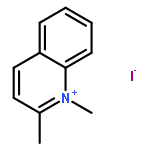
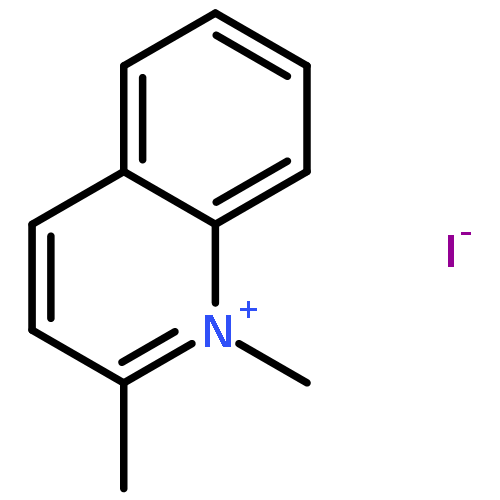
Co-reporter:Yan Zhang, Deying Zeng, Jiaojiao Cao, Mingxue Wang, Bing Shu, Guotao Kuang, Tian-Miao Ou, Jia-Heng Tan, Lian-Quan Gu, Zhi-Shu Huang, Ding Li
Biochimica et Biophysica Acta (BBA) - General Subjects 2017 Volume 1861, Issue 12(Issue 12) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.bbagen.2017.09.015
•Telomeric repeat–containing RNA (TERRA) is a component of telomeric heterochromatin.•Telomeric repeat factor 2 (TRF2) is a component of Shelterin protecting chromosome.•Quindoline derivative CK1-14 could bind to and stabilize TERRA G-quadruplex.•The above complex could inhibit TRF2, resulting in its dissociation from telomere.•CK1-14 could activate DNA-damage response and induce tumor cell apoptosis.BackgroundTelomeric repeat–containing RNA (TERRA) is a large non-coding RNA in mammalian cells, which forms an integral component of telomeric heterochromatin. TERRA can bind to an allosteric site of telomeric repeat factor 2 (TRF2), a key component of Shelterin that protect chromosome termini. Both TERRA and TRF2 have been recognized as promising new therapeutic targets for cancer treatment.MethodsOur methods include FRET assay, SPR, CD, microscale thermophoresis (MST), enzyme-linked immunosorbent assay (ELISA), chromatin immunoprecipitation (ChIP), colony formation assays, Western blot, immunofluorescence, cell cycle arrest and apoptosis detection, and xCELLigence real-time cell analysis (RTCA).ResultsIn our routine screening of small molecule libraries, we found that a Quindoline derivative, CK1-14 could bind to and stabilize TERRA G-quadruplex structure, which could bind more tightly with an allosteric site of a telomeric binding protein TRF2, resulting in dissociation of TRF2 from telomeric DNA. Further in cellular studies indicated that the above effect of CK1-14 on TERRA G-quadruplex could activate DNA-damage response and cause cell cycle arrest, resulting in inhibition of U2OS cell proliferation and causing cell apoptosis.ConclusionsOur mechanistic studies indicated that interaction of CK1-14 with TERRA induces telomeric DNA-damage response in U2OS cancer cells through inhibition of TRF2. CK1-14 could be further developed as a promising lead compound targeting telomere for cancer treatment.General significanceOur present study provides the first evidence that allosteric modulation of TRF2 by TERRA G-quadruplex with a binding ligand could become a promising new strategy for cancer treatment especially for ALT tumor cells.Download high-res image (59KB)Download full-size image
Co-reporter:Jun Qiu, Jinggong Liu, Shuobin Chen, Tian-Miao Ou, Jia-Heng Tan, Lian-Quan Gu, Zhi-Shu Huang, and Ding Li
Organic Letters 2015 Volume 17(Issue 18) pp:4584-4587
Publication Date(Web):September 2, 2015
DOI:10.1021/acs.orglett.5b02310
The promoter of hnRNP K oncogene was found to contain a G/C-rich sequence on the same DNA strand, which can form interconvertible G-quadruplex, i-motif, and hairpin structures. Protein CNBP could bind and stabilize the G-quadruplex, inducing transformation of the hairpin into the G-quadruplex, resulting in down-regulation of hnRNP K transcription. In contrast, Corticosterone could bind and stabilize the hairpin, inducing transformation of the G-quadruplex into the hairpin, resulting in up-regulation of hnRNP K gene transcription.
Co-reporter:Shang-Shi Zhang, Chun-Yong Jiang, Jia-Qiang Wu, Xu-Ge Liu, Qingjiang Li, Zhi-Shu Huang, Ding Li and Honggen Wang
Chemical Communications 2015 vol. 51(Issue 50) pp:10240-10243
Publication Date(Web):14 May 2015
DOI:10.1039/C5CC03187G
Cp*Rh(III)- and Cp*Ir(III)-catalysed direct C–H arylation with quinone diazides as efficient coupling partners is disclosed. This redox-neutral protocol offers a facile, operationally simple and environmentally benign access to arylated phenols. The reaction represents the first example of Cp*Ir(III)-catalysed C–H direct arylation reaction.
Co-reporter:Hu Ge, Jinggong Liu, Wenxia Zhao, Yu Wang, Qingqing He, Ruibo Wu, Ding Li and Jun Xu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 27) pp:4941-4951
Publication Date(Web):17 Apr 2014
DOI:10.1039/C4OB00589A
In the present study, we found that three enzymes, MVK, MDD and FPPS, in the mevalonate pathway (MVP) of cholesterol biosynthesis, can be simultaneously inhibited by two green tea polyphenols ((−)-epicatechin-3-gallate, ECG; (−)-epigallocatechin-3-gallate, EGCG). Molecular dynamics simulations and pharmacophore studies were carried out to elucidate the tri-targeted inhibition mechanisms. Our results indicate that similar triangular binding pockets exist in all three enzymes, which is essential for their binding with polyphenols. Two distinct binding poses for ECG and EGCG were observed in our MD simulations. These results shed light on the potential for further selective and multi-targeted inhibitor design for the treatment of hyperlipidemia.
