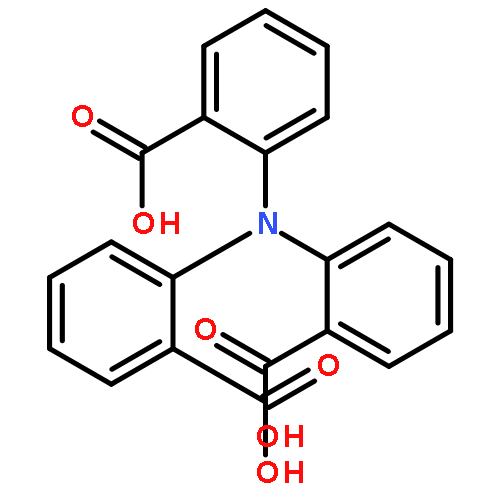
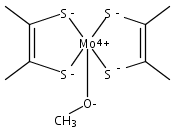
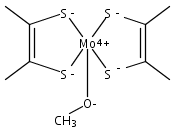
Co-reporter:Matthias Hofmann, Farooq Ahmad Kiani
Journal of Organometallic Chemistry 2009 694(11) pp: 1666-1670
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.11.014
Co-reporter:Matthias Hofmann
Inorganic Chemistry 2008 Volume 47(Issue 13) pp:5546-5548
Publication Date(Web):June 6, 2008
DOI:10.1021/ic800519d
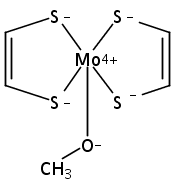
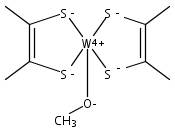
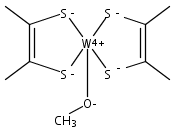
Computations suggest that in contrast with small models the active site geometry of reduced dimethyl sulfoxide reductase might prefer a triplet over a singlet electronic state.
Co-reporter:FarooqAhmad Kiani Dr. Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 9) pp:2886-2893
Publication Date(Web):
DOI:10.1002/chem.200701369
Abstract
Macropolyhedral borane clusters are concave polyhedra constituting fused convex simple polyhedra. They are formally obtained by condensation of simple polyhedral boranes under elimination of between one and four BH3 or isoelectronic units. The number of eliminated vertexes from simple polyhedra equals the number of shared vertexes in macropolyhedral boranes. For each of the eight classes with general formulae ranging from BnHn−4 to BnHn+10, more than one structure type is possible, differing in the number of shared vertexes and in the types of the two combined cluster fragments. However, only one type of “potential structures” is represented by experimentally known examples and is found to be favored by theoretical calculations. A sophisticated system exists among the favored macropolyhedral borane structures. For each class of macropolyhedral boranes, the number of skeletal electron pairs is directly related to the general formula, the number of shared vertexes and the type of fused cluster fragments. In order to predict the distribution of vertexes among the fused fragments, we propose the concept of preferred fragments. Preferred fragments are those usually present in the thermodynamically most stable structure of a given class of macropolyhedral boranes and are also frequently observed in the experimentally known structures. This allows us to completely predict the cluster framework of the thermodynamically most stable macropolyhedral borane isomers.
Co-reporter:Farooq Ahmad Kiani and Matthias Hofmann
Dalton Transactions 2007 (Issue 12) pp:1207-1213
Publication Date(Web):20 Feb 2007
DOI:10.1039/B700270J
Various two vertex sharing macropolyhedral boranes were computed at the B3LYP/6-311 + G**//B3LYP/6-31G* level of theory to determine the preferred fragments for the thermodynamically most stable isomers. These are nido-10 and arachno-9 vertex fragments for neutral macropolyhedral boranes. The thermodynamically most stable isomers of the nido:nido-, arachno:nido- and arachno:arachno-macropolyhedral borane classes are structurally related to each other by the successive removal of one open face vertex as in the case of simple polyhedral boranes. For these classes, the stabilities of the thermodynamically most stable macropolyhedra relative to isomeric simple polyhedra follow similar trends with respect to the number of skeletal electrons.
