Co-reporter:Johannes Kirchmair, Mark J. Williamson, Avid M. Afzal, Jonathan D. Tyzack, Alison P. K. Choy, Andrew Howlett, Patrik Rydberg, and Robert C. Glen
Journal of Chemical Information and Modeling November 25, 2013 Volume 53(Issue 11) pp:
Publication Date(Web):November 12, 2013
DOI:10.1021/ci400503s
FAst MEtabolizer (FAME) is a fast and accurate predictor of sites of metabolism (SoMs). It is based on a collection of random forest models trained on diverse chemical data sets of more than 20 000 molecules annotated with their experimentally determined SoMs. Using a comprehensive set of available data, FAME aims to assess metabolic processes from a holistic point of view. It is not limited to a specific enzyme family or species. Besides a global model, dedicated models are available for human, rat, and dog metabolism; specific prediction of phase I and II metabolism is also supported. FAME is able to identify at least one known SoM among the top-1, top-2, and top-3 highest ranked atom positions in up to 71%, 81%, and 87% of all cases tested, respectively. These prediction rates are comparable to or better than SoM predictors focused on specific enzyme families (such as cytochrome P450s), despite the fact that FAME uses only seven chemical descriptors. FAME covers a very broad chemical space, which together with its inter- and extrapolation power makes it applicable to a wide range of chemicals. Predictions take less than 2.5 s per molecule in batch mode on an Ultrabook. Results are visualized using Jmol, with the most likely SoMs highlighted.
Co-reporter:Jonathan D. Tyzack, Mark J. Williamson, Rubben Torella, and Robert C. Glen
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 6) pp:1294-1305
Publication Date(Web):May 24, 2013
DOI:10.1021/ci400058s
Metabolism of xenobiotic and endogenous compounds is frequently complex, not completely elucidated, and therefore often ambiguous. The prediction of sites of metabolism (SoM) can be particularly helpful as a first step toward the identification of metabolites, a process especially relevant to drug discovery. This paper describes a reactivity approach for predicting SoM whereby reactivity is derived directly from the ground state ligand molecular orbital analysis, calculated using Density Functional Theory, using a novel implementation of the average local ionization energy. Thus each potential SoM is sampled in the context of the whole ligand, in contrast to other popular approaches where activation energies are calculated for a predefined database of molecular fragments and assigned to matching moieties in a query ligand. In addition, one of the first descriptions of molecular dynamics of cytochrome P450 (CYP) isoforms 3A4, 2D6, and 2C9 in their Compound I state is reported, and, from the representative protein structures obtained, an analysis and evaluation of various docking approaches using GOLD is performed. In particular, a covalent docking approach is described coupled with the modeling of important electrostatic interactions between CYP and ligand using spherical constraints. Combining the docking and reactivity results, obtained using standard functionality from common docking and quantum chemical applications, enables a SoM to be identified in the top 2 predictions for 75%, 80%, and 78% of the data sets for 3A4, 2D6, and 2C9, respectively, results that are accessible and competitive with other recently published prediction tools.
Co-reporter:Johannes Kirchmair, Andrew Howlett, Julio E. Peironcely, Daniel S. Murrell, Mark J. Williamson, Samuel E. Adams, Thomas Hankemeier, Leo van Buren, Guus Duchateau, Werner Klaffke, and Robert C. Glen
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 2) pp:354-367
Publication Date(Web):January 25, 2013
DOI:10.1021/ci300487z
Understanding which physicochemical properties, or property distributions, are favorable for successful design and development of drugs, nutritional supplements, cosmetics, and agrochemicals is of great importance. In this study we have analyzed molecules from three distinct chemical spaces (i) approved drugs, (ii) human metabolites, and (iii) traditional Chinese medicine (TCM) to investigate four aspects determining the disposition of small organic molecules. First, we examined the physicochemical properties of these three classes of molecules and identified characteristic features resulting from their distinctive biological functions. For example, human metabolites and TCM molecules can be larger and more hydrophobic than drugs, which makes them less likely to cross membranes. We then quantified the shifts in physicochemical property space induced by metabolism from a holistic perspective by analyzing a data set of several thousand experimentally observed metabolic trees. Results show how the metabolic system aims to retain nutrients/micronutrients while facilitating a rapid elimination of xenobiotics. In the third part we compared these global shifts with the contributions made by individual metabolic reactions. For better resolution, all reactions were classified into phase I and phase II biotransformations. Interestingly, not all metabolic reactions lead to more hydrophilic molecules. We were able to identify biotransformations leading to an increase of logP by more than one log unit, which could be used for the design of drugs with enhanced efficacy. The study closes with the analysis of the physicochemical properties of metabolites found in the bile, faeces, and urine. Metabolites in the bile can be large and are often negatively charged. Molecules with molecular weight >500 Da are rarely found in the urine, and most of these large molecules are charged phase II conjugates.
