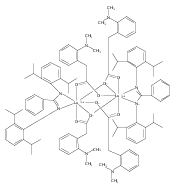
Co-reporter:Fangjun Zhang, Jie ZhangXigeng Zhou
Inorganic Chemistry 2017 Volume 56(Issue 4) pp:
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.inorgchem.6b02747
Treatment of the yttrium dialkyl complex TpMe2Y(CH2Ph)2(THF) (TpMe2 = tri(3,5 dimethylpyrazolyl)borate, THF = tetrahydrofuran) with S8 in a 1:1 molar ratio in THF at room temperature afforded a yttrium pentasulfide TpMe2Y(κ4-S5) (THF) (1) in 93% yield. The yttrium monoalkyl complex TpMe2CpYCH2Ph(THF) reacted with S8 in a 1:0.5 molar ratio under the same conditions to give another yttrium pentasulfide [(TpMe2)2Y]+[Cp2Y(κ4-S5)]− (10) in low yield. Further investigations indicated that the S52– anion facilely turned into the corresponding thioethers or organic disulfides, and released the redundant S8, when it reacted with some electrophilic reagents. The mechanism for the formation of the S52– ligand has been investigated by the controlling of the reaction stoichiometric ratios and the stepwise reactions.
Co-reporter:Jianquan Hong, Zhenhua Li, Zhening Chen, Linhong Weng, Xigeng Zhou and Lixin Zhang
Dalton Transactions 2016 vol. 45(Issue 15) pp:6641-6649
Publication Date(Web):02 Mar 2016
DOI:10.1039/C6DT00314A
Diverse reactivity patterns of mixed tetramethyl/methylidene rare-earth complexes bearing bulky benzamidinate coligands L3Ln3(μ2-Me)3(μ3-Me)(μ3-CH2) [L = [PhC(NC6H3iPr2-2,6)2]−; Ln = Y(1a), Lu(1b)] with PhCN, alkynes, and CS2 have been established. Reaction of complexes 1 with PhCN gave the μ3-CH2 addition complexes (NCNdipp)3Lu3(μ2-Me)3(μ3-Me)[μ–η1:η1:η3-CH2C(Ph)N] [Ln = Y(2a), Lu(2b)]. Treatment of complexes 1 with phenylacetylene afforded unexpected alkenyl dianion complexes L3Ln3(μ2-Me)3(μ3-Me)(μ–η1:η3-PhCCMe) [Ln = Y(3a), Lu(3b)] through the insertion of rare earth methylidene into a C–H bond in a reductive fashion. However, reaction of complexes 1 and HCCSiMe3 gave μ3-Me protonolysis complexes L3Ln3(μ2-Me)3(μ3-CCSiMe3)(μ3-CH2) [Ln = Y (4a), Lu (4b)] in excellent yields. Treatment of complexes 1 with CS2 led to the formation of the methyl activation complexes L3Ln3(μ2-Me)2(μ3-CH2)(μ3–η1:η2:η2-S2CCH2) [Ln = Y(5a), Lu(5b)]. All the new complexes were fully characterized.
Co-reporter:Bin Liu, Chunlei Zhang, Xigeng Zhou
Tetrahedron 2016 Volume 72(Issue 50) pp:8282-8286
Publication Date(Web):15 December 2016
DOI:10.1016/j.tet.2016.10.065
The palladium-catalyzed three-component cycloaminocarbonylation of 2-formylaryl tosylates with hydrazines and carbon monoxide has been established, which provides an efficient method for synthesis of substituted phthalazinones. In addition, by applying this protocol as the key step, Hydralazine can easily be synthesized in 65% yield.
Co-reporter:Bin Liu, Yan Wang, Benren Liao, Chunlei Zhang, Xigeng Zhou
Tetrahedron Letters 2015 Volume 56(Issue 42) pp:5776-5780
Publication Date(Web):14 October 2015
DOI:10.1016/j.tetlet.2015.08.088
The palladium-catalyzed cycloaminocarbonylation of 2-(aminomethyl)aryl tosylates with CO has been established, by which a variety of salicylaldehyde derived 2-(aminomethyl)aryl tosylates may be cyclocarbonylated in the presence of CO, to afford the corresponding substituted isoindolinones in moderate to excellent yields. Furthermore, the method is also effective for the synthesis of isoindoline-1,3-diones and 2-alkyl-1H-benzo[e]isoindol-3(2H)-ones from 2-(N-alkylcarbamoyl)aryl tosylates and 1-(aminomethyl)naphthalene-2-yl tosylates, respectively.
Co-reporter:Shujian Huang, Yinlin Shao, Ruiting Liu, Xigeng Zhou
Tetrahedron 2015 Volume 71(Issue 24) pp:4219-4226
Publication Date(Web):17 June 2015
DOI:10.1016/j.tet.2015.04.080
A simple, cheap, efficient, and metal-free method for one-step synthesis of a library of 1,3-diheteroatom five-membered heterocycles with exocyclic CN and CC double bonds was presented. The convenient reactions proceed via the direct three-component halocyclization of propargylamines, heterocumulenes and I2 (NBS).
Co-reporter:Shujian Huang;Yinlin Shao;Dr. Lixin Zhang;Dr. Xigeng Zhou
Angewandte Chemie 2015 Volume 127( Issue 48) pp:14660-14664
Publication Date(Web):
DOI:10.1002/ange.201508442
Abstract
The first catalytic cycloamidination of aminoalkenes with nitriles has been achieved by using rare-earth complexes. This reaction is equivalent to the desired intramolecular hydroamination of alkenylamidines, and allows a new direct access to substituted 2-imidazolines and tetrahydropyrimidines in high yields under operationally simple reaction conditions. Moreover, the methodology is also efficient for synthesis of symmetric and unsymmetric bridged diimidazolines. Compared with the traditional stepwise-mediated synthetic approaches, the present method avoids the use of additives and harsh reaction conditions, and thus leads to a completely different product distribution. Mechanistic data suggest that the reaction involves the initial NH activation by lanthanide complex followed by nitrile insertion into a LnN bond to form an amidinate lanthanide intermediate which undergoes the cyclization.
Co-reporter:Shujian Huang;Yinlin Shao;Dr. Lixin Zhang;Dr. Xigeng Zhou
Angewandte Chemie International Edition 2015 Volume 54( Issue 48) pp:14452-14456
Publication Date(Web):
DOI:10.1002/anie.201508442
Abstract
The first catalytic cycloamidination of aminoalkenes with nitriles has been achieved by using rare-earth complexes. This reaction is equivalent to the desired intramolecular hydroamination of alkenylamidines, and allows a new direct access to substituted 2-imidazolines and tetrahydropyrimidines in high yields under operationally simple reaction conditions. Moreover, the methodology is also efficient for synthesis of symmetric and unsymmetric bridged diimidazolines. Compared with the traditional stepwise-mediated synthetic approaches, the present method avoids the use of additives and harsh reaction conditions, and thus leads to a completely different product distribution. Mechanistic data suggest that the reaction involves the initial NH activation by lanthanide complex followed by nitrile insertion into a LnN bond to form an amidinate lanthanide intermediate which undergoes the cyclization.
Co-reporter:Yin Zhang, Jie Zhang, Jianquan Hong, Fangjun Zhang, Linhong Weng, and Xigeng Zhou
Organometallics 2014 Volume 33(Issue 24) pp:7052-7058
Publication Date(Web):December 2, 2014
DOI:10.1021/om500500b
The reactions of β-diketiminatoyttrium dialkyl complex LY(CH2Ph)2(THF) (1, L = [{N(2,6-iPr2C6H3)C(Me)}2CH]−) with a series of aromatic N-heterocycles such as 2-phenylpyridine, benzothiazole, and benzoxazole were studied and displayed discrete reactivity including C–H activation, C–C coupling, ring-opening/insertion, and dearomatization. The reaction of 1 with 2-phenylpyridine in 1:2 molar ratio in THF at 30 °C for 14 days afforded a structurally characterized metal complex, LY(η2-N,C-C5H4NC6H4-2)[C5H4N(CH2Ph-4)Ph] (2), in 73% isolated yield, indicating the occurrence of phenyl ring C(sp2)–H activation and pyridine ring 1,4-addition/dearomatization. However, when this reaction was done at 5 °C for 7 days, it gave the pyridine ring 1,2-addition product LY(η2-N,C-C5H4NC6H4-2)[C5H4N(CH2Ph-2)Ph] (3) in 54% isolated yield. Further investigations revealed that complex 2 is the thermodynamic controlled product and complex 3 is the kinetically controlled product; 3 converted slowly into 2, as confirmed by 1H NMR spectroscopy. The equimolar reaction of 1 with benzothiazole or benzoxazole produced two C–C coupling/ring-opening/insertion products, LY[η2-S,N-SC6H4NCH(CH2Ph)2](THF) (4) and {LY[μ-η2:η1-O,N-OC6H4NCH(CH2Ph)2]}2 (5), in 84% and 78% isolated yields, respectively.
Co-reporter:Fangjun Zhang, Jie Zhang, Yin Zhang, Jianquan Hong, and Xigeng Zhou
Organometallics 2014 Volume 33(Issue 21) pp:6186-6192
Publication Date(Web):October 13, 2014
DOI:10.1021/om5008665
A high-efficiency and atom-economic route for the synthesis of N-aryl-substituted propiolamidines was established through the addition of terminal alkynes with aryl-substituted symmetrical or unsymmetrical carbodiimides catalyzed by mixed TpMe2/Cp rare-earth-metal alkyl complexes (TpMe2)CpLnCH2Ph(THF) (1Ln). Moreover, the gadolinium alkyl complex 1Gd can also serve as a catalyst for the double addition of dialkynes with carbodiimides. Mechanism studies indicated that the variable coordination modes (κ3 or κ2) of the TpMe2 ligand on the rare-earth-metal species may play an important role in the catalytic cycles.
Co-reporter:Dr. Weiyin Yi;Dr. Jie Zhang;Shujian Huang;Dr. Linhong Weng ;Dr. Xigeng Zhou
Chemistry - A European Journal 2014 Volume 20( Issue 3) pp:867-876
Publication Date(Web):
DOI:10.1002/chem.201303608
Abstract
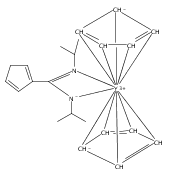
Unusual chemical transformations such as three-component combination and ring-opening of N-heterocycles or formation of a carbon–carbon double bond through multiple C–H activation were observed in the reactions of TpMe2-supported yttrium alkyl complexes with aromatic N-heterocycles. The scorpionate-anchored yttrium dialkyl complex [TpMe2Y(CH2Ph)2(THF)] reacted with 1-methylimidazole in 1:2 molar ratio to give a rare hexanuclear 24-membered rare-earth metallomacrocyclic compound [TpMe2Y(μ-N,C-Im)(η2-N,C-Im)]6 (1; Im=1-methylimidazolyl) through two kinds of C–H activations at the C2- and C5-positions of the imidazole ring. However, [TpMe2Y(CH2Ph)2(THF)] reacted with two equivalents of 1-methylbenzimidazole to afford a C–C coupling/ring-opening/C–C coupling product [TpMe2Y{η3-(N,N,N)-N(CH3)C6H4NHCHC(Ph)CN(CH3)C6H4NH}] (2). Further investigations indicated that [TpMe2Y(CH2Ph)2(THF)] reacted with benzothiazole in 1:1 or 1:2 molar ratio to produce a C–C coupling/ring-opening product {(TpMe2)Y[μ-η2:η1-SC6H4N(CHCHPh)](THF)}2 (3). Moreover, the mixed TpMe2/Cp yttrium monoalkyl complex [(TpMe2)CpYCH2Ph(THF)] reacted with two equivalents of 1-methylimidazole in THF at room temperature to afford a trinuclear yttrium complex [TpMe2CpY(μ-N,C-Im)]3 (5), whereas when the above reaction was carried out at 55 °C for two days, two structurally characterized metal complexes [TpMe2Y(Im-TpMe2)] (7; Im-TpMe2=1-methyl-imidazolyl-TpMe2) and [Cp3Y(HIm)] (8; HIm=1-methylimidazole) were obtained in 26 and 17 % isolated yields, respectively, accompanied by some unidentified materials. The formation of 7 reveals an uncommon example of construction of a CC bond through multiple C–H activations.
Co-reporter:Longcheng Hong;Yinlin Shao;Lixin Zhang;Dr. Xigeng Zhou
Chemistry - A European Journal 2014 Volume 20( Issue 28) pp:8551-8555
Publication Date(Web):
DOI:10.1002/chem.201402701
Abstract
A lanthanide-catalyzed sequential insertion of CN and CC into an NH bond is presented. The convenient reaction, which proceeds under mild conditions, is an efficient method for preparing 1,2,4-trisubstituted imidazoles directly from readily available propargylamines and nitriles.
Co-reporter:HouCai Zhang;RuiTing Liu
Science China Chemistry 2014 Volume 57( Issue 2) pp:282-288
Publication Date(Web):2014 February
DOI:10.1007/s11426-013-5042-2
