;Dr. Alexer Kros;Dr. Gilles Bruylants;Dr. Sylvestre Bonnet
Chemistry - A European Journal 2014 Volume 20( Issue 24) pp:7429-7438
Publication Date(Web):
DOI:10.1002/chem.201400377
Abstract
The interaction between the ruthenium polypyridyl complex [Ru(terpy)(dcbpy)(H2O)]2+ (terpy=2,2′;6′,2“-terpyridine, dcbpy=6,6′-dichloro-2,2′-bipyridine) and phospholipid membranes containing either thioether ligands or cholesterol were investigated using UV–visible spectroscopy, Langmuir–Blodgett monolayer surface pressure measurements, and isothermal titration calorimety (ITC). When embedded in a membrane, the thioether ligand coordinated to the dicationic metal complex only when the phospholipids of the membrane were negatively charged, that is, in the presence of attractive electrostatic interaction. In such a case coordination is much faster than in homogeneous conditions. A two-step model for the coordination of the metal complex to the membrane-embedded sulfur ligand is proposed, in which adsorption of the complex to the negative surface of the monolayers or bilayers occurs within minutes, whereas formation of the coordination bond between the surface-bound metal complex and ligand takes hours. Finally, adsorption of the aqua complex to the membrane is driven by entropy. It does not involve insertion of the metal complex into the hydrophobic lipid layer, but rather simple electrostatic adsorption at the water–bilayer interface.
Co-reporter:Azadeh Bahreman, Bart Limburg, Maxime A. Siegler, Elisabeth Bouwman, and Sylvestre Bonnet
Inorganic Chemistry 2013 Volume 52(Issue 16) pp:9456-9469
Publication Date(Web):August 2, 2013
DOI:10.1021/ic401105v
In this work the thermal and photochemical reactivity of a series of ruthenium complexes [Ru(terpy)(N–N)(L)](X)2 (terpy = 2,2′;6′,2″-terpyridine, L = 2-(methylthio)ethanol (Hmte) or water, and X is Cl– or PF6–) with four different bidentate chelates N–N = bpy (2,2′-bipyridine), biq (2,2′-biquinoline), dcbpy (6,6′-dichloro-2,2′-bipyridine), or dmbpy (6,6′-dimethyl-2,2′-bipyridine), is described. For each chelate N–N the thermodynamic constant of the dark equilibrium between the aqua- and Hmte- complexes, the Hmte photosubstitution quantum yield, and the rate constants of the thermal interconversion between the aqua and Hmte complexes were measured at room temperature. By changing the steric hindrance and electronic properties of the spectator N–N ligand along the series bpy, biq, dcbpy, dmbpy the dark reactivity clearly shifts from a nonlabile equilibrium with N–N = bpy to a very labile thermal equilibrium with N–N = dmbpy. According to variable-temperature rate constant measurements in the dark near pH = 7 the activation enthalpies for the thermal substitution of H2O by Hmte are comparable for all ruthenium complexes, whereas the activation entropies are negative for bpy and biq, and positive for dcbpy and dmbpy complexes. These data are indicative of a change in the substitution mechanism, being interchange associative with nonhindered or poorly hindered chelates (bpy, biq), and interchange dissociative for more bulky ligands (dcbpy, dmbpy). For the most labile dmbpy system, the thermal equilibrium is too fast to allow significant modification of the composition of the mixture using light, and for the nonhindered bpy complex the photosubstitution of Hmte by H2O is possible but thermal binding of Hmte to the aqua complex does not occur at room temperature. By contrast, with N–N = biq or dcbpy the thermodynamic and kinetic parameters describing the formation and breakage of the Ru–S bond lie in a range where the bond forms spontaneously in the dark, but is efficiently cleaved under light irradiation. Thus, the ratio between the aqua and Hmte complex in solution can be efficiently controlled at room temperature using visible light irradiation.