Co-reporter:Farooq Ahmad Kiani and Matthias Hofmann
Dalton Transactions 2006 (Issue 46) pp:5515-5520
Publication Date(Web):06 Oct 2006
DOI:10.1039/B610138K
Cluster increments derived for individual cluster fragments reproduce the DFT computed relative stabilities of macropolyhedral boranes usually within ±6 kcal mol−1. A simple summation procedure helps to select the best partner for a given cluster fragment in order to construct the thermodynamically most stable macropolyhedral borane. Cluster increments are considerably smaller for nido-cluster fragments with an even number of vertexes than for odd nido-cluster fragments pointing towards high thermodynamic stability of macropolyhedral boranes with even numbered nido-units.
Co-reporter:Farooq Ahmad Kiani and Matthias Hofmann
Dalton Transactions 2006 (Issue 5) pp:686-692
Publication Date(Web):22 Nov 2005
DOI:10.1039/B512700A
The relative thermodynamic stabilities of ortho-, meta- and para-isomers of 12-vertex closo-heteroboranes and -borates with different p-block heteroatoms were determined using density functional theory. More electronegative (smaller) heteroatoms tend to occupy non-adjacent, whereas less electronegative (larger) heteroatoms tend to occupy adjacent vertices in the thermodynamically most stable closo-diheterododecaborane isomers. The computed relative stabilities agree perfectly with experimental observations. The energy differences of para- and meta- relative to ortho-isomers of 12-vertex closo-heteroboranes generally depend on the extent of electron localization by a given heteroatom and show highly periodic trends, i.e. increase along the period and decrease down the group. The energy penalties for the HetHet structural feature (two heteroatoms adjacent to each other) for the 12-vertex closo-cluster are apparently significantly different from those for the 11-vertex nido-cluster. Reformulating two 11-vertex nido-structural features, i.e. Het5k(2) and HetHet, in terms of connection increments along with the additional structural feature HetHetm give the relative stabilities of various isomeric 11-vertex nido- as well as 12-vertex closo-heteroboranes and -borates, using one unique set of increments.
Co-reporter:Farooq A. Kiani
European Journal of Inorganic Chemistry 2005 Volume 2005(Issue 12) pp:
Publication Date(Web):15 JUN 2005
DOI:10.1002/ejic.200400982
A structural increment system, i.e. quantitative rules that govern the relative stabilities of 10-vertex nido-boranes and-carboranes, has been determined. Density functional theory computations at the B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level with ZPE corrections were carried out for 81 different boron hydride and carborane structures from [B10H12]2– to C3B7H11 to determine their relative stabilities. A set of eleven disfavored geometrical features that destabilize a cluster structure relative to a hypothetical ideal situation were identified and weighted by so-called energy penalties. The latter show good additive behavior and allow us to reproduce the DFT computed relative energies mostly with an accuracy of 6.0 kcal mol–1. Some unknown 10-vertex nido-carboranes that are thermodynamically more stable than their known isomers are also identified. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2005)
Co-reporter:Farooq Ahmad Kiani and Matthias Hofmann
Dalton Transactions 2007(Issue 12) pp:NaN1213-1213
Publication Date(Web):2007/02/20
DOI:10.1039/B700270J
Various two vertex sharing macropolyhedral boranes were computed at the B3LYP/6-311 + G**//B3LYP/6-31G* level of theory to determine the preferred fragments for the thermodynamically most stable isomers. These are nido-10 and arachno-9 vertex fragments for neutral macropolyhedral boranes. The thermodynamically most stable isomers of the nido:nido-, arachno:nido- and arachno:arachno-macropolyhedral borane classes are structurally related to each other by the successive removal of one open face vertex as in the case of simple polyhedral boranes. For these classes, the stabilities of the thermodynamically most stable macropolyhedra relative to isomeric simple polyhedra follow similar trends with respect to the number of skeletal electrons.