Iron-catalyzed direct reduction of allylic halides with benzylic alcohol was achieved, providing a new, simple, and efficient method for conducting highly regioselective hydrodehalogenation. This method not only features a readily available reductant, an inexpensive catalyst, simple manipulation, and good tolerance of functional groups including nitriles, nitro, esters, and methoxyl groups, it also has mild reaction conditions and shows complete regioselectivity in that only halides sited at the allylic position are reduced. Alternatively, this method can be applied in the selective transformation of benzylic alcohols to aromatic aldehydes without overoxidation to carboxylic acids.
Co-reporter:YuFeng Jing;RuiTing Liu;YangHui Lin
Science China Chemistry 2014 Volume 57( Issue 8) pp:1117-1125
Publication Date(Web):2014 August
DOI:10.1007/s11426-014-5149-0
La[N(SiMe3)2]3 proves to be an efficient catalyst system for the cyclocarbonylation of 1,2-disubstituted benzenes with isocyanates. In this approach, aryl/alkyl isocyanates react with o-phenylenediamine, o-aminophenol, o-aminothiophenol, catechols and anilines ortho-substituted by CH2NH2 and CONH2 to form, respectively, the corresponding benzimidazolones, benzoxazolones, benzothiazolones, benzodioxolones, 3,4-dihydroquinazolin-2(1H)-one, and quinazolinediones. These results represent the first example of lanthanide-catalyzed carbonylation. This methodology is also applicable for the preparation of various benzannulated 1,3-diheteroatom cyclic thioketones starting from aryl/alkyl isothiocyanates or CS2 in good to excellent yields. Based on the results of experiments performed using an o-aminobenzamido dianion lanthanide complex, a general mechanism, involving the tandem reaction of two lanthanide-ligand bonds with one heterocumulene molecule, is proposed as well.
Co-reporter:Ruiting Liu and Xigeng Zhou
Chemical Communications 2013 vol. 49(Issue 31) pp:3171-3187
Publication Date(Web):14 Dec 2012
DOI:10.1039/C2CC35637F
Cyclopentadienyl and substituted cyclopentadienyl ligands are observed in a wide range of organometallic complexes. In addition to serving as ancillary ligands, these ligands have come into their own as intermediates in organometallic reactions, and shown many unique reaction modes involving ring C–H, C–C and CC bond cleavages. This feature article summarizes the progressive development of cyclopentadienyl-based reactions of metallocene complexes of transition metals and rare-earth metals, with the aim of further developing the fundamental modes of reactivity of such systems together with their synthetic applications.
Co-reporter:Longcheng Hong, Weijia Lin, Fangjun Zhang, Ruiting Liu and Xigeng Zhou
Chemical Communications 2013 vol. 49(Issue 49) pp:5589-5591
Publication Date(Web):30 Apr 2013
DOI:10.1039/C3CC42534G
The first example of rare earth metal-catalyzed cycloaddition of terminal alkynes to azides resulting in the formation of 1,5-disubstituted 1,2,3-triazoles is described. Preliminary studies revealed that the present cycloaddition shows unprecedented mechanistic features involving a tandem anionic cascade cyclization and anti-addition across the CC triple bond.
Co-reporter:Dr. Weiyin Yi;Jie Zhang;Dr. Fangjun Zhang;Yin Zhang;Dr. Zhenxia Chen;Dr. Xigeng Zhou
Chemistry - A European Journal 2013 Volume 19( Issue 36) pp:11975-11983
Publication Date(Web):
DOI:10.1002/chem.201300610
Abstract
A series of unusual chemical-bond transformations were observed in the reactions of high active yttriumdialkyl complexes with unsaturated small molecules. The reaction of scorpionate-anchored yttriumdibenzyl complex [TpMe2Y(CH2Ph)2(thf)] (1, TpMe2=tri(3,5-dimethylpyrazolyl)borate) with phenyl isothiocyanate led to CS bond cleavage to give a cubane-type yttrium–sulfur cluster, {TpMe2Y(μ3-S)}4 (2), accompanied by the elimination of PhNC(CH2Ph)2. However, compound 1 reacted with phenyl isocyanate to afford a C(sp3)H activation product, [TpMe2Y(thf){μ-η1:η3-OC(CHPh)NPh}{μ-η3:η2-OC(CHPh)NPh}YTpMe2] (3). Moreover, compound 1 reacted with phenylacetonitrile at room temperature to produce γ-deprotonation product [(TpMe2)2Y]+[TpMe2Y(N=CCHPh)3]− (6), in which the newly formed NCCHPh ligands bound to the metal through the terminal nitrogen atoms. When this reaction was carried out in toluene at 120 °C, it gave a tandem γ-deprotonation/insertion/partial-TpMe2-degradation product, [(TpMe2Y)2(μ-Pz)2{μ-η1:η3-NC(CH2Ph)CHPh}] (7, Pz=3,5-dimethylpyrazolyl).
Co-reporter:Dr. Jianquan Hong;Dr. Lixin Zhang;Kai Wang;Yin Zhang; Linhong Weng ; Xigeng Zhou
Chemistry - A European Journal 2013 Volume 19( Issue 24) pp:
Publication Date(Web):
DOI:10.1002/chem.201390085
Co-reporter:Jianquan Hong, Lixin Zhang, Kai Wang, Zhenxia Chen, Limin Wu, and Xigeng Zhou
Organometallics 2013 Volume 32(Issue 24) pp:7312-7322
Publication Date(Web):December 2, 2013
DOI:10.1021/om400787j
Three kinds of solvated lithium amidinates with different coordination models were obtained via recrystallization of [PhC(NC6H4iPr2-2,6)2]Li(THF) (1a) in different solvents. Treatment of o-Me2NC6H4CH2Li with LLnCl2(THF)n (2; L = [PhC(NC6H4iPr2-2,6)2]− (NCNdipp), [o-Me2NC6H4CH2C(NC6H4iPr2-2,6)2]− (NCNdipp′)) formed in situ from reaction of LnCl3(THF)x with LLi(THF) gave the rare-earth-metal bis(aminobenzyl) complexes LLn(CH2C6H4NMe2-o)2 (L = NCNdipp, Ln = Sc (3a), Y (3b), Lu (3c); L = NCNdipp′, Ln = Sc (3d), Lu (3e)) in high yields. Reactions of complexes 3 with CO2, PhNCO, 2,6-diisopropylaniline, and S have been explored. CO2 inserted into each Ln–C bond of complexes 3a–c to form the dual-core complexes [(NCNdipp)Sc(μ-η1:η1-O2CCH2C6H4NMe2-o)2]2 (4a) and [(NCNdipp)Ln(μ-η1:η2-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)]2 (Ln = Y (4b), Lu (4c)). The reaction of 3b,c,e with PhNCO produced LLu[OC(CH2C6H4NMe2-o)NPh]2(thf) (L = NCNdipp, Ln = Y (5b), Lu (5c); L = NCNdipp′, Ln = Lu (5e)). Protonolysis of 3a,b by 2,6-diisopropylaniline formed straightforwardly the μ2-imido complexes [(NCNdipp)Ln(μ-NC6H4iPr2-2,6)]2 (Ln = Sc (6a), Lu (6c)). Reaction of 3e with S8 afforded the sulfur insertion products (NCNdipp′)Lu(CH2C6H4NMe2-o)(SCH2C6H4NMe2-o)(thf) (7e) and (NCNdipp′)Lu(SCH2C6H4NMe2-o)2(thf)2 (7f) in high yields, respectively, depending on the stoichiometric ratio. All of these complexes were fully characterized by elemental analysis, NMR spectroscopy, and X-ray structural determinations.
Co-reporter:Weiyin Yi ; Shujian Huang ; Jie Zhang ; Zhenxia Chen
Organometallics 2013 Volume 32(Issue 19) pp:5409-5415
Publication Date(Web):September 12, 2013
DOI:10.1021/om400698j
The TpMe2-supported yttrium dialkyl TpMe2Y(CH2Ph)2(THF) (TpMe2 = tri(3,5-dimethylpyrazolyl)borate) reacted with 1 equiv of ArNH2 in THF at room temperature to afford the yttrium alkyl primary amido complexes TpMe2YNHAr(CH2Ph)(THF) (Ar = Ph (1), C6H3-iPr2-2,6 (2)) in 84% and 88% isolated yields, respectively. Complex 1 reacted with iPrN═C═NiPr in THF at room temperature to give a yttrium dianionic guanidinate complex, TpMe2Y[(iPrN)2C═NPh](THF)2 (3, 74%). However, the reaction of 1 with ArN═C═NAr (Ar = C6H3-iPr2-2,6) in the same conditions produced a Y–C bond insertion product, TpMe2Y[(ArN)2CCH2Ph](NHPh) (4, 87%). Moreover, treatment of 2 with 1 equiv of iPrN═C═NiPr in THF at room temperature afforded two yttrium complex, TpMe2Y[(iPrN)C═NAr](THF) (5) and TpMe2Y[(iPrN)2CCH2Ph](NHAr) (6), in 58% and 19% isolated yields, respectively. These results indicated that carbodiimide can selectively insert into the Y–CH2Ph and Y–NHAr σ-bonds of TpMe2-supported yttrium alkyl primary amido complexes TpMe2YNHAr(CH2Ph)(THF), and this selectivity depends on the steric hindrance of the substituent groups R of cabodiimides and the primary amido ligands. All these new complexes were characterized by elemental analysis and spectroscopic methods, and their solid-state structures except 1 were also confirmed by single-crystal X-ray diffraction analysis.
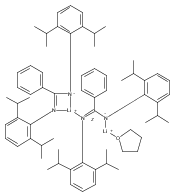
Co-reporter:Dr. Jianquan Hong;Dr. Lixin Zhang;Kai Wang;Yin Zhang; Linhong Weng ; Xigeng Zhou
Chemistry - A European Journal 2013 Volume 19( Issue 24) pp:7865-7873
Publication Date(Web):
DOI:10.1002/chem.201300440
Abstract
Three new patterns of reactivity of rare-earth metal methylidene complexes have been established and thus have resulted in access to a wide variety of imido rare-earth metal complexes [L3Ln3(μ2-Me)3(μ3-Me)(μ-NR)] (L=[PhC(NC6H3iPr2-2,6)2]−; R=Ph, Ln=Y (2 a), Lu (2 b); R=2,6-Me2C6H3, Ln=Y (3 a), Lu (3 b); R=p-ClC6H4, Ln=Y (4 a), Lu (4 b); R=p-MeOC6H4, Ln=Y (5 a), Lu (5 b); R=Me2CHCH2CH2, Ln=Y (6 a), Lu (6 b)) and [{L3Lu3(μ2-Me)3(μ3-Me)}2(μ-NR′N)] (R′=(CH2)6 (7 b), (C6H4)2 (8 b)). Complex 2 b was treated with an excess of CO2 to give the corresponding carboxylate complex [L3Lu3(μ-η1:η1-O2CCH3)3(μ-η1:η2-O2C-CH3)(μ-η1:η1:η2-O2CNPh)] (9 b) easily. Complex 2 a could undergo the selective μ3-Me abstraction reaction with phenyl acetylene to give the mixed imido/alkynide complex [L3Y3(μ2-Me)3(μ3-η1:η1:η3-NPh)(μ3-CCPh)] (10 a) in high yield. Treatment of 2 with one equivalent of thiophenol gave the selective μ3-methyl-abstracted products [L3Ln3(μ2-Me)3(μ3-η1:η1:η3-NPh)(μ3-SPh)] (Ln=Y (11 a); Lu (11 b). All new complexes have been characterized by elemental analysis, NMR spectroscopy, and most of the structures confirmed by X-ray diffraction.
Co-reporter:Weiyin Yi, Jie Zhang, Zhenxia Chen, and Xigeng Zhou
Inorganic Chemistry 2012 Volume 51(Issue 20) pp:10631-10638
Publication Date(Web):September 24, 2012
DOI:10.1021/ic3008552
A novel TpMe2-supported (TpMe2 = tri(3,5-dimethylpyrazolyl)borate) rare earth metal complex promoted Me–Si cleavage of the bis(trimethylsilyl) amide ligand ([(Me3Si)2N]−) was observed. Reaction of TpMe2LnCl2 with 2 equiv of K[(RN)2CN(SiMe3)2] (KGua) gave the methylamidinate complexes TpMe2Ln[(RN)2CMe][N(SiMe3)2] (R = isopropyl, Ln = Y (1Y), Er (1Er); R = cyclohexyl, Ln = Y (2Y)) in moderate yields. In contrast, TpMe2YCl2(THF) reacted with 1 equiv of KGua to afford a C–N cleavage product TpMe2Y(Cl)N(SiMe3)2(THF) (4), indicating that this guanidinate ligand is not stable in the yttrium complex with the TpMe2 ligand, and a carbodiimide deinsertion takes place easily. The mechanism for the formation of complexes 1 and 2 was also studied by controlling the substrate stoichiometry and the reaction sequence and revealed that the bis(trimethylsilyl)amine anion N(SiMe3)2– can undergo two routes of γ-methyl deprotonation and Si–Me cleavage for its functionalizations. All these new complexes were characterized by elemental analysis and spectroscopic methods, and their solid-state structures were also confirmed by single-crystal X-ray diffraction.
Co-reporter:Weiyin Yi, Jie Zhang, Zhenxia Chen, and Xigeng Zhou
Organometallics 2012 Volume 31(Issue 20) pp:7213-7221
Publication Date(Web):October 2, 2012
DOI:10.1021/om3007773