Co-reporter:Xiaojun Liu, Long Wu, Guisheng Deng, Gong Chen, Nan Li, Xiusheng Chu, Ding Li
Bioorganic Chemistry 2013 Volume 47() pp:1-8
Publication Date(Web):April 2013
DOI:10.1016/j.bioorg.2012.12.001
Short/branched chain acyl-CoA dehydrogenase (SBCAD), isovaleryl-CoA dehydrogenase (IVD), and isobutyryl-CoA dehydrogenase (IBD) are involved in metabolism of isoleucine, leucine, and valine, respectively. These three enzymes all belong to acyl-CoA dehydrogenase (ACD) family, and catalyze the dehydrogenation of monomethyl branched-chain fatty acid (mmBCFA) thioester derivatives. In the present work, the catalytic properties of rat SBCAD, IVD, and IBD, including their substrate specificity, isomerase activity, and enzyme inhibition, were comparatively studied. Our results indicated that SBCAD has its catalytic properties relatively similar to those of straight-chain acyl-CoA dehydrogenases in terms of their isomerase activity and enzyme inhibition, while IVD and IBD are different. IVD has relatively broader substrate specificity than those of the other two enzymes in accommodating various substrate analogs. The present study increased our understanding for the metabolism of monomethyl branched-chain fatty acids (mmBCFAs) and branched-chain amino acids (BCAAs), which should also be useful for selective control of a particular reaction through the design of specific inhibitors.Graphical abstractHighlights► SBCAD, IVD, and IBD are involved in metabolism of isoleucine, leucine, and valine. ► These three enzymes all belong to acyl-CoA dehydrogenase (ACD) family. ► The catalytic properties of rat SBCAD, IVD, and IBD, were comparatively studied. ► SBCAD has its catalytic properties similar to those of straight-chain ACDs. ► IVD has relatively broader substrate specificity than those of the other two enzymes.
Co-reporter:Zeng Li, Jia-Heng Tan, Jin-Hui He, Yi Long, Tian-Miao Ou, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2012 Volume 47() pp:299-311
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.10.057
A series of 2,4-disubstituted quinazoline derivatives found to be a new type of highly selective ligand to bind with telomeric G-quadruplex DNA, and their biological properties were reported for the first time.Their interactions with telomeric G-quadruplex DNA were evaluated by using fluorescence resonance energy transfer (FRET) melting assay, circular dichroism (CD) spectroscopy, surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), and molecular modeling. Our results showed that these derivatives could well recognize G-quadruplex and have high selectivity toward G-quadruplex over duplex DNA. The structure–activity relationships (SARs) study revealed that the disubstitution of quinazoline and the length of the amide side chain were important for its interaction with the G-quadruplex. Furthermore, telomerase inhibition of the quinazoline derivatives and their cellular effects were studied.A series of quinazoline derivatives as novel telomeric G-quadruplex ligands were synthesized and evaluated. These derivatives could well recognize G-quadruplex DNA and have significant cellular biological activity.Highlights► 17 quinazoline derivatives as novel G-quadruplex DNA ligands were synthesized. ► They showed high binding affinity and selectivity with telomeric G-quadruplex DNA. ► 11d could significantly inhibit cellular biological activity.
Co-reporter:Yuqin Qiao, Jinbo Gao, Yongge Qiu, Long Wu, Fei Guo, Kenneth Kam-Wing Lo, Ding Li
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 6) pp:2264-2273
Publication Date(Web):June 2011
DOI:10.1016/j.ejmech.2011.03.007
Farnesyltransferase (FTase) and geranylgeranyltransferase type-I (GGTase-I) both catalyze the prenylation of protein substrate containing a typical –CAAX motif at the carboxyl terminus. The inhibitors for these two enzymes have been widely studied as potential cancer chemotherapeutic agents. In the present study, various piperazinedione derivatives were designed and synthesized as a new type of peptide mimetic compounds, which were characterized and found to be dual protein inhibitors for both FTase and GGTase-I. These compounds have similar chemical and physical properties to –CAAX motif of the protein substrate, which may facilitate their transfer to appropriate drug target in vivo. The best inhibitor compound 26b was found to occupy both isoprenoid and peptide substrate binding sites through kinetics and computer molecular docking studies.Various piperazinedione derivatives were designed and synthesized as a new type of peptide mimetic compounds, which were characterized and found to be dual protein inhibitors for both Farnesyltransferase and Geranylgeranyltransferase-I.Highlights► Various piperazinedione derivatives were designed and synthesized as a new type of peptide mimetic compounds, which were characterized and found to be dual protein inhibitors for both FTase and GGTase-I. ► These compounds have similar chemical and physical properties to –CAAX motif of the protein substrate, which may facilitate their transfer to appropriate drug target in vivo. ► The best inhibitor compound 26b was found to occupy both isoprenoid and peptide substrate binding sites through kinetics and computer molecular docking studies.