Co-reporter:Alexios Koutsoukas, Robert Lowe, Yasaman KalantarMotamedi, Hamse Y. Mussa, Werner Klaffke, John B. O. Mitchell, Robert C. Glen, and Andreas Bender
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 8) pp:1957-1966
Publication Date(Web):July 8, 2013
DOI:10.1021/ci300435j
In this study, two probabilistic machine-learning algorithms were compared for in silico target prediction of bioactive molecules, namely the well-established Laplacian-modified Naïve Bayes classifier (NB) and the more recently introduced (to Cheminformatics) Parzen-Rosenblatt Window. Both classifiers were trained in conjunction with circular fingerprints on a large data set of bioactive compounds extracted from ChEMBL, covering 894 human protein targets with more than 155,000 ligand-protein pairs. This data set is also provided as a benchmark data set for future target prediction methods due to its size as well as the number of bioactivity classes it contains. In addition to evaluating the methods, different performance measures were explored. This is not as straightforward as in binary classification settings, due to the number of classes, the possibility of multiple class memberships, and the need to translate model scores into “yes/no” predictions for assessing model performance. Both algorithms achieved a recall of correct targets that exceeds 80% in the top 1% of predictions. Performance depends significantly on the underlying diversity and size of a given class of bioactive compounds, with small classes and low structural similarity affecting both algorithms to different degrees. When tested on an external test set extracted from WOMBAT covering more than 500 targets by excluding all compounds with Tanimoto similarity above 0.8 to compounds from the ChEMBL data set, the current methodologies achieved a recall of 63.3% and 66.6% among the top 1% for Naïve Bayes and Parzen-Rosenblatt Window, respectively. While those numbers seem to indicate lower performance, they are also more realistic for settings where protein targets need to be established for novel chemical substances.
Co-reporter:Johannes Kirchmair;Andrew Howlett;Julio Peironcely
Journal of Cheminformatics 2013 Volume 5( Issue 1 Supplement) pp:
Publication Date(Web):2013 March
DOI:10.1186/1758-2946-5-S1-O12
Co-reporter:Johannes Kirchmair, Mark J. Williamson, Jonathan D. Tyzack, Lu Tan, Peter J. Bond, Andreas Bender, and Robert C. Glen
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 3) pp:617-648
Publication Date(Web):February 17, 2012
DOI:10.1021/ci200542m
Metabolism of xenobiotics remains a central challenge for the discovery and development of drugs, cosmetics, nutritional supplements, and agrochemicals. Metabolic transformations are frequently related to the incidence of toxic effects that may result from the emergence of reactive species, the systemic accumulation of metabolites, or by induction of metabolic pathways. Experimental investigation of the metabolism of small organic molecules is particularly resource demanding; hence, computational methods are of considerable interest to complement experimental approaches. This review provides a broad overview of structure- and ligand-based computational methods for the prediction of xenobiotic metabolism. Current computational approaches to address xenobiotic metabolism are discussed from three major perspectives: (i) prediction of sites of metabolism (SOMs), (ii) elucidation of potential metabolites and their chemical structures, and (iii) prediction of direct and indirect effects of xenobiotics on metabolizing enzymes, where the focus is on the cytochrome P450 (CYP) superfamily of enzymes, the cardinal xenobiotics metabolizing enzymes. For each of these domains, a variety of approaches and their applications are systematically reviewed, including expert systems, data mining approaches, quantitative structure–activity relationships (QSARs), and machine learning-based methods, pharmacophore-based algorithms, shape-focused techniques, molecular interaction fields (MIFs), reactivity-focused techniques, protein–ligand docking, molecular dynamics (MD) simulations, and combinations of methods. Predictive metabolism is a developing area, and there is still enormous potential for improvement. However, it is clear that the combination of rapidly increasing amounts of available ligand- and structure-related experimental data (in particular, quantitative data) with novel and diverse simulation and modeling approaches is accelerating the development of effective tools for prediction of in vivo metabolism, which is reflected by the diverse and comprehensive data sources and methods for metabolism prediction reviewed here. This review attempts to survey the range and scope of computational methods applied to metabolism prediction and also to compare and contrast their applicability and performance.
Co-reporter:Robert Charles Glen
Journal of Computer-Aided Molecular Design 2012 Volume 26( Issue 1) pp:47-49
Publication Date(Web):2012 January
DOI:10.1007/s10822-011-9501-6
Computers have changed the way we do science. Surrounded by a sea of data and with phenomenal computing capacity, the methodology and approach to scientific problems is evolving into a partnership between experiment, theory and data analysis. Given the pace of change of the last twenty-five years, it seems folly to speculate on the future, but along with unpredictable leaps of progress there will be a continuous evolution of capability, which points to opportunities and improvements that will certainly appear as our discipline matures.