The mixed TpMe2/Cp-supported yttrium monoalkyl (TpMe2)CpYCH2Ph(THF) (1) reacted with 1 equiv of PhCN in THF at room temperature to afford the imine–enamine tautomer (TpMe2)CpY(N(H)C(Ph)═CHPh)(THF) (2) and the insertion product (TpMe2)CpY(N═C(CH2Ph)Ph)(THF) (3), in 61% and 12% isolated yields, respectively. 2 further reacted with PhCN in toluene at 120 °C to give the N–H bond addition product (TpMe2)CpY(N(H)C(Ph)NC(Ph)═CHPh) (4). Treatment of 1 with 1 equiv of anthranilonitrile produced the dimer [(TpMe2)CpY(μ-NHC6H4CN)]2 (5). The monomer product (TpMe2)CpY(NHC6H4CN)(HMPA) (6) can be obtained through the coordination of HMPA (hexamethylphosphoric triamide). The reaction of 5 with 1 in THF at room temperature gave the cyano group insertion product [(TpMe2)CpY(THF)]2(μ-NHC6H4C(CH2Ph)═N) (7). However, this reaction under the heating conditions gave an unexpected rearrangement product, (TpMe2)CpY(THF)(η2-NHC6H4C(CH2Ph)═NH) (8). 5 further reacted with o-aminobenzonitrile at 120 °C to afford the nucleophilic addition/cyclization product TpMe2Y[κ3-(4-NH═(C8N2H4)(2-NHC6H4)](HMPA) (9), accompanied with the elimination of the Cp ring. These results indicated that the yttrium alkyl complex exhibits high activity toward organic nitriles and reveals some unusual transformations during the insertion process. All these new complexes were characterized by elemental analysis and spectroscopic methods, and their solid-state structures were also confirmed by single-crystal X-ray diffraction analysis.
Co-reporter:Xiuli Bu, Longcheng Hong, Ruiting Liu, Jianquan Hong, Zhengxing Zhang, Xigeng Zhou
Tetrahedron 2012 68(38) pp: 7960-7965
Publication Date(Web):
DOI:10.1016/j.tet.2012.07.007
Co-reporter:Zhen Li, Jianquan Hong, Linhong Weng, Xigeng Zhou
Tetrahedron 2012 68(5) pp: 1552-1559
Publication Date(Web):
DOI:10.1016/j.tet.2011.12.003
Co-reporter:Xiuli Bu;Jianquan Hong
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 11-12) pp:2111-2118
Publication Date(Web):
DOI:10.1002/adsc.201100065
Abstract
A convenient and efficient method for the synthesis of polysubstituted indenes has been developed by the iron(III) trichloride-catalyzed tandem mono- and dibenzylation/cyclization reactions of benzylic alcohols with alkynes. This method is featured with the easily available starting materials, cheap catalyst, simple manipulation and mild conditions.
Co-reporter:Weiyin Yi ; Jie Zhang ; Meng Li ; Zhenxia Chen
Inorganic Chemistry 2011 Volume 50(Issue 22) pp:11813-11824
Publication Date(Web):October 27, 2011
DOI:10.1021/ic2019499
The structurally characterized TpMe2-supported rare earth metal monoalkyl complex (TpMe2)CpYCH2Ph(THF) (1) was synthesized via the salt-metathesis reaction of (TpMe2)CpYCl(THF) with KCH2Ph in THF at room temperature. Treatment of 1 with 1 equiv of PhC≡CH under the same conditions afforded the corresponding alkynyl complex (TpMe2)CpYC≡CPh(THF) (2). Complex 1 exhibits high activity toward carbodiimides, isocyanate, isothiocyanate, and CS2; treatment of 1 with such substrates led to the formation of a series of the corresponding Y–C(benzyl) σ-bond insertion products (TpMe2)CpY[(RN)2CCH2Ph] (R = iPr(3a), Cy(3b), 2,6-iPr–C6H3(3c)), (TpMe2)CpY[SC(CH2Ph)NPh] (4), (TpMe2)CpY[OC(CH2Ph)NPh] (5), and (TpMe2)CpY(S2CCH2Ph) (6) in 40–70% isolated yields. Carbodiimides and isothiocyanate can also insert into the Y–C(alkynyl) σ bond of 2 to yield complexes (TpMe2)CpY[(RN)2CC≡CPh] (R = iPr(7a), Cy(7b)) and (TpMe2)CpY[SC(C≡CPh)NPh] (9). Further investigation results indicated that 1 can effectively catalyze the cross-coupling reactions of phenylacetylene with carbodiimides. However, treatment of o-allylaniline with a catalytic amount of 1 gave only the benzyl abstraction product (TpMe2)CpY(NHC6H4CH2CH═CH2-o)(THF) (10), without observation of the expected organic hydroamination/cyclization product. All of these new complexes were characterized by elemental analysis and spectroscopic properties, and their solid-state structures were also confirmed by single-crystal X-ray diffraction analysis.
Co-reporter:Zhen Li, Longcheng Hong, Ruiting Liu, Jianzhong Shen, Xigeng Zhou
Tetrahedron Letters 2011 Volume 52(Issue 12) pp:1343-1347
Publication Date(Web):23 March 2011
DOI:10.1016/j.tetlet.2011.01.052
The copper-catalyzed chalcogenoamination of 2-alkynylanilines with dichalcogenides has been achieved under mild reaction conditions using the weak base Cs2CO3 in combination with air oxidant, providing a convenient and efficient method for synthesis of 3-sulfenylindoles and 3-selenylindoles, including complex products bearing two attached 3-sulfenylindole rings.The copper-catalyzed chalcogenoamination of 2-alkynylanilines with dichalcogenides has been established, providing a convenient and efficient method for synthesis of 3-sulfenylindoles and 3-selenylindoles, including complex products bearing two attached 3-sulfenylindole rings.
Co-reporter:Ruiting Liu, Zhen Li, Weiyin Yi, Zhenxia Chen, Xigeng Zhou
Journal of Organometallic Chemistry 2011 696(13) pp: 2648-2653
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.04.016
Co-reporter:Jianquan Hong;Dr. Lixin Zhang;Xiaying Yu;Meng Li;Dr. Zhengxing Zhang;Dr. Pengzhi Zheng;Dr. Masayoshi Nishiura;Dr. Zhaomin Hou;Dr. Xigeng Zhou
Chemistry - A European Journal 2011 Volume 17( Issue 7) pp:2130-2137
Publication Date(Web):
DOI:10.1002/chem.201002670
Abstract
Unsolvated, trinuclear, homometallic, rare-earth-metal multimethyl methylidene complexes [{(NCN)Ln(μ2-CH3)}3(μ3-CH3)(μ3-CH2)] (NCN=L=[PhC{NC6H4(iPr-2,6)2}2]−; Ln=Sc (2 a), Lu (2 b)) have been synthesized by treatment of [(L)Ln{CH2C6H4N(CH3)2-o}2] (Ln=Sc (1 a), Lu (1 b)) with two equivalents of AlMe3 in toluene at ambient temperature in good yields. Treatment of 1 with three equivalents of AlMe3 gives the heterometallic trinuclear complexes [(L)Ln(AlMe4)2] (Ln=Sc (3 a), Lu (3 b)) in good yields. Interestingly, 2 can also be generated by recrystallization of 3 in THF/toluene, thereby indicating that the THF molecule can also induce CH bond activation of 2. Reaction of 2 with one equivalent of ketones affords the trinuclear homometallic oxo–trimethyl complexes [{(L)Ln(μ2-CH3)}3(μ3-CH3)(μ3-O)] (Ln=Sc(4 a), Lu(4 b)) in high yields. Complex 4 b reacts with one equivalent of cyclohexanone to give the methyl abstraction product [{(L)Lu(μ2-CH3)}3(μ3-OC6H9)(μ3-O)] (5 b), whereas reaction of 4 b with acetophenone forms the insertion product [{(L)Lu(μ2-CH3)}3{μ3-OCPh(CH3)2}(μ3-O)] (6 b). Complex 4 a is inert to ketone under the same conditions. All these new complexes have been characterized by elemental analysis, NMR spectroscopy, and confirmed by X-ray diffraction determination.
Co-reporter:Zhen Li, Jianquan Hong, Xigeng Zhou
Tetrahedron 2011 67(20) pp: 3690-3697
Publication Date(Web):
DOI:10.1016/j.tet.2011.03.067
Co-reporter:Weiyin Yi, Jie Zhang, Longcheng Hong, Zhenxia Chen, and Xigeng Zhou
Organometallics 2011 Volume 30(Issue 21) pp:5809-5814
Publication Date(Web):October 10, 2011
DOI:10.1021/om2006722
The synthesis, structure, and reactivity of organoyttrium phosphides toward phenyl isocyanate (PhNCO) and phenyl isothiocyanate (PhNCS) are described. Reaction of (TpMe2)CpYCH2Ph(THF) (TpMe2 = tris(3,5-dimethylpyrazolyl)borate; Cp = C5H5) with 1 equiv of HPPh2 in THF at ambient temperature gives an organoyttrium phosphide (TpMe2)CpYPPh2(THF) (1). Treatment of 1 with 1 equiv of PhNCO in THF at ambient temperature results in monoinsertion of PhNCO into the Y–P σ-bond to yield complex (TpMe2)CpY[OC(PPh2)NPh](THF) (2), whereas reaction of 1 with 2 equiv of PhNCO affords the PhNCO diinsertion product (TpMe2)CpY[OC(PPh2)N(Ph)C(O)NPh] (4). However, reaction of 1 with PhNCS under the same conditions is independent of the stoichiometric ratio and gives only the monoinsertion product (TpMe2)CpY[SC(PPh2)NPh] (3). Moreover, 1 can effectively catalyze the cyclotrimerization of PhNCO under mild conditions, but does not catalyze the cyclotrimerization of PhNCS. In addition, the reaction of Cp2LnPPh2(THF) with PhNCS affords the insertion products Cp2Ln[SC(PPh2)NPh](THF) (Ln = Y (6), Er (7), Dy (8)). All new complexes were characterized by elemental analysis, IR, and/or 1H, 13C and 31P NMR, and their solid-state structures, except 4, were determined through single-crystal X-ray diffraction analysis. These reactions represent the first example of isocyanate and isothiocyanate insertions into the Ln–P σ-bond and provide an efficient method for the construction of phosphaureido, phosphadiureido, and phosphathioureido ligands.
Co-reporter:Chengfu Pi, Xiaoqing Li, Lili Zhang, Ruiting Liu, Linhong Weng and Xigeng Zhou
Inorganic Chemistry 2010 Volume 49(Issue 17) pp:7632-7634
Publication Date(Web):August 13, 2010
DOI:10.1021/ic101346b
Unusual insertion of carbodiimide into the Y−Cp (Cp = C5H5) bond and isomerization of the resulting cyclopentadienyl (Cp)-substituted amidinate complex to the amidino-substituted Cp complex have been established, representing an efficient and simple method for ring modification of sensitive metallocenes. All products, including the rare four-center interaction precursor of the insertion, have been characterized by X-ray structural analyses.
Co-reporter:Zhengxing Zhang ; Lixin Zhang ; Yanrong Li ; Longcheng Hong ; Zhenxia Chen
Inorganic Chemistry 2010 Volume 49(Issue 12) pp:5715-5722
Publication Date(Web):May 27, 2010
DOI:10.1021/ic100617n
The treatment of [(Me3Si)2NC(NCy)2]2Ln(μ-Cl)2Li(THF)2 with 1 equiv of BnK (Bn = benzyl) in toluene affords [(Me3Si)2NC(NCy)2]2LnBn [Ln = Er (1-Er), Y (1-Y)] in good yields. Similarly, [(Me3Si)2NC(NCy)2]2LntBu [Ln = Er (2-Er), Yb (2-Yb)] are obtained in satisfactory yields by the reaction of [(Me3Si)2NC(NCy)2]2Ln(μ-Cl)2Li(THF)2 with tBuLi in hexane. 1 reacts with 1/8 equiv of S8 in toluene to form the sulfur insertion products {[(Me3Si)2NC(NCy)2]2Ln(μ-SBn)}2 [Ln = Er (3-Er), Y (3-Y)], while the reaction of 2 with elemental sulfur under the same conditions affords the oxidation products {[(Me3Si)2NC(NCy)2]2Ln}2(μ-η2:η2-S2) [Ln = Er (4-Er), Yb (4-Yb)] regardless of the equivalency of S8 employed. Disulfide complexes 4 can also be obtained by the reaction of 3 with 1/4 equiv of S8. Furthermore, the treatment of [(Me3Si)2NC(NCy)2]2Ln(μ-Cl)2Li(THF)2 with 1 equiv of nBuLi in hexane, followed by reaction with 1/8 equiv of S8, affords the dinuclear thiolate complexes {[(Me3Si)2NC(NCy)2]2Ln(μ-SnBu)}2 [Ln = Y (5-Y), Er (5-Er)] in good yields. However, under the same conditions, [(Me3Si)2NC(NCy)2]2Yb(μ-Cl)2Li(THF)2 reacts with nBuLi and S8 to give {[(Me3Si)2NC(NCy)2]2Yb}2(μ-η2:η2-S2) (4-Yb) as the main metal-containing product. [(Me3Si)2NC(NCy)2]2LnPh (generated in situ from [(Me3Si)2NC(NCy)2]2Ln(μ-Cl)2Li(THF)2 and PhLi) also undergoes sulfur insertion, affording {[(Me3Si)2NC(NCy)2]2Ln(μ-SPh)}2 [Ln = Er (6-Er), Yb (6-Yb)] in good yields. All of the complexes were characterized by spectroscopic and elemental analyses. The structures of all of these compounds, except 3-Y, are also determined by single-crystal X-ray diffraction analysis. Surprisingly, 3, 5, and 6 bear the same space group and very similar cell parameters, despite the different thiolate ligands.
Co-reporter:Fuyan Han ; Jie Zhang ; Weiyin Yi ; Zhengxing Zhang ; Jingyi Yu ; Linhong Weng
Inorganic Chemistry 2010 Volume 49(Issue 6) pp:2793-2798
Publication Date(Web):February 9, 2010
DOI:10.1021/ic902296d