Co-reporter:Elwin Molenbroek, Natan Straathof, Sebastian Dück, Zahid Rashid, Joop H. van Lenthe, Martin Lutz, Aurore Gandubert, Robertus J. M. Klein Gebbink, Luisa De Cola and Sylvestre Bonnet
Dalton Transactions 2013 vol. 42(Issue 8) pp:2973-2984
Publication Date(Web):30 Nov 2012
DOI:10.1039/C2DT32488A
In this work, the complexation of the bapbpy ligand to zinc dichloride is described (bapbpy = 6,6′-bis(2-aminopyridyl)-2,2′-bipyridine). The water-soluble, colorless complex [Zn(bapbpy)Cl]Cl·2H2O (compound 2·H2O) was synthesized; its X-ray crystal structure shows a mononuclear, pentacoordinated geometry with one chloride ligand in apical position. Upon excitation of its lowest-energy absorption band (375 nm) compound 2 shows intense emission (Φ = 0.50) at 418 nm in aqueous solution, and an excited state lifetime of 5 ns at room temperature. Photophysical measurements, DFT, and TD-DFT calculations prove that emission arises from vibronically coupled Ligand-to-Ligand Charge Transfer singlet excited states, characterized by electron density flowing from the lone pairs of the non-coordinated NH bridges to the π* orbitals of the pyridine rings. Monofunctionalization of the ligand with one long alkyl chain was realized to afford ligand3, which can be inserted into dimyristoylphosphatidylglycerol (DMPG) or dimyristoylphosphatidylcholine (DMPC) unilamellar vesicles. For negatively charged DMPG membranes the addition of a zinc salt to the vesicles leads to an enhancement of the fluorescence due to zinc coordination to the membrane-embedded tetrapyridyl ligand. No changes were observed for the zwitterionic DMPC lipids, where binding of the Zn ions does not take place. A modest binding constant was found (5 × 106 M−1) for the coordination of zinc cations to bapbpy-functionalized DMPG membranes, which allows for the detection of micromolar zinc concentrations in aqueous solution. The influence of chloride concentration and other transition metal ions on the zinc binding was evaluated, and the potential of liposome-supported metal chelators such as ligand3 for zinc detection in biological media is discussed.
Co-reporter:Bart Limburg, Elisabeth Bouwman, Sylvestre Bonnet
Coordination Chemistry Reviews 2012 Volume 256(15–16) pp:1451-1467
Publication Date(Web):August 2012
DOI:10.1016/j.ccr.2012.02.021
Water is a widely available source of “cheap electrons”, and water oxidation a chemical challenge that was tackled by the community of homogeneous catalysis decades ago. Although catalytic activity was initially the major bottleneck, when the blue dimer was one of the few available homogeneous catalysts that could perform water oxidation, major breakthroughs have been realized in the past few years. These advances have now resulted in a situation where stability, turnover numbers, and price of the catalysts have become the major issues. In this field of research, chemical oxidation using large excesses of CeIV salts or [Ru(bpy)3]3+ has become a standard to test the catalytic activity of new water oxidation catalysts. In such highly oxidizing conditions however, the stability of the catalyst is seriously challenged, and most known catalysts deactivate after some time. Currently the available information concerning the main decomposition pathways is rather scattered, and sometimes contradictory. The present review sums up the evolution of water oxidation catalysts in terms of stability, and the available experimental studies dealing with their decomposition pathways. It also gathers the rare examples of photochemical systems capable of performing water oxidation, and discusses the relation between photochemical and chemical water oxidation regarding catalyst stability.Graphical abstractHighlights► Standard conditions to test new homogeneous water oxidation catalysts are very harsh. ► Most active catalysts based on Mn, Ru, Co, and even Ir, decompose. ► Ligand design can improve catalyst stability. ► Oxidizing water photochemically may be milder than using an excess of CeIV.