Co-reporter:Long Wu, Yuqin Qiao, Jinbo Gao, Guisheng Deng, Wenhua Yu, Gong Chen, Ding Li
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 22) pp:6667-6673
Publication Date(Web):15 November 2011
DOI:10.1016/j.bmcl.2011.09.062
Glutaryl-CoA dehydrogenase catalyzes the oxidative decarboxylation of the γ-carboxylate of the substrate, glutaryl-CoA, to yield crotonyl-CoA and CO2. The enzyme is a member of the acyl-CoA dehydrogenase (ACD) family of flavoproteins. In the present study, the catalytic properties of this enzyme, including its substrate specificity, isomerase activity, and interactions with inhibitors, were systematically studied. Our results indicated that the enzyme has its catalytic properties very similar to those of short-chain and medium-chain acyl-CoA dehydrogenase except its additional decarboxylation reaction. Therefore, the inhibitors of fatty acid oxidation targeting straight chain acyl-CoA dehydrogenase could also function as inhibitors for amino acid metabolism of lysine, hydroxylysine, and tryptophan.The catalytic properties of glutaryl-CoA dehydrogenase, including its substrate specificity, isomerase activity, and interactions with inhibitors, were systematically studied.
Co-reporter:Jinbo Gao, Xiusheng Chu, Yongge Qiu, Long Wu, Yuqin Qiao, Jiasheng Wu and Ding Li
Chemical Communications 2010 vol. 46(Issue 29) pp:5340-5342
Publication Date(Web):14 Jun 2010
DOI:10.1039/C0CC00992J
The mevalonate pathway is an important drug target for the treatment of cancer and cardiovascular disease. We synthesized and studied a new type of nitrogen-containing bisphosphonate analogs and developed a sensitive end point assay method for enzyme FPPS, which was used for inhibitor screening. One potent FPPS inhibitor was discovered, and the structure–activity relationship of bisphosphonates for the enzyme inactivation was studied.
Co-reporter:Hu Ge, Jinggong Liu, Wenxia Zhao, Yu Wang, Qingqing He, Ruibo Wu, Ding Li and Jun Xu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 27) pp:NaN4951-4951
Publication Date(Web):2014/04/17
DOI:10.1039/C4OB00589A
In the present study, we found that three enzymes, MVK, MDD and FPPS, in the mevalonate pathway (MVP) of cholesterol biosynthesis, can be simultaneously inhibited by two green tea polyphenols ((−)-epicatechin-3-gallate, ECG; (−)-epigallocatechin-3-gallate, EGCG). Molecular dynamics simulations and pharmacophore studies were carried out to elucidate the tri-targeted inhibition mechanisms. Our results indicate that similar triangular binding pockets exist in all three enzymes, which is essential for their binding with polyphenols. Two distinct binding poses for ECG and EGCG were observed in our MD simulations. These results shed light on the potential for further selective and multi-targeted inhibitor design for the treatment of hyperlipidemia.
Co-reporter:Shang-Shi Zhang, Chun-Yong Jiang, Jia-Qiang Wu, Xu-Ge Liu, Qingjiang Li, Zhi-Shu Huang, Ding Li and Honggen Wang
Chemical Communications 2015 - vol. 51(Issue 50) pp:NaN10243-10243
Publication Date(Web):2015/05/14
DOI:10.1039/C5CC03187G
Cp*Rh(III)- and Cp*Ir(III)-catalysed direct C–H arylation with quinone diazides as efficient coupling partners is disclosed. This redox-neutral protocol offers a facile, operationally simple and environmentally benign access to arylated phenols. The reaction represents the first example of Cp*Ir(III)-catalysed C–H direct arylation reaction.
Co-reporter:Jinbo Gao, Xiusheng Chu, Yongge Qiu, Long Wu, Yuqin Qiao, Jiasheng Wu and Ding Li
Chemical Communications 2010 - vol. 46(Issue 29) pp:NaN5342-5342
Publication Date(Web):2010/06/14
DOI:10.1039/C0CC00992J
The mevalonate pathway is an important drug target for the treatment of cancer and cardiovascular disease. We synthesized and studied a new type of nitrogen-containing bisphosphonate analogs and developed a sensitive end point assay method for enzyme FPPS, which was used for inhibitor screening. One potent FPPS inhibitor was discovered, and the structure–activity relationship of bisphosphonates for the enzyme inactivation was studied.