TpMe2LnCl2 (1) reacts with 2 equiv of KN(SiMe3)2 in tetrahydrofuran at room temperature to yield the ligand redistribution/γ-deprotonation products [(TpMe2)2Ln]+[((Me3Si)2N)2Ln(CH2)SiMe2N(SiMe3)]− [Ln = Er (2), Y (3)]. Complex 2 can also be obtained by reacting [(Me3Si)2N]2ErCl with KTpMe2. However, 1 reacts with 1.5 and 1 equiv of KN(SiMe3)2 to yield [(TpMe2)2Er]+[((Me3Si)2N)3ErCl]− (4) and [(TpMe2)2Er]+{[(Me3Si)2N)TpMe2ErCl]2(μ-Cl)2K}− (5), respectively. Furthermore, it is found that 2 reacts with 2 equiv of CyN═C═NCy (Cy = cyclohexyl) to give the tandem HN(SiMe3)2 elimination and Ln−C insertion product (TpMe2)Er[(CyN)2CCH2SiMe2N(SiMe3)] (6) in 71% isolated yield. The results reveal that the γ-deprotonation degree of advancement increases with an increase of the steric hindrance around the central metal ion. All new complexes have been characterized by elemental analysis and spectroscopic properties, and their solid-state structures have also been determined through single-crystal X-ray diffraction analysis.
Co-reporter:Yan Sun, Zhengxing Zhang, Xu Wang, Xiaoqing Li, Linhong Weng and Xigeng Zhou
Dalton Transactions 2010 vol. 39(Issue 1) pp:221-226
Publication Date(Web):03 Nov 2009
DOI:10.1039/B918181D
[Cp2LnNHPy]2 (Py = 2-pyridyl) (1a–e) react with phenyl isocyanate to form the N–H diinsertion products Cp2Ln[η2:η1-PyNCON(Ph)CONHPh](THF) (Ln = Yb (3a), Er (3b), Y (3c), Dy (3d), Gd (3e)). It has been proven that nPr2NH can abstract one PhNCO unit from 3c to form Cp2Y[η3-OC(NHPh)NPy] (2c) and nPr2NHCONHPh (4), representing a rare example of selective release of a functional group of ligands in organolanthanide chemistry. Hydrolysis of 2c gives the organic nitrogen-containing product PyNHCONHPh (5). Moreover, 3c can also be obtained by the reaction of 2c with PhNCO. These results demonstrate that the diinsertion of PhNCO into the N–H bond of coordinated amino ligands might proceed in a stepwise manner. All the compounds were characterized by elemental analysis and spectroscopic properties. The structures of compounds 3a–e and 4 are also determined through X-ray single-crystal diffraction analysis.
Co-reporter:Xiaoqing Li, Jianquan Hong, Ruiting Liu, Linhong Weng, and Xigeng Zhou
Organometallics 2010 Volume 29(Issue 20) pp:4606-4610
Publication Date(Web):September 30, 2010
DOI:10.1021/om100755s
Insertion of benzophenone into the Y−Cp (Cp = C5H5) bond and two new reactivity patterns of the Cp-substituted alkoxide complexes have been established, by which an efficient and convenient method for conversion of the unsubstituted cyclopentadienyl group to the single-carbon-bridged ansa-cyclopentadienyl/alkoxyl ligand by using a simple ketone as the functionalizing reagent is developed. All products including the four-center interaction precursor of the insertion have been characterized by X-ray structural analyses.
Co-reporter:Xiuli Bu, Zhengxing Zhang and Xigeng Zhou
Organometallics 2010 Volume 29(Issue 16) pp:3530-3534
Publication Date(Web):July 27, 2010
DOI:10.1021/om100402h
Y[N(TMS)2]3/FeCl3 has been found to be an efficient bimetallic catalyst system for the cyclotrimerization of terminal alkynes, which cannot be achieved by either trivalent iron or trivalent lanthanide catalysts. Furthermore, this reaction also occurs efficiently in the presence of Fe[N(TMS)2]3 and YCl3. Both aromatic and aliphatic alkynes are compatible with this catalytic system. It is postulated that the catalytic cyclotrimerization proceeds through a tandem intermolecular diinsertion of alkynes into the yttrium−alkynyl bond and intramolecular electrophilic addition of a π-coordinated alkyne moiety, and the π-coordination of Fe3+ to alkyne may play an important role in controlling the insertion degree of advancement and selectivity. The observed catalytic reaction is sharply in contrast with the cyclotrimerization of alkynes, known to proceed through a typical metallacyclopentadiene intermediate.
Co-reporter:Xiaoqing Li, Ruiting Liu, Zhengxing Zhang, Xiaoming Mu, Linhong Weng and Xigeng Zhou
Organometallics 2010 Volume 29(Issue 15) pp:3298-3302
Publication Date(Web):July 14, 2010
DOI:10.1021/om1002045
Unprecedented tandem insertion/cycloaddition/isomerization reactions of Cp3Y with diphenylketene have been revealed, by which a general method for assembling one or two anionic side chains to the cyclopentadienyl ring bound to lanthanide metals in a one-pot procedure is established.
Co-reporter:Pengzhi Zheng, Jianquan Hong, Ruiting Liu, Zhengxing Zhang, Zhen Pang, Linhong Weng and Xigeng Zhou
Organometallics 2010 Volume 29(Issue 5) pp:1284-1289
Publication Date(Web):February 1, 2010
DOI:10.1021/om9010615
The treatment of Cp3Ln with 1 equiv of N,N′-dicyclohexyl-N′′-phenylguanidine followed by reacting with butyllithium yields a series of novel guanidinate dianion complexes of heterobimetallic lanthanide−lithium with formula Cp2Ln[(CyN)2CNPh]Li(THF)3 (Ln = Yb (1a), Er (1b), Y (1c), Dy (1d)). Reactivities of these dianionic guanidinate complexes toward various electrophiles have been investigated. Reaction of 1 with Me2RSiCl produced tetrasubstituted guanidinate monoanion complexes Cp2Ln[(CyN)2CN(Ph)SiRMe2] ((R = Me, Ln = Yb (2a), Er (2b), Y (2c); R = tBu, Ln = Yb (3a), Er (3b)), indicating that the Li−N bond is preferred to couple with chlorosilanes. In contrast, the regioselective functionalization of the NCy group bonded to the lanthanide ion was achieved by reaction of 1a with Me2SiCl2 to produce Me2Si(CyN)2C═NPh (4) and Cp2YbCl(THF) (5). Significantly, treatment of 1d with PhCOCl leads to the cleavage of one C−N bond of the dianionic guanidinate, giving the acylamino complex [Cp2Dy(OC(Ph)NCy)]2 (6). These results have shown that the active site of the dianionic guanidinate ligand is tunable due to the delocalization of the two negative charges on the three N atoms. All the compounds were characterized by elemental analysis and spectroscopic methods. The structures of compounds 1−6 are also determined through X-ray single-crystal diffraction analysis.
Co-reporter:ChuanFeng Wang;ZhengXing Zhang;LiLi Zhang;FuYou Li
Science China Chemistry 2010 Volume 53( Issue 10) pp:2079-2082
Publication Date(Web):2010 October
DOI:10.1007/s11426-010-4098-5
Self-assembly of cadmium ions and the rigid bridging ligand 4,4′-methylenebis(3-hydroxy-2-naphthoic acid) (pamoic acid, H4PA) leads to a one-dimensional metal-organic framework with open cube-like M2(H2PA)2 cages within its backbone, and exhibiting interesting yellow fluorescence.
Co-reporter:Fuyan Han ; Jie Zhang ; Yanan Han ; Zhengxing Zhang ; Zhenxia Chen ; Linhong Weng
Inorganic Chemistry 2009 Volume 48(Issue 4) pp:1774-1781
Publication Date(Web):January 13, 2009
DOI:10.1021/ic802094q
Reaction of Cp2LnCl with 1 equiv of KTpMe2 in toluene gives the mixed TpMe2/Cp lanthanide complexes Cp2Ln(TpMe2) (Ln = Yb (1a), Er (1b), Dy (1c)), while unexpected complexes CpLn(TpMe2)Cl(THF) (Ln = Yb (2a), Er (2b·THF), Dy (2c), Y (2d)) are obtained when the reactions are carried out in THF. Complex 2b can also be formed by the reaction of CpErCl2(THF)3 with 1 equiv of KTpMe2 in THF. Moreover, complex 1a can also be obtained from the reaction of Cp3Yb and KTpMe2. The results not only represent an efficient and versatile method for the synthesis of mixed Cp/TpMe2 lanthanide complexes but also provide new insight into the reactivity of Cp2LnCl. Furthermore, the reactivities of complexes 1a−c toward proton-donating reagents are examined. It has been found that 1b reacts with benzotriazole (C6H4NHN2) in THF to yield a lanthanide metallomacrocyclic complex [(TpMe2)CpEr(μ-N3C6H4)]3 (3), while the reaction of 1a with 1 equiv of 2-aminopyridine in THF gives an unexpected oxide complex [(TpMe2)Yb(2-HNC5H4N)]2(μ-O) (4). Presumably, the oxide ligand of compound 4 results from adventitious water. In addition, treatment of 1c with 2 equiv of 3,5-dimethylpyrazole yields a completely Cp-abstracted product (TpMe2)Dy(PzMe2)2(THF) (5), which can also be directly obtained from a three-component reaction of Cp2DyCl, KTpMe2, and 3,5-dimethylpyrazole in THF. These results further indicate that the new mixed TpMe2/Cp lanthanide complexes are practical and versatile precursors for the synthesis of poly(pyrazolyl)borate lanthanide derivatives. All new compounds have been characterized by elemental analysis and spectroscopic methods. The structures of complexes 1a,b and 2−5 have also been determined through single-crystal X-ray diffraction analysis.
Co-reporter:Jie Zhang, Liping Ma, Yanan Han, Fuyan Han Zhengxing Zhang, Ruifang Cai, Zhenxia Chen and Xigeng Zhou
Dalton Transactions 2009 (Issue 17) pp:3298-3305
Publication Date(Web):04 Feb 2009
DOI:10.1039/B813711K
The reaction of N,N′-dicyclohexylcarbodiimide (DCCI) with [Cp2Yb(o-H2NC6H4S)]2 (Cp = C5H5) (1) forms the monomer product Cp2Yb[SC6H4NC(NHCy)2] (2), indicating that the adjacent NH2group can add to the CN double bonds of carbodiimide to construct a neutral guanidine group. When DCCI reacts with [Cp2Y(o-H2NC6H4S)]2·2THF (4), a dimer product [CpY(μ-η2:η1-SC6H4NC(NHCy)NCy)(THF)]2 (5) was isolated, through the amino group addition and cyclopentadienyl elimination. Interestingly, on treatment of 4 with one or two equivalent of iPrNCNiPr at the same conditions gave an amino group partial addition product CpY(THF)[μ-η2:η1-SC6H4NC(NHiPr)NiPr)](μ-η2:η1-SC6H4NH2)YCp2·THF (6), where only one NH2group can add to the CN double bonds of carbodiimide molecule, another one is remained. However, when we extended this reaction to the gadolinium complex, a novel co-crystalline compound {Cp2Gd[SC6H4NC(NHCy)2]}·{CpGd(THF)[μ-η2:η1-SC6H4NC(NHCy)NCy)][μ-η2:η1-SC6H4NH2]GdCp2·THF} (8) was obtained from the reaction of [Cp2Gd(o-H2NC6H4S)]2 (7) with DCCI. In order to investigate the sequence of addition and the elimination of the cyclopentadienyl group, a deprotonation reaction of the addition product has also been studied. Reaction of CpYb[SC6H4NC(NHiPr)2]2(THF) (9), formed by reaction of Cp3Yb with two equivalent of o-aminothiophenol, and subsequently with 2 equiv. of iPrNCNiPr, with one equiv. of Cp3Yb gave a cyclopentadienyl elimination product [CpYb(μ-η2:η1-SC6H4NC(NHiPr)NiPr)(THF)]2 (3). This result reveals that addition of the NH2group to carbodiimide is prior to the elimination of cyclopentadienyl group. All of new compounds have been characterized by elemental analysis and spectroscopic properties. The solid-state structures of complexes 2, and 5–9 were determined by single-crystal X-ray diffraction.
Co-reporter:Wen Huang, Longcheng Hong, Pengzhi Zheng, Ruiting Liu, Xigeng Zhou
Tetrahedron 2009 65(18) pp: 3603-3610
Publication Date(Web):
DOI:10.1016/j.tet.2009.03.007
Co-reporter:Jialiang Wang, Lixin Zhang, Yufeng Jing, Wen Huang, Xigeng Zhou
Tetrahedron Letters 2009 50(35) pp: 4978-4982
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.06.070
Co-reporter:Yan Sun, Zhengxing Zhang, Xu Wang, Xiaoqing Li, Linhong Weng and Xigeng Zhou
Organometallics 2009 Volume 28(Issue 21) pp:6320-6330
Publication Date(Web):September 30, 2009
DOI:10.1021/om900631j