Co-reporter:Azadeh Bahreman;Bart Limburg;Dr. Maxime A. Siegler;Dr. Roman Koning; Abraham J. Koster;Dr. Sylvestre Bonnet
Chemistry - A European Journal 2012 Volume 18( Issue 33) pp:10271-10280
Publication Date(Web):
DOI:10.1002/chem.201200624
Abstract
The new ruthenium complex [Ru(terpy)(dcbpy)(Hmte)](PF6)2 ([2](PF6)2; dcbpy=6,6′-dichloro-2,2′-bipyridine, terpy=2,2′;6′,2“-terpyridine, Hmte=2-(methylthio)ethanol) was synthesized. In the crystal structure, this complex is highly distorted, revealing steric congestion between dcbpy and Hmte. In water, [2]2+ forms spontaneously by reacting Hmte and the aqua complex [Ru(terpy)(dcbpy)(OH2)]2+ ([1]2+), with a second-order rate constant of 0.025 s−1 M−1 at 25 °C. In the dark, the RuS bond of [2]2+ is thermally unstable and partially hydrolyzes; in fact, [1]2+ and [2]2+ are in an equilibrium characterized by an equilibrium constant K of 151 M−1. When exposed to visible light, the RuS bond is selectively broken to release [1]2+, that is, the equilibrium is shifted by visible-light irradiation. The light-induced equilibrium shifts were repeated four times without major signs of degradation; the RuS coordination bond in [2]2+ can be described as a robust, light-sensitive, supramolecular bond in water. To demonstrate the potential of this system in supramolecular chemistry, a new thioether–cholesterol conjugate (4), which inserts into lipid bilayers through its cholesterol moiety and coordinates to ruthenium through its sulfur atom, was synthesized. Thioether-functionalized, anionic, dimyristoylphosphatidylglycerol (DMPG), lipid vesicles, to which aqua complex [1]2+ efficiently coordinates, were prepared. Upon exposure of the Ru-decorated vesicles to visible light, the RuS bond is selectively broken, thus releasing [1]2+ that stays at the water-bilayer interface. When the light is switched off, the metal complex spontaneously coordinates back to the membrane-embedded thioether ligands without a need to heat the system. This process was repeated four times at 35 °C, thus achieving light-triggered hopping of the metal complex at the water-bilayer interface.
Co-reporter:Zulema Arcis-Castíllo;Sipeng Zheng;Dr. Maxime A. Siegler;Dr. Olivier Roubeau;Salma Bedoui;Dr. Sylvestre Bonnet
Chemistry - A European Journal 2011 Volume 17( Issue 52) pp:14826-14836
Publication Date(Web):
DOI:10.1002/chem.201101301
Abstract
In this study, we show that 1) different isomers of the same mononuclear iron(II) complex give materials with different spin-crossover (hereafter SCO) properties, and 2) minor modifications of the bapbpy (bapbpy=N6,N6′-di(pyridin-2-yl)-2,2′-bipyridine-6,6′-diamine) ligand allows SCO to be obtained near room temperature. We also provide a qualitative model to understand the link between the structure of bapbpy-based ligands and the SCO properties of their iron(II) compounds. Thus, seven new trans-[Fe{R2(bapbpy)}(NCS)2] compounds were prepared, in which the R2bapbpy ligand bears picoline (9–12), quin-2-oline (13), isoquin-3-oline (14), or isoquin-1-oline (15) substituents. From this series, three compounds (12, 14, and 15) have SCO properties, one of which (15) occurs at 288 K. The crystal structures of compounds 11, 12, and 15 show that the intermolecular interactions in these materials are similar to those found in the parent compound [Fe(bapbpy)(NCS)2] (1), in which each iron complex interacts with its neighbors through weak NH⋅⋅⋅S hydrogen bonding and π–π stacking. For compounds 12 and 15, hindering groups located near the NH bridges weaken the NS intermolecular interactions, which is correlated to non-cooperative SCO. For compound 14, the substitution is further away from the NH bridges, and the SCO remains cooperative as in 1 with a hysteresis cycle. Optical microscopy photographs show the strikingly different spatio-temporal evolution of the phase transition in the noncooperative SCO compound 12 relative to that found in 1. Heat-capacity measurements were made for compounds 1, 12, 14, and 15 and fitted to the Sorai domain model. The number n of like-spin SCO centers per interacting domain, which is related to the cooperativity of the spin transition, was found high for compounds 1 and 14 and low for compounds 12 and 15. Finally, we found that although both pairs of compounds 11/12 and 14/15 are pairs of isomers their SCO properties are surprisingly different.