![11H-PYRIDO[2,1-B]QUINAZOLIN-11-ONE, 2,3-DIFLUORO-6,7,8,9-TETRAHYDRO-](http://img.cochemist.com/ccimg/642500/642492-07-7.png)
![11H-PYRIDO[2,1-B]QUINAZOLIN-11-ONE, 2,3-DIFLUORO-6,7,8,9-TETRAHYDRO-](http://img.cochemist.com/ccimg/642500/642492-07-7_b.png)













































![Pyrrolo[2,1-b]quinazolin-9(1H)-one, 6,7-difluoro-2,3-dihydro-](/data/chemimg/2702800/642491-85-8.png)
![Pyrrolo[2,1-b]quinazolin-9(1H)-one, 6,7-difluoro-2,3-dihydro-](/data/chemimg/2702800/642491-85-8_b.png)


![1,2-Benzenediamine, N-[2-(4-methyl-1-piperazinyl)ethyl]-](http://img.cochemist.com/ccimg/600800/600725-24-4.png)
![1,2-Benzenediamine, N-[2-(4-methyl-1-piperazinyl)ethyl]-](http://img.cochemist.com/ccimg/600800/600725-24-4_b.png)
![1,2-BENZENEDIAMINE, N-[4-(DIMETHYLAMINO)BUTYL]-](http://img.cochemist.com/ccimg/600800/600725-11-9.png)
![1,2-BENZENEDIAMINE, N-[4-(DIMETHYLAMINO)BUTYL]-](http://img.cochemist.com/ccimg/600800/600725-11-9_b.png)
![3-[4-(2,5-Dimethoxy-phenyl)-5-mercapto-4H-[1,2,4]triazol-3-yl]-phenol](http://img.cochemist.com/ccimg/488100/488088-81-9.png)
![3-[4-(2,5-Dimethoxy-phenyl)-5-mercapto-4H-[1,2,4]triazol-3-yl]-phenol](http://img.cochemist.com/ccimg/488100/488088-81-9_b.png)






![(3-fluorophenyl)[4-(furan-2-ylmethyl)piperazin-1-yl]methanone](http://img.cochemist.com/ccimg/355900/355813-18-2.png)
![(3-fluorophenyl)[4-(furan-2-ylmethyl)piperazin-1-yl]methanone](http://img.cochemist.com/ccimg/355900/355813-18-2_b.png)