Treatment of [Cp2LnNHPy]2 (Py = 2-pyridyl) (1) with 4-nitrophenyl isocyanate gives the unexpected complexes Cp2Ln[η2:η1-PyNCON(C6H4NO2-4)CONHC6H4NO2-4] (Ln = Yb (5a), Er (5b)) as the main product regardless of the equivalency of isocyanate reagent employed. The more bulky isocyanate (2,6-iPr2C6H3NCO) also undergoes double insertion into 1, affording Cp2Ln[η2:η1-PyNCON(C6H3iPr2-2,6)CONHC6H3iPr2-2,6] (Ln = Yb (6a), Er (6b)). Reaction of 1 with 4 equiv of ClCH2CH2CH2NCO affords Cp2Ln[η2:η1-PyNCON(CH2CH2CH2Cl)CONH(CH2)3Cl] (Ln = Yb (7a), Er (7b)) in good yields. Moreover, [(C5H4Me)2LnNHPy]2 (Ln = Yb (2a), Er (2b), Y (2c)) are also reactive toward isocyanate diinsertion, giving (C5H4Me)2Ln[η2:η1-PyNCON(Ph)CONHPh] (Ln = Yb (8a), Er (8b), Y (8c)) in almost quantitative yields. Furthermore, it is found that the presence of electron-withdrawing and electron-donating substituents on the pyridyl ring does not appear to alter the product selectivity and yields. The diinsertion of PhNCO into [Cp2LnNHPyMe]2 (PyMe = 4-methyl-2-pyridyl) (3) and [Cp2LnNHPyCl]2 (PyCl = 5-chloro-2-pyridyl) (4) leads to the formation of Cp2Ln[η2:η1-PyMeNCON(Ph)CONHPh] (Ln = Er (9a), Y (9b)) and Cp2Ln[η2:η1-PyClNCON(Ph)CONHPh] (Ln = Yb (10a), Er (10b), Y (10c)), respectively. In addition, the monoinsertion intermediate Cp2Yb[η2-N(Py)CONHC6H3iPr2-2,6](HMPA) (11·HMPA) can be trapped during this diinsertion process by adding HMPA. Interestingly, the mixed diinsertion complex Cp2Yb[η2:η1-PyNCON(C6H3iPr2-2,6)CONHPh] (12) can be prepared by allowing 2,6-iPr2C6H3NCO to react first with PyNH2 and then with Cp3Yb followed by inserting with PhNCO or by the reaction of 11 with PhNCO. On the other hand, treatment of 6a with excess PhNCO leads to the replacement of 2,6-iPr2C6H3NCO units inserted into the N−H bond by PhNCO molecules, wherein the newly formed ligand has been structurally characterized in its protonated form PyNHCON(Ph)CONHPh (13). Similarly, PyNHCON(C6H3iPr2-2,6)CONHC6H3iPr2-2,6 (14) could also be obtained by reaction of Cp2Yb[PyNCON(Ph)CONHPh] with excess 2,6-iPr2C6H3NCO followed by hydrolysis. All the complexes were characterized by spectroscopic analysis. The structures of compounds 5−14 are also determined through X-ray single-crystal diffraction analysis.
Co-reporter:Jialiang Wang, Wen Huang, Zhengxing Zhang, Xu Xiang, Ruiting Liu and Xigeng Zhou
The Journal of Organic Chemistry 2009 Volume 74(Issue 9) pp:3299-3304
Publication Date(Web):April 6, 2009
DOI:10.1021/jo900070q
Iron chloride has been found to be an efficient catalyst for the disproportionation of allylic alcohols, which provides a convenient method for selective transformation of allylic alcohols to alkenes and α,β-unsaturated ketones. Furthermore, this catalytic system is also effective for highly selective allylic reduction of allylic alcohols, allylic ethers, and allylic acetates with benzyl alcohol under neutral and convenient reaction conditions.
Co-reporter:Jie Zhang ; Yanan Han ; Fuyan Han ; Zhenxia Chen ; Linhong Weng
Inorganic Chemistry 2008 Volume 47(Issue 13) pp:5552-5554
Publication Date(Web):June 13, 2008
DOI:10.1021/ic800782r
Compounds Cp2Ln[κ3-(4-NH(C8N2H4)(2-NH2C6H4)] [Cp = C5H5; Ln = Er (1), Y (2)] were synthesized by the reaction of Cp2LnNiPr2(THF) with anthranilonitrile, indicating a novel organolanthanide-mediated intermolecular nucleophilic addition/cyclization of anthranilonitrile. To trap the intermediate I, a probe reaction of Cp2ErNiPr2(THF) with anthranilonitrile and carbodiimide has also been investigated.
Co-reporter:Wen HUANG;Quan-Sheng SHEN;Jia-Liang WANG ;Xi-Geng ZHOU
Chinese Journal of Chemistry 2008 Volume 26( Issue 4) pp:729-735
Publication Date(Web):
DOI:10.1002/cjoc.200890136
Abstract
An efficient and highly selective Yb(OTf)3-catalyzed direct substitution of the hydroxy group at the allylic and propargylic positions with a variety of heteroatom- and carbon-centered nucleophiles, such as alcohols, thiols, amines, amides and active methylene compounds has been developed. The advantages of the present catalytic system are wide availability of the starting materials, especially for tolerance to thiols, no need for dried solvents and additives, mild conditions, short time of reaction, simple manipulation and environmentally friendly catalyst that can be recovered and reused at least ten times without significant reduction of activity.
Co-reporter:Xu Xiang, Quansheng Shen, Jialiang Wang, Zhenyu Zhu, Wen Huang and Xigeng Zhou
Organometallics 2008 Volume 27(Issue 8) pp:1959-1962
Publication Date(Web):March 20, 2008
DOI:10.1021/om8000313
We have found that the combination of dysprosium diiodide and dichloromethane can serve as an effective methylene transfer reagent for cyclopropanation of unfunctionalized alkenes. Furthermore, a Dy/I2 system has also proved to be effective in the cyclopropanation of alkenes and CH2Cl2.
Co-reporter:Quansheng Shen, Wen Huang, Jialiang Wang and Xigeng Zhou
Organometallics 2008 Volume 27(Issue 2) pp:301-303
Publication Date(Web):January 4, 2008
DOI:10.1021/om700891k
A mild and highly selective Ln[N(SiMe3)2]3/n-BuNH2 catalyzed addition of terminal alkynes to nitriles has been established, revealing a novel reactivity pattern of alkynes with nitriles and providing a new, economical, and mild method for the synthesis of conjugated ynones.
Co-reporter:Chengfu Pi, Zhenyu Zhu, Linhong Weng, Zhenxia Chen and Xigeng Zhou
Chemical Communications 2007 (Issue 21) pp:2190-2192
Publication Date(Web):05 Mar 2007
DOI:10.1039/B618151A
Unprecedented disproportionation and deprotonation of the linked diguanidinate ligand have been established, demonstrating, for the first time, that the number and distribution of negative charges on the diguanidinate ligand are tunable, and representing a new and efficient method for synthesis of linked diguanidinate di- and tri-anion complexes.
Co-reporter:Zhenyu Zhu, Chuanfeng Wang, Xu Xiang, Chengfu Pi and Xigeng Zhou
Chemical Communications 2006 (Issue 19) pp:2066-2068
Publication Date(Web):04 Apr 2006
DOI:10.1039/B602883G
An efficient method for the formation of SiCl3 radicals by the reaction of abundant and cheaper chlorosilanes with DyI2 has been established, not only demonstrating new distinctive reactivities of solvated DyI2 but also suggesting that the presence of lanthanide ions can improve the selectivity of some silyl radical-catalyzed reactions.
Co-reporter:Chunmei Zhang, Yanghui Lin, Zhenxia Chen, Xegeng Zhou
Journal of Rare Earths 2006 Volume 24(Issue 1) pp:9-14
Publication Date(Web):February 2006
DOI:10.1016/S1002-0721(06)60056-2
The reactivity of lanthanocene thiolates [Cp2Ln(μ-SEt)]2(Ln = Er, Y, Yb) with PhEtCCO and with PhNCO was examined. Treatment of [Cp2Ln(μ-SEt)]2 with PhEtCCO gives the α-thiolate-substituted enolate complexes [Cp2Ln (μ-η1:η3-OC(SEt)CPhEt)]2(Ln = Yb, Y), while [Cp2Ln(μ-SEt)]2 react with PhNCO to give the lanthanocene amido derivatives [Cp2Ln(OC(SEt)NPh)]2(Ln = Er, Yb). In both cases, the insertion reaction is independent of the stoichiometric ratio and the nature of lanthanides, demonstrating that lanthanocene thiolates are high reactive toward PhEtCCO and PhNCO under the conditions involved. All complexes are characterized by elemental analysis and spectroscopic properties, of which the structures of ytterbium enolate and erbium amido are also determined by X-ray single crystal diffraction analysis, indicating that both of them are centrosymmetric binuclear structures.
Co-reporter:Chun-Mei Zhang;Rui-Ting Liu;Zhen-Xia Chen;Xi-Geng Zhou
Chinese Journal of Chemistry 2006 Volume 24(Issue 2) pp:
Publication Date(Web):13 FEB 2006
DOI:10.1002/cjoc.200690044
[Cp2Ln(μ-SR)]2 was reacted with Ph2CCO to yield ketene mono-insertion products [Cp2Ln(μ-η1:η2-OC(SR)=CPh2)]2 [R=Bn, Ln=Yb (1), Er (2), Y (3) and R=Ph, Ln=Yb (4)], indicating that the reactions of organolanthanide thiolates with ketenes are independent of the nature of the thiolate ligand and the ketene as well as the reaction condition. These reactions could provide an efficient method for the synthesis of organolanthanide complexes with the α-thiolate-substituted enolate ligand. All these complexes were characterized by elemental analysis and spectroscopic properties and the structure of complex 1 was determined through X-ray single crystal diffraction analysis.
Co-reporter:Ruiting Liu;Chunmei Zhang;Zhenyu Zhu;Jun Luo Dr. ;Linhong Weng
Chemistry - A European Journal 2006 Volume 12(Issue 26) pp:
Publication Date(Web):6 JUN 2006
DOI:10.1002/chem.200600161
This paper presents some unusual types of reactions of lanthanocene amide complexes with ketenes, and demonstrates that these reactions are dependent on the nature of amide ligands and ketenes as well as the stoichiometric ratio under the conditions involved. The reaction of [{Cp2LnNiPr2}2] with four equivalents of Ph2CCO in toluene affords the unexpected enolization dearomatization products [Cp2Ln(OC{2,5-C6H5(CPhCONiPr2-4)}CPh2)] (Ln = Yb (1 a), Er (1 b)) in good yields, representing an unprecedented conjugate electrophilic addition to a non-coordinated benzenoid nucleus. Treatment of [{Cp2LnNiPr2}2] with four equivalents of PhEtCCO under the same conditions gives the unexpected enolization dearomatization/rearomatization products [{Cp2Ln(OC{C6H4(p-CHEtCONiPr2)}CEtPh)}2] (Ln = Yb (2 a), Er (2 b), Dy (2 c)). However, reaction of [{Cp2YbNiPr2}2] with PhEtCCO in THF forms only the mono-insertion product [Cp2Yb{OC(NiPr2)CEtPh}](THF) (3). Hydrolysis of 2 afforded aryl ketone PhEtCHCOC6H4(p-CHEtCONiPr2) (4) and the overall formation of aryl ketone 4 provides an alternative route to the acylation of aromatic compounds. Moreover, reaction of [{Cp2LnNHPh}2] with excess of PhEtCCO or Ph2CCO in toluene affords only the products from a formal insertion of the CC bond of the ketene into the NH bond, [(Cp2Ln{OC(CHEtPh)NPh})2] (Ln = Yb (5 a), Y (5 b)) or [(Cp2Er{OC(CHPh2)NPh})2] (6), respectively, indicating that an isomerization involving a 1,3-hydrogen shift occurs more easily than the conjugate electrophilic addition reaction, along with the initial amide attack on the ketene carbonyl carbon. [{Cp2ErNHEt}2] reacts with an excess of PhEtCCO to give [(Cp2Er{PhEtCHCON(Et)COCEtPh})2] (7), revealing another unique pattern of double-insertion of ketenes into the metal–ligand bond without bond formation between two ketene molecules. All complexes were characterized by elemental analysis and by their spectroscopic properties. The structures of complexes 1 b, 2 a, 2 b, 5 a, 5 b, 6, and 7 were also determined through X-ray single-crystal diffraction analysis.
Co-reporter:Xigeng Zhou, Ming Zhu, Libei Zhang, Zhenyu Zhu, Chengfu Pi, Zhen Pang, Linhong Weng and Ruifang Cai
Chemical Communications 2005 (Issue 18) pp:2342-2344
Publication Date(Web):15 Mar 2005
DOI:10.1039/B417609J
Two unusual regioselective O2 oxidation reactions of air-sensitive lanthanocene thiolates and a thioether chelate are described, revealing a novel oxygenation pattern of thiolate ligands.
Co-reporter:Luo Jun;Zhou Xi-Geng;Hou Xiu-Feng;Wu Hui-Xia;Weng Lin-Hong;Li Yan-Rong
Chinese Journal of Chemistry 2005 Volume 23(Issue 3) pp:
Publication Date(Web):4 APR 2005
DOI:10.1002/cjoc.200590310
Two new dicyanamide coordination polymers, {Mn(dmpz)[N(CN)2]2}2 (1) and {Cu(dmpz)[N(CN)2]2}2 (2) (dmpz=3,5-dimethylpyrazole), were synthesized and characterized by single crystal X-ray diffraction analysis and IR spectroscopy. In 1 and 2 the metal ions have two different coordination modes, where one is coordinated to four dicyanamide anions and two monodentate dmpz molecules to form a slightly distorted octahedral geometry, while the other adopts octahedral geometry, surrounded by four nitrile N atoms and two amide N atoms of the dicyanamide anions. Both complexes contain two alternating chains that are parallel to each other.
Co-reporter:Yinghua Chen, Zhenyu Zhu, Jie Zhang, Jianzhong Shen, Xigeng Zhou