Co-reporter:Roosmarijn E. Goldbach;Isabel Rodriguez-Garcia;Joop H. vanLenthe;Maxime A. Siegler
Chemistry - A European Journal 2011 Volume 17( Issue 36) pp:9924-9929
Publication Date(Web):
DOI:10.1002/chem.201101541
Co-reporter:Sylvestre Bonnet ; Bart Limburg ; Johannes D. Meeldijk ; Robertus J. M. Klein Gebbink ;J. Antoinette Killian
Journal of the American Chemical Society 2010 Volume 133(Issue 2) pp:252-261
Publication Date(Web):December 16, 2010
DOI:10.1021/ja105025m
Electrostatic forces play an important role in the interaction between large transition metal complexes and lipid bilayers. In this work, a thioether-cholestanol hybrid ligand (4) was synthesized, which coordinates to ruthenium(II) via its sulfur atom and intercalates into lipid bilayers via its apolar tail. By mixing its ruthenium complex [Ru(terpy)(bpy)(4)]2+ (terpy = 2,2′;6′,2′′-terpyridine; bpy = 2,2′-bipyridine) with either the negatively charged lipid dimyristoylphosphatidylglycerol (DMPG) or with the zwitterionic lipid dimyristoylphosphatidylcholine (DMPC), large unilamellar vesicles decorated with ruthenium polypyridyl complexes are formed. Upon visible light irradiation the ruthenium−sulfur coordination bond is selectively broken, releasing the ruthenium fragment as the free aqua complex [Ru(terpy)(bpy)(OH2)]2+. The photochemical quantum yield under blue light irradiation (452 nm) is 0.0074(8) for DMPG vesicles and 0.0073(8) for DMPC vesicles (at 25 °C), which is not significantly different from similar homogeneous systems. Dynamic light scattering and cryo-TEM pictures show that the size and shape of the vesicles are not perturbed by light irradiation. Depending on the charge of the lipids, the cationic aqua complex either strongly interacts with the membrane (DMPG) or diffuses away from it (DMPC). Back coordination of [Ru(terpy)(bpy)(OH2)]2+ to the thioether-decorated vesicles takes place only at DMPG bilayers with high ligand concentrations (25 mol %) and elevated temperatures (70 °C). During this process, partial vesicle fusion was also observed. We discuss the potential of such ruthenium-decorated vesicles in the context of light-controlled molecular motion and light-triggered drug delivery.
Co-reporter:Sven H. C. Askes, Néstor López Mora, Rolf Harkes, Roman I. Koning, Bram Koster, Thomas Schmidt, Alexander Kros and Sylvestre Bonnet
Chemical Communications 2015 - vol. 51(Issue 44) pp:NaN9140-9140
Publication Date(Web):2015/04/27
DOI:10.1039/C5CC02197A
Red-to-blue triplet–triplet annihilation upconversion was obtained in giant unilamellar vesicles. The upconverted light was homogeneously distributed across the membrane and could be utilized for the imaging of individual giant vesicles in three dimensions. These results show the great potential of TTA-UC for imaging applications under anoxic conditions.