![SODIUM (2S)-5-AMINO-5-OXO-2-[(PHENYLACETYL)AMINO]PENTANOATE PHENY<WBR />LACETATE (2:1:1)](http://img.cochemist.com/ccimg/303300/303224-88-6.png)
![SODIUM (2S)-5-AMINO-5-OXO-2-[(PHENYLACETYL)AMINO]PENTANOATE PHENY<WBR />LACETATE (2:1:1)](http://img.cochemist.com/ccimg/303300/303224-88-6_b.png)





![Quinoline, 2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](/data/chemimg/3761900/190437-90-2.png)
![Quinoline, 2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](/data/chemimg/3761900/190437-90-2_b.png)
![Ethanone, 1-[4-(2-pyridinyl)phenyl]-](http://img.cochemist.com/ccimg/173700/173681-56-6.png)
![Ethanone, 1-[4-(2-pyridinyl)phenyl]-](http://img.cochemist.com/ccimg/173700/173681-56-6_b.png)









![1,2-Benzenediamine, N-[3-(diethylamino)propyl]-](http://img.cochemist.com/ccimg/92000/91971-96-9.png)
![1,2-Benzenediamine, N-[3-(diethylamino)propyl]-](http://img.cochemist.com/ccimg/92000/91971-96-9_b.png)











![Quinoline, 2-[(1E)-2-(2-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/77700/77669-18-2.png)
![Quinoline, 2-[(1E)-2-(2-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/77700/77669-18-2_b.png)







![Benzofuro[3,2-b]quinoline, 11-chloro-](/data/chemimg/2543200/69764-19-8.png)
![Benzofuro[3,2-b]quinoline, 11-chloro-](/data/chemimg/2543200/69764-19-8_b.png)

![Benzamide, N-[2-(aminocarbonyl)phenyl]-2-nitro-](/data/chemimg/1917700/67718-36-9.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-2-nitro-](/data/chemimg/1917700/67718-36-9_b.png)


![1,2-Benzenediamine, N-[3-(4-morpholinyl)propyl]-](http://img.cochemist.com/ccimg/62600/62553-49-5.png)
![1,2-Benzenediamine, N-[3-(4-morpholinyl)propyl]-](http://img.cochemist.com/ccimg/62600/62553-49-5_b.png)
![Benzo[h]quinoline, 4-chloro-2-methyl-](http://img.cochemist.com/ccimg/61800/61773-04-4.png)
![Benzo[h]quinoline, 4-chloro-2-methyl-](http://img.cochemist.com/ccimg/61800/61773-04-4_b.png)






![1,2-BENZENEDIAMINE, 4-CHLORO-N1-[3-(DIMETHYLAMINO)PROPYL]-](http://img.cochemist.com/ccimg/56800/56756-22-0.png)
![1,2-BENZENEDIAMINE, 4-CHLORO-N1-[3-(DIMETHYLAMINO)PROPYL]-](http://img.cochemist.com/ccimg/56800/56756-22-0_b.png)











![BENZENAMINE, N,N-DIMETHYL-4-[(1E)-2-(2-QUINOLINYL)ETHENYL]-](http://img.cochemist.com/ccimg/48200/48189-64-6.png)
![BENZENAMINE, N,N-DIMETHYL-4-[(1E)-2-(2-QUINOLINYL)ETHENYL]-](http://img.cochemist.com/ccimg/48200/48189-64-6_b.png)




![Quinoline, 2-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1815300/38101-93-8.png)
![Quinoline, 2-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1815300/38101-93-8_b.png)











![2-N-[3-(DIMETHYLAMINO)PROPYL]BENZENE-1,2-DIAMINE](http://img.cochemist.com/ccimg/21700/21627-59-8.png)
![2-N-[3-(DIMETHYLAMINO)PROPYL]BENZENE-1,2-DIAMINE](http://img.cochemist.com/ccimg/21700/21627-59-8_b.png)

![2-(Benzo[d]oxazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/15400/15344-56-6.png)
![2-(Benzo[d]oxazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/15400/15344-56-6_b.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)





















![2-(1-Methyl-1H-benzo[d]imidazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/2800/2735-62-8.png)
![2-(1-Methyl-1H-benzo[d]imidazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/2800/2735-62-8_b.png)