Journal of Organometallic Chemistry 2005 Volume 690(Issue 16) pp:3783-3789
Publication Date(Web):15 August 2005
DOI:10.1016/j.jorganchem.2005.05.013
Treatment of mesoporous silicate SBA-15 with Sm[N(SiMe3)2]3 led to the formation of a novel organolanthanide/inorganic hybrid material [SBA-15]Sm[N(SiMe3)2]x via abstraction of N(SiMe3)2 by terminal silanol groups and subsequent surface silylation. The hybrid material was characterized by elemental analyses, IR spectroscopy, X-ray diffraction, and nitrogen sorption, indicating a successful tailoring inside the silicate SBA-15 and the maintenance of the well-ordered mesostructure. This hybrid material is a promising heterogeneous catalyst for the Tishchenko reaction, where it is superior to the homogeneous correspondent in deactivation behavior, reusability and relative tolerance to oxygen, particularly in the control of selectivity of mixed Tishchenko reaction due to the steric hindrance and the diffusion control derived from the surface confinement.Treatment of mesoporous silicate SBA-15 with Sm[N(SiMe3)2]3 led to the formation of a novel organolanthanide/inorganic hybrid material [SBA-15]Sm[N(SiMe3)2]x, which is an efficient and mild catalyst for Tishchenko reaction.
Co-reporter:Ming Zhu;Li-Bei Zhang;Ying-Hua Chen;Xi-Geng Zhou;Rui-Fang Cai;Lin-Hong Weng
Chinese Journal of Chemistry 2004 Volume 22(Issue 9) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20040220912
Six new ethylthioethylcyclopentadienyl containing organolanthanide complexes CpLnCl [Ln=Gd (1), Dy (2)] and Cp2LnCpTh [Cp=C5H5, Ln=Yb (3), Sm (4), Dy (5), Y (6)] were synthesized by the reaction of ethylthioethyl-cyclopentadienyl (CpTh) sodium salt with LnCl3 or Cp2LnCl in THF. Complexes 1–6 were characterized by elemental analyses, infrared and mass spectroscopies. The molecular structures of complexes 1–3 were also determined by the X-ray single crystal diffraction. The results show that the side-chain sulfur atom on the ethylthioethylcyclopentadienyl ring can form intramolecular chelating coordination to the central lanthanide ion, improving the stability of organolanthanide complexes and reducing the number of coordinated THF molecules.
Co-reporter:Jie Zhang, Ruifang Cai, Linhong Weng, Xigeng Zhou
Journal of Organometallic Chemistry 2003 Volume 672(1–2) pp:94-99
Publication Date(Web):14 April 2003
DOI:10.1016/S0022-328X(03)00147-5
The synthesis and structures of three new lanthanide complexes incorporating tetra-substituted guanidinate ligand [iPrN∴C(NiPr2)∴NiPr] are described. Treatment of Cp2LnNiPr2(THF) (Ln=Yb, Dy, Gd) with N,N′-di-isopropyl-carbodiimide results in mono-insertion of carbodiimide into the LnN σ-bond to yield Cp2Ln[iPrN∴C(NiPr2)∴NiPr] (Ln=Yb(1), Dy(2), Gd(3)), providing an efficient method for the synthesis of organolanthanide guanidinate complexes. It was found that an excess of N,N′-di-isopropyl-carbodiimide did not affect the nature of the final product. Complexes 1–3 were characterized by elemental analysis, IR and mass spectroscopies. Complexes 1 and 2 were determined by the X-ray single crystal diffraction analysis.N,N′-Di-isopropyl-carbodiimide can be inserted into the LnN σ-bond of Cp2LnNiPr2, which provides an efficient method for synthesis of lanthanide guanidinate complexes.
Co-reporter:Ruyi Ruan, Jie Zhang, Xigeng Zhou and Ruifang Cai
Chemical Communications 2002 (Issue 5) pp:538-539
Publication Date(Web):13 Feb 2002
DOI:10.1039/B200107A
In the presence of Cp2LnX–HgCl2, the treatment of RCCCH2Br with Mg leads to the formation of benzene derivatives C6H4R2-1,2 (R = H, Ph) in moderate yield, which provides a new method for the construction of the benzene ring skeleton.
Co-reporter:Xi-Geng Zhou, Li-Bei Zhang, Rui-Fang Cai, Qiang-Jin Wu, Lin-Hong Weng, Zu-En Huang
Journal of Organometallic Chemistry 2000 Volume 604(Issue 2) pp:260-266
Publication Date(Web):16 June 2000
DOI:10.1016/S0022-328X(00)00281-3
A new phosphaazaallene Mes*PCNAr (Mes*=2,4,6-tBu3C6H2, Ar=p-ClC6H4) (1) has been synthesized. Complex 1 reacts with Mes*PHSiMe2tBu to form the phosphaazaallene insertion product Mes*PC(PHMes*)NAr(SiMe2tBu) (2). The treatment of 1 with an excess amount of YCl3 in THF leads to the isolation of the symmetrical dimer Mes*P(μ-CNAr)2PMes* (3), indicating both of the PC and CN bonds of 1 are reactive sites. X-ray structure analysis of 1 indicates that the distance of the PC bond, 1.642(5) Å, is markedly shorter than those of isolated PC bonds (1.66–1.72 Å), and the NCP unit slightly deviates from linearity with the PCN angle of 170.8(4)°. The structure determination of 2 revealed the molecule adopts a planar conformation about PC bond and the mutual orientation of two bulky Mes* moieties is almost orthogonal (92.8°). Complex 3 is a symmetrical four-membered ring structure in which the Mes* rings lie nearly perpendicular to the planes of the Ar ring and the P2C2 ring (84.8 and 75.6°, respectively), while the Ar ring is almost parallel to the plane of the P2C2 ring (25.2°).
Co-reporter:Ruiting Liu and Xigeng Zhou
Chemical Communications 2013 - vol. 49(Issue 31) pp:NaN3187-3187
Publication Date(Web):2012/12/14
DOI:10.1039/C2CC35637F
Cyclopentadienyl and substituted cyclopentadienyl ligands are observed in a wide range of organometallic complexes. In addition to serving as ancillary ligands, these ligands have come into their own as intermediates in organometallic reactions, and shown many unique reaction modes involving ring C–H, C–C and CC bond cleavages. This feature article summarizes the progressive development of cyclopentadienyl-based reactions of metallocene complexes of transition metals and rare-earth metals, with the aim of further developing the fundamental modes of reactivity of such systems together with their synthetic applications.
Co-reporter:Longcheng Hong, Weijia Lin, Fangjun Zhang, Ruiting Liu and Xigeng Zhou
Chemical Communications 2013 - vol. 49(Issue 49) pp:NaN5591-5591
Publication Date(Web):2013/04/30
DOI:10.1039/C3CC42534G
The first example of rare earth metal-catalyzed cycloaddition of terminal alkynes to azides resulting in the formation of 1,5-disubstituted 1,2,3-triazoles is described. Preliminary studies revealed that the present cycloaddition shows unprecedented mechanistic features involving a tandem anionic cascade cyclization and anti-addition across the CC triple bond.
Co-reporter:Chengfu Pi, Zhenyu Zhu, Linhong Weng, Zhenxia Chen and Xigeng Zhou
Chemical Communications 2007(Issue 21) pp:NaN2192-2192
Publication Date(Web):2007/03/05
DOI:10.1039/B618151A
Unprecedented disproportionation and deprotonation of the linked diguanidinate ligand have been established, demonstrating, for the first time, that the number and distribution of negative charges on the diguanidinate ligand are tunable, and representing a new and efficient method for synthesis of linked diguanidinate di- and tri-anion complexes.
Co-reporter:Jianquan Hong, Zhenhua Li, Zhening Chen, Linhong Weng, Xigeng Zhou and Lixin Zhang
Dalton Transactions 2016 - vol. 45(Issue 15) pp:NaN6649-6649
Publication Date(Web):2016/03/02
DOI:10.1039/C6DT00314A
Diverse reactivity patterns of mixed tetramethyl/methylidene rare-earth complexes bearing bulky benzamidinate coligands L3Ln3(μ2-Me)3(μ3-Me)(μ3-CH2) [L = [PhC(NC6H3iPr2-2,6)2]−; Ln = Y(1a), Lu(1b)] with PhCN, alkynes, and CS2 have been established. Reaction of complexes 1 with PhCN gave the μ3-CH2 addition complexes (NCNdipp)3Lu3(μ2-Me)3(μ3-Me)[μ–η1:η1:η3-CH2C(Ph)N] [Ln = Y(2a), Lu(2b)]. Treatment of complexes 1 with phenylacetylene afforded unexpected alkenyl dianion complexes L3Ln3(μ2-Me)3(μ3-Me)(μ–η1:η3-PhCCMe) [Ln = Y(3a), Lu(3b)] through the insertion of rare earth methylidene into a C–H bond in a reductive fashion. However, reaction of complexes 1 and HCCSiMe3 gave μ3-Me protonolysis complexes L3Ln3(μ2-Me)3(μ3-CCSiMe3)(μ3-CH2) [Ln = Y (4a), Lu (4b)] in excellent yields. Treatment of complexes 1 with CS2 led to the formation of the methyl activation complexes L3Ln3(μ2-Me)2(μ3-CH2)(μ3–η1:η2:η2-S2CCH2) [Ln = Y(5a), Lu(5b)]. All the new complexes were fully characterized.
Co-reporter:Yan Sun, Zhengxing Zhang, Xu Wang, Xiaoqing Li, Linhong Weng and Xigeng Zhou
Dalton Transactions 2010 - vol. 39(Issue 1) pp:NaN226-226
Publication Date(Web):2009/11/03
DOI:10.1039/B918181D
[Cp2LnNHPy]2 (Py = 2-pyridyl) (1a–e) react with phenyl isocyanate to form the N–H diinsertion products Cp2Ln[η2:η1-PyNCON(Ph)CONHPh](THF) (Ln = Yb (3a), Er (3b), Y (3c), Dy (3d), Gd (3e)). It has been proven that nPr2NH can abstract one PhNCO unit from 3c to form Cp2Y[η3-OC(NHPh)NPy] (2c) and nPr2NHCONHPh (4), representing a rare example of selective release of a functional group of ligands in organolanthanide chemistry. Hydrolysis of 2c gives the organic nitrogen-containing product PyNHCONHPh (5). Moreover, 3c can also be obtained by the reaction of 2c with PhNCO. These results demonstrate that the diinsertion of PhNCO into the N–H bond of coordinated amino ligands might proceed in a stepwise manner. All the compounds were characterized by elemental analysis and spectroscopic properties. The structures of compounds 3a–e and 4 are also determined through X-ray single-crystal diffraction analysis.
Co-reporter:Jie Zhang, Liping Ma, Yanan Han, Fuyan Han Zhengxing Zhang, Ruifang Cai, Zhenxia Chen and Xigeng Zhou
Dalton Transactions 2009(Issue 17) pp:NaN3305-3305
Publication Date(Web):2009/02/04
DOI:10.1039/B813711K
The reaction of N,N′-dicyclohexylcarbodiimide (DCCI) with [Cp2Yb(o-H2NC6H4S)]2 (Cp = C5H5) (1) forms the monomer product Cp2Yb[SC6H4NC(NHCy)2] (2), indicating that the adjacent NH2group can add to the CN double bonds of carbodiimide to construct a neutral guanidine group. When DCCI reacts with [Cp2Y(o-H2NC6H4S)]2·2THF (4), a dimer product [CpY(μ-η2:η1-SC6H4NC(NHCy)NCy)(THF)]2 (5) was isolated, through the amino group addition and cyclopentadienyl elimination. Interestingly, on treatment of 4 with one or two equivalent of iPrNCNiPr at the same conditions gave an amino group partial addition product CpY(THF)[μ-η2:η1-SC6H4NC(NHiPr)NiPr)](μ-η2:η1-SC6H4NH2)YCp2·THF (6), where only one NH2group can add to the CN double bonds of carbodiimide molecule, another one is remained. However, when we extended this reaction to the gadolinium complex, a novel co-crystalline compound {Cp2Gd[SC6H4NC(NHCy)2]}·{CpGd(THF)[μ-η2:η1-SC6H4NC(NHCy)NCy)][μ-η2:η1-SC6H4NH2]GdCp2·THF} (8) was obtained from the reaction of [Cp2Gd(o-H2NC6H4S)]2 (7) with DCCI. In order to investigate the sequence of addition and the elimination of the cyclopentadienyl group, a deprotonation reaction of the addition product has also been studied. Reaction of CpYb[SC6H4NC(NHiPr)2]2(THF) (9), formed by reaction of Cp3Yb with two equivalent of o-aminothiophenol, and subsequently with 2 equiv. of iPrNCNiPr, with one equiv. of Cp3Yb gave a cyclopentadienyl elimination product [CpYb(μ-η2:η1-SC6H4NC(NHiPr)NiPr)(THF)]2 (3). This result reveals that addition of the NH2group to carbodiimide is prior to the elimination of cyclopentadienyl group. All of new compounds have been characterized by elemental analysis and spectroscopic properties. The solid-state structures of complexes 2, and 5–9 were determined by single-crystal X-ray diffraction.