Co-reporter:Sven H. C. Askes, Miroslav Kloz, Gilles Bruylants, John T. M. Kennis and Sylvestre Bonnet
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 41) pp:NaN27390-27390
Publication Date(Web):2015/09/22
DOI:10.1039/C5CP04352B
Upconversion is a promising way to trigger high-energy photochemistry with low-energy photons. However, combining upconversion schemes with non-radiative energy transfer is challenging because bringing several photochemically active components in close proximity results in complex multi-component systems where quenching processes may deactivate the whole assembly. In this work, PEGylated liposomes were prepared that contained three photoactive components: a porphyrin dye absorbing red light, a perylene moiety emitting in the blue, and a light-activatable ruthenium prodrug sensitive to blue light. Time-dependent spectroscopic studies demonstrate that singlet perylene excited states are non-radiatively transferred to the nearby ruthenium complex by Förster resonance energy transfer (FRET). Under red-light irradiation of the three-component membranes, triplet–triplet annihilation upconversion (TTA-UC) occurs followed by FRET, which results in a more efficient activation of the ruthenium prodrug compared to a physical mixture of two-component upconverting liposomes and liposomes containing only the ruthenium complex. This work represents a rare example where TTA-UC and Förster resonance energy transfer are combined to achieve prodrug activation in the phototherapeutic window.
Co-reporter:Azadeh Bahreman, Jordi-Amat Cuello-Garibo and Sylvestre Bonnet
Dalton Transactions 2014 - vol. 43(Issue 11) pp:NaN4505-4505
Publication Date(Web):2013/12/05
DOI:10.1039/C3DT52643G
The ruthenium complex [Ru(terpy)(bpy)(Hmte)]2+ ([1]2+), where terpy is 2,2′;6′,2′′-terpyridine, bpy is 2,2′-bipyridine, and Hmte is 2-methylthioethan-1-ol, poorly absorbs yellow light, and although its quantum yield for the photosubstitution of Hmte by water is comparable at 570 nm and at 452 nm (0.011(4) vs. 0.016(4) at 298 K at neutral pH), the photoreaction using yellow photons is very slow. Complex [1]2+ was thus functionalized with rhodamine B, an organic dye known for its high extinction coefficient for yellow light. Complex [Ru(Rterpy)(bpy)(Hmte)]3+ ([2]3+) was synthesized, where Rterpy is a terpyridine ligand covalently bound to rhodamine B via a short saturated linker. [2]Cl3 shows a very high extinction coefficient at 570 nm (44000 M−1 cm−1), but its luminescence upon irradiation at 570 nm is completely quenched in aqueous solution. The quantum yield for the photosubstitution of Hmte by water in [2]3+ was comparable to that in [1]2+ at 570 nm (0.0085(6) vs. 0.011(4), respectively), which, in combination with the much better photon collection, resulted in a higher photosubstitution rate constant for [2]3+ than for [1]2+. The energy of yellow photons is thus transferred efficiently from the rhodamine antenna to the ruthenium center, leading to efficient photosubstitution of Hmte. These results bring new opportunities for extending the photoactivation of polypyridyl ruthenium complexes towards longer wavelengths.
Co-reporter:Elwin Molenbroek, Natan Straathof, Sebastian Dück, Zahid Rashid, Joop H. van Lenthe, Martin Lutz, Aurore Gandubert, Robertus J. M. Klein Gebbink, Luisa De Cola and Sylvestre Bonnet
Dalton Transactions 2013 - vol. 42(Issue 8) pp:NaN2984-2984
Publication Date(Web):2012/11/30
DOI:10.1039/C2DT32488A
In this work, the complexation of the bapbpy ligand to zinc dichloride is described (bapbpy = 6,6′-bis(2-aminopyridyl)-2,2′-bipyridine). The water-soluble, colorless complex [Zn(bapbpy)Cl]Cl·2H2O (compound 2·H2O) was synthesized; its X-ray crystal structure shows a mononuclear, pentacoordinated geometry with one chloride ligand in apical position. Upon excitation of its lowest-energy absorption band (375 nm) compound 2 shows intense emission (Φ = 0.50) at 418 nm in aqueous solution, and an excited state lifetime of 5 ns at room temperature. Photophysical measurements, DFT, and TD-DFT calculations prove that emission arises from vibronically coupled Ligand-to-Ligand Charge Transfer singlet excited states, characterized by electron density flowing from the lone pairs of the non-coordinated NH bridges to the π* orbitals of the pyridine rings. Monofunctionalization of the ligand with one long alkyl chain was realized to afford ligand3, which can be inserted into dimyristoylphosphatidylglycerol (DMPG) or dimyristoylphosphatidylcholine (DMPC) unilamellar vesicles. For negatively charged DMPG membranes the addition of a zinc salt to the vesicles leads to an enhancement of the fluorescence due to zinc coordination to the membrane-embedded tetrapyridyl ligand. No changes were observed for the zwitterionic DMPC lipids, where binding of the Zn ions does not take place. A modest binding constant was found (5 × 106 M−1) for the coordination of zinc cations to bapbpy-functionalized DMPG membranes, which allows for the detection of micromolar zinc concentrations in aqueous solution. The influence of chloride concentration and other transition metal ions on the zinc binding was evaluated, and the potential of liposome-supported metal chelators such as ligand3 for zinc detection in biological media is discussed.