Co-reporter:Alexios Koutsoukas, Benjamin Simms, Johannes Kirchmair, Peter J. Bond, Alan V. Whitmore, Steven Zimmer, Malcolm P. Young, Jeremy L. Jenkins, Meir Glick, Robert C. Glen, Andreas Bender
Journal of Proteomics 2011 Volume 74(Issue 12) pp:2554-2574
Publication Date(Web):18 November 2011
DOI:10.1016/j.jprot.2011.05.011
Given the tremendous growth of bioactivity databases, the use of computational tools to predict protein targets of small molecules has been gaining importance in recent years. Applications span a wide range, from the ‘designed polypharmacology’ of compounds to mode-of-action analysis. In this review, we firstly survey databases that can be used for ligand-based target prediction and which have grown tremendously in size in the past. We furthermore outline methods for target prediction that exist, both based on the knowledge of bioactivities from the ligand side and methods that can be applied in situations when a protein structure is known. Applications of successful in silico target identification attempts are discussed in detail, which were based partly or in whole on computational target predictions in the first instance. This includes the authors' own experience using target prediction tools, in this case considering phenotypic antibacterial screens and the analysis of high-throughput screening data. Finally, we will conclude with the prospective application of databases to not only predict, retrospectively, the protein targets of a small molecule, but also how to design ligands with desired polypharmacology in a prospective manner.Highlights► Public bioactivity databases are increasing in size at a tremendous rate. ► Integration of chemical and biological data is next key step; first examples exist. ► Target prediction and mode-of-action analysis help us rationalize compound action. ► Incorporating pathway information potentially enables multi-target drug design.
Co-reporter:Dr. N. J. Maximilian Macaluso;Dr. Sarah L. Pitkin;Dr. Janet J. Maguire;Dr. Anthony P. Davenport; Robert C. Glen
ChemMedChem 2011 Volume 6( Issue 6) pp:1017-1023
Publication Date(Web):
DOI:10.1002/cmdc.201100069
Abstract
The apelin receptor (APJ) is a class A G-protein-coupled receptor (GPCR) and is a putative target for the treatment of cardiovascular and metabolic diseases. Apelin-13 (NH2-QRPRLSHKGPMPF-COOH) is a vasoactive peptide and one of the most potent endogenous inotropic agents identified to date. We report the design and discovery of a novel APJ antagonist. By using a bivalent ligand approach, we have designed compounds with two ′affinity′ motifs and a short series of linker groups with different conformational and non-bonded interaction properties. One of these, cyclo(1–6)CRPRLC-KH-cyclo(9–14)CRPRLC is a competitive antagonist at APJ. Radioligand binding in CHO cells transfected with human APJ gave a Ki value of 82 nM, competition binding in human left ventricle gave a KD value of 3.2 μM, and cAMP accumulation assays in CHO-K1-APJ cells gave a KD value of 1.32 μM.
Co-reporter:N.J. Maximilian Macaluso ;RobertC. Glen
ChemMedChem 2010 Volume 5( Issue 8) pp:1247-1253
Publication Date(Web):
DOI:10.1002/cmdc.201000061
Co-reporter:Andreas Bender and Robert C. Glen
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 22) pp:3204-3218
Publication Date(Web):14 Oct 2004
DOI:10.1039/B409813G
Molecular Informatics utilises many ideas and concepts to find relationships between molecules. The concept of similarity, where molecules may be grouped according to their biological effects or physicochemical properties has found extensive use in drug discovery. Some areas of particular interest have been in lead discovery and compound optimisation. For example, in designing libraries of compounds for lead generation, one approach is to design sets of compounds ‘similar’ to known active compounds in the hope that alternative molecular structures are found that maintain the properties required while enhancing e.g. patentability, medicinal chemistry opportunities or even in achieving optimised pharmacokinetic profiles. Thus the practical importance of the concept of molecular similarity has grown dramatically in recent years. The predominant users are pharmaceutical companies, employing similarity methods in a wide range of applications e.g. virtual screening, estimation of absorption, distribution, metabolism, excretion and toxicity (ADME/Tox) and prediction of physicochemical properties (solubility, partitioning etc.). In this perspective, we discuss the representation of molecular structure (descriptors), methods of comparing structures and how these relate to measured properties. This leads to the concept of molecular similarity, its various definitions and uses and how these have evolved in recent years. Here, we wish to evaluate and in some cases challenge accepted views and uses of molecular similarity. Molecular similarity, as a paradigm, contains many implicit and explicit assumptions in particular with respect to the prediction of the binding and efficacy of molecules at biological receptors. The fundamental observation is that molecular similarity has a context which both defines and limits its use. The key issues of solvation effects, heterogeneity of binding sites and the fundamental problem of the form of similarity measure to use are addressed.