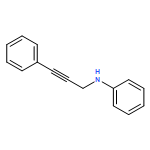
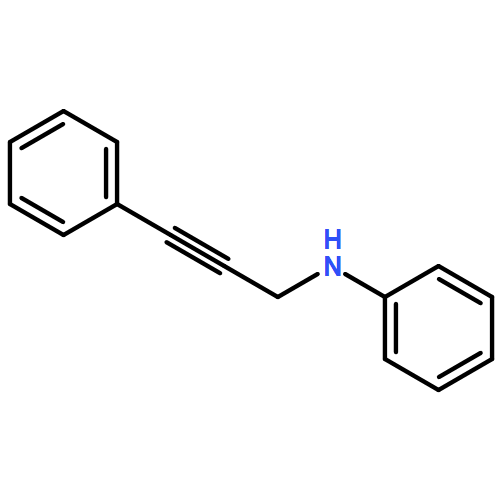

![Benzenemethanamine, N-[3-(trimethylsilyl)-2-propynyl]-](http://img.cochemist.com/ccimg/102000/101911-12-0.png)
![Benzenemethanamine, N-[3-(trimethylsilyl)-2-propynyl]-](http://img.cochemist.com/ccimg/102000/101911-12-0_b.png)





![Benzaldehyde, 2-[[(4-methylphenyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/19900/19820-56-5.png)
![Benzaldehyde, 2-[[(4-methylphenyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/19900/19820-56-5_b.png)