Co-reporter:V. H. S. van Rixel, B. Siewert, S. L. Hopkins, S. H. C. Askes, A. Busemann, M. A. Siegler and Sylvestre Bonnet
Chemical Science (2010-Present) 2016 - vol. 7(Issue 8) pp:
Publication Date(Web):
DOI:10.1039/C6SC00167J
Co-reporter:B. Limburg, E. Bouwman and S. Bonnet
Chemical Communications 2015 - vol. 51(Issue 96) pp:NaN17131-17131
Publication Date(Web):2015/10/07
DOI:10.1039/C5CC07745A
Unidirectional photocatalytic electron transfer from a hydrophilic electron donor encapsulated in the interior of a liposome, to a hydrophilic electron acceptor on the other side of the membrane, has been achieved using the simple membrane-soluble electron relay 1-methoxy-N-methylphenazinium (MMP+). The total amount of photoproduct (>140 nmol) exceeds the number of moles of MMP+ present (125 nmol), thus showing that the transport of electrons is catalytic.
Co-reporter:Hyo Jin Jang, Samantha L. Hopkins, Maxime A. Siegler and Sylvestre Bonnet
Dalton Transactions 2017 - vol. 46(Issue 30) pp:NaN9980-9980
Publication Date(Web):2017/07/20
DOI:10.1039/C7DT01540B
The synthesis and characterization of [Ru(tpy)(R2bpy)(L)](X)n complexes (tpy = 2,2′:6′,2′′-terpyridine, R2bpy = 4,4′-dimethyl-2,2′-bipyridine (dmbpy), or 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (tfmbpy), X = Cl− or PF6−, and n = 1 or 2) are described. The dmbpy and tfmbpy bidentate ligands allow for investigating the effects of electron-donating and electron-withdrawing ligands, respectively, on the frontier orbital energetics as well as the photoreactivity of these ruthenium polypyridyl complexes for five prototypical monodentate ligands L = Cl−, H2O, CH3CN, 2-(methylthio)ethanol (Hmte), or pyridine. According to spectroscopic and electrochemical studies, the dmbpy analogues displayed a singlet metal-to-ligand charge transfer (1MLCT) transition at higher energy than the tfmbpy analogues. The shift of the 1MLCT to higher energy results from the lowest unoccupied molecular orbital (LUMO) for the dmbpy analogues being tpy-based, whereas for the tfmbpy analogues orbital inversion occurs resulting in a tfmbpy-based LUMO. The energy level of the highest occupied molecular orbital (HOMO) was considerably affected by the nature of the monodentate ligand. Visible light irradiation of the complexes demonstrated that the tfmbpy analogue increased the rate and quantum yields of photosubstitution reactions, compared to the dmbpy analogue, suggesting that the electron-withdrawing substituents allowed better thermal accessibility of the triplet metal-centered (3MC) state from the photochemically generated triplet metal-to-ligand charge transfer (3MLCT) excited state. A correlation between the photolability of the monodentate ligands and the electrochemical reversibility of the metal-based oxidation is also reported.