![Benzene, 1,1'-[(1E)-1-buten-3-yne-1,4-diyl]bis-](http://img.cochemist.com/ccimg/13400/13343-79-8.png)
![Benzene, 1,1'-[(1E)-1-buten-3-yne-1,4-diyl]bis-](http://img.cochemist.com/ccimg/13400/13343-79-8_b.png)















![Benzamide, 2-[[(4-methylphenyl)sulfonyl]oxy]-N-phenyl-](http://img.cochemist.com/ccimg/858500/858478-93-0.png)
![Benzamide, 2-[[(4-methylphenyl)sulfonyl]oxy]-N-phenyl-](http://img.cochemist.com/ccimg/858500/858478-93-0_b.png)








![1,2-dihydro-2-methyl-3H-Benz[e]isoindol-3-one](http://img.cochemist.com/ccimg/95500/95469-25-3.png)
![1,2-dihydro-2-methyl-3H-Benz[e]isoindol-3-one](http://img.cochemist.com/ccimg/95500/95469-25-3_b.png)





![Thiophene, 2,2'-[(1Z)-1,2-dimethyl-1,2-ethenediyl]bis-](http://img.cochemist.com/ccimg/68800/68728-21-2.png)
![Thiophene, 2,2'-[(1Z)-1,2-dimethyl-1,2-ethenediyl]bis-](http://img.cochemist.com/ccimg/68800/68728-21-2_b.png)







![Benzene, 1,1'-[(1Z)-1,2-dimethyl-1,2-ethenediyl]bis[4-methoxy-](/data/chemimg/1847300/54953-13-8.png)
![Benzene, 1,1'-[(1Z)-1,2-dimethyl-1,2-ethenediyl]bis[4-methoxy-](/data/chemimg/1847300/54953-13-8_b.png)








![Benzene, 1,1'-[(1E)-1,2-diethyl-1,2-ethenediyl]bis-](/data/chemimg/1816700/38443-18-4.png)
![Benzene, 1,1'-[(1E)-1,2-diethyl-1,2-ethenediyl]bis-](/data/chemimg/1816700/38443-18-4_b.png)











![Benzene, 1,1'-[2,3-bis(phenylmethyl)-2-butene-1,4-diyl]bis-](http://img.cochemist.com/ccimg/19800/19754-02-0.png)
![Benzene, 1,1'-[2,3-bis(phenylmethyl)-2-butene-1,4-diyl]bis-](http://img.cochemist.com/ccimg/19800/19754-02-0_b.png)












![Benzene,1-[cyclohexylidene(4-methoxyphenyl)methyl]-4-methoxy- (9CI)](http://img.cochemist.com/ccimg/10300/10218-57-2.png)
![Benzene,1-[cyclohexylidene(4-methoxyphenyl)methyl]-4-methoxy- (9CI)](http://img.cochemist.com/ccimg/10300/10218-57-2_b.png)


![[CYCLOPENTYLIDENE(PHENYL)METHYL]BENZENE](http://img.cochemist.com/ccimg/7800/7714-72-9.png)
![[CYCLOPENTYLIDENE(PHENYL)METHYL]BENZENE](http://img.cochemist.com/ccimg/7800/7714-72-9_b.png)



















![1-ISOPROPOXY-3-[(4-METHYLPHENYL)AMINO]-2-PROPANOL](http://img.cochemist.com/ccimg/34800/34706-60-0.png)
![1-ISOPROPOXY-3-[(4-METHYLPHENYL)AMINO]-2-PROPANOL](http://img.cochemist.com/ccimg/34800/34706-60-0_b.png)






















































































































































































































































































































































































































































































































































































































































































































































































































































![Benzenamine, 2-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/157900/157869-15-3.png)
![Benzenamine, 2-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/157900/157869-15-3_b.png)
![1-(phenylmethyl)-1H-Pyrrolo[2,3-b]pyridine](http://img.cochemist.com/ccimg/153000/152955-68-5.png)
![1-(phenylmethyl)-1H-Pyrrolo[2,3-b]pyridine](http://img.cochemist.com/ccimg/153000/152955-68-5_b.png)















































![Imidazo[1,2-a]pyridine,6-chloro-2-(4-chlorophenyl)-](http://img.cochemist.com/ccimg/89000/88964-99-2.png)
![Imidazo[1,2-a]pyridine,6-chloro-2-(4-chlorophenyl)-](http://img.cochemist.com/ccimg/89000/88964-99-2_b.png)




![2-(4-Chloro-phenyl)-7-methyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-62-7.png)
![2-(4-Chloro-phenyl)-7-methyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-62-7_b.png)
![IMIDAZO[1,2-A]PYRIDINE, 7-METHYL-2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/66000/65964-61-6.png)
![IMIDAZO[1,2-A]PYRIDINE, 7-METHYL-2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/66000/65964-61-6_b.png)
![2-(p-Tolyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-60-5.png)
![2-(p-Tolyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-60-5_b.png)




![2-(4-chlorophenyl)imidazo[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/57000/56921-86-9.png)
![2-(4-chlorophenyl)imidazo[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/57000/56921-86-9_b.png)









































![Imidazo[1,2-a]pyrimidine, 2-phenyl-](http://img.cochemist.com/ccimg/15800/15764-47-3.png)
![Imidazo[1,2-a]pyrimidine, 2-phenyl-](http://img.cochemist.com/ccimg/15800/15764-47-3_b.png)













![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)











![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6.png)
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6_b.png)