Co-reporter:Jordi-Amat Cuello-Garibo, Michael S. Meijer and Sylvestre Bonnet
Chemical Communications 2017 - vol. 53(Issue 50) pp:NaN6771-6771
Publication Date(Web):2017/06/05
DOI:10.1039/C7CC03469E
In metal-based photoactivated chemotherapy (PACT), two photoproducts are generated by light-triggered photosubstitution of a metal-bound ligand: the free ligand itself and an aquated metal complex. By analogy with cisplatin, the aquated metal complex is usually presented as the biologically active species, as it can typically bind to DNA. In this work, we show that this qualitative assumption is not necessarily valid by comparing the biological activity, logP, and cellular uptake of three ruthenium-based PACT complexes: [Ru(bpy)2(dmbpy)]2+, [Ru(bpy)2(mtmp)]2+, and [Ru(Ph2phen)2(mtmp)]2+. For the first complex, the photoreleased dmbpy ligand is responsible for the observed phototoxicity, whereas the second complex is not phototoxic, and for the third complex it is the ruthenium bis-aqua photoproduct that is the sole cytotoxic species.

![3-{[(2,3-dihydroxypropoxy)(hydroxy)phosphoryl]oxy}-2-(tetradecanoyloxy)propyl tetradecanoate](http://img.cochemist.com/ccimg/61400/61361-72-6.png)
![3-{[(2,3-dihydroxypropoxy)(hydroxy)phosphoryl]oxy}-2-(tetradecanoyloxy)propyl tetradecanoate](http://img.cochemist.com/ccimg/61400/61361-72-6_b.png)
![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7.png)
![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7_b.png)
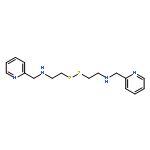
![Ethanol, 2-(methyl[2,2':6',2''-terpyridin]-4'-ylamino)-](http://img.cochemist.com/ccimg/194000/193945-29-8.png)
![Ethanol, 2-(methyl[2,2':6',2''-terpyridin]-4'-ylamino)-](http://img.cochemist.com/ccimg/194000/193945-29-8_b.png)


![[2,2'-Bipyridin]-6-amine](/data/chemimg/1757100/178039-84-4.png)
![[2,2'-Bipyridin]-6-amine](/data/chemimg/1757100/178039-84-4_b.png)




![[2,2'-Bipyridine]-6,6'-diamine](http://img.cochemist.com/ccimg/93200/93127-75-4.png)
![[2,2'-Bipyridine]-6,6'-diamine](http://img.cochemist.com/ccimg/93200/93127-75-4_b.png)

![Platinum, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/540800/31248-39-2.png)
![Platinum, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/540800/31248-39-2_b.png)
![Zinc(4+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,chloride (1:4), (SP-4-1)-](http://img.cochemist.com/ccimg/28900/28850-44-4.png)
![Zinc(4+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,chloride (1:4), (SP-4-1)-](http://img.cochemist.com/ccimg/28900/28850-44-4_b.png)
![Palladium, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/134800/119654-64-7.png)
![Palladium, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/134800/119654-64-7_b.png)




![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)


![3,5,9-Trioxa-4-phosphatetracosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxopentadecyl)oxy]-, inner salt,4-oxide, (7R)-](http://img.cochemist.com/ccimg/3400/3355-27-9.png)
![3,5,9-Trioxa-4-phosphatetracosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxopentadecyl)oxy]-, inner salt,4-oxide, (7R)-](http://img.cochemist.com/ccimg/3400/3355-27-9_b.png)
![Sodium 2,3-dihydroxypropyl [(2r)-2,3-di(tetradecanoyloxy)propyl] Phosphate](http://img.cochemist.com/ccimg/200900/200880-40-6.png)
![Sodium 2,3-dihydroxypropyl [(2r)-2,3-di(tetradecanoyloxy)propyl] Phosphate](http://img.cochemist.com/ccimg/200900/200880-40-6_b.png)