![Spiro[2H-1-benzopyran-2,4'-piperidine], 4-(4-methylphenyl)-](http://img.cochemist.com/ccimg/850200/850175-54-1.png)
![Spiro[2H-1-benzopyran-2,4'-piperidine], 4-(4-methylphenyl)-](http://img.cochemist.com/ccimg/850200/850175-54-1_b.png)
![Benzonitrile, 2-[(4-amino-1,3,5-triazin-2-yl)amino]-5-chloro-](http://img.cochemist.com/ccimg/880100/880084-59-3.png)
![Benzonitrile, 2-[(4-amino-1,3,5-triazin-2-yl)amino]-5-chloro-](http://img.cochemist.com/ccimg/880100/880084-59-3_b.png)
![Phenol, 4-spiro[2H-1-benzopyran-2,4'-piperidin]-4-yl-](http://img.cochemist.com/ccimg/850200/850175-57-4.png)
![Phenol, 4-spiro[2H-1-benzopyran-2,4'-piperidin]-4-yl-](http://img.cochemist.com/ccimg/850200/850175-57-4_b.png)
![Benzamide, N,N-diethyl-2-spiro[2H-1-benzopyran-2,4'-piperidin]-4-yl-](http://img.cochemist.com/ccimg/850200/850176-28-2.png)
![Benzamide, N,N-diethyl-2-spiro[2H-1-benzopyran-2,4'-piperidin]-4-yl-](http://img.cochemist.com/ccimg/850200/850176-28-2_b.png)


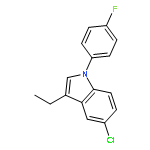
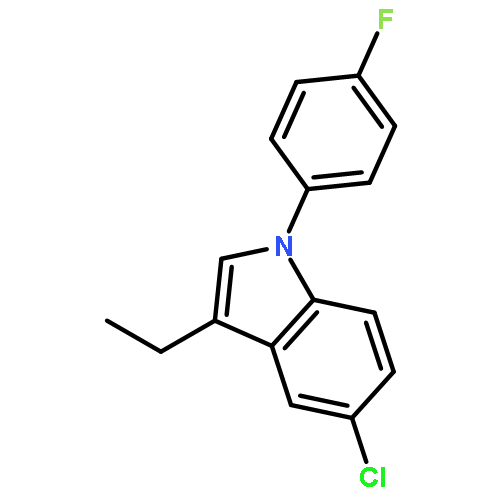
![1-Propanone, 1-[5-chloro-1-(4-fluorophenyl)-1H-indol-3-yl]-](http://img.cochemist.com/ccimg/573000/572913-88-3.png)
![1-Propanone, 1-[5-chloro-1-(4-fluorophenyl)-1H-indol-3-yl]-](http://img.cochemist.com/ccimg/573000/572913-88-3_b.png)
![FORMAMIDE, N-[4-[4-(ETHYLHEPTYLAMINO)BUTYL]PHENYL]-](http://img.cochemist.com/ccimg/880600/880554-75-6.png)
![FORMAMIDE, N-[4-[4-(ETHYLHEPTYLAMINO)BUTYL]PHENYL]-](http://img.cochemist.com/ccimg/880600/880554-75-6_b.png)


![2-Oxazolidinone, 4-[[3-(2-aminoethyl)-1H-indol-5-yl]methyl]-](http://img.cochemist.com/ccimg/331500/331441-79-3.png)
![2-Oxazolidinone, 4-[[3-(2-aminoethyl)-1H-indol-5-yl]methyl]-](http://img.cochemist.com/ccimg/331500/331441-79-3_b.png)


![ETHANONE, 1-[5-CHLORO-1-(4-FLUOROPHENYL)-1H-INDOL-3-YL]-](http://img.cochemist.com/ccimg/573000/572913-91-8.png)
![ETHANONE, 1-[5-CHLORO-1-(4-FLUOROPHENYL)-1H-INDOL-3-YL]-](http://img.cochemist.com/ccimg/573000/572913-91-8_b.png)


![BENZOIC ACID, 4-[5-CHLORO-3-(1-METHYLETHYL)-1H-INDOL-1-YL]-, METHYL ESTER](http://img.cochemist.com/ccimg/573000/572913-81-6.png)
![BENZOIC ACID, 4-[5-CHLORO-3-(1-METHYLETHYL)-1H-INDOL-1-YL]-, METHYL ESTER](http://img.cochemist.com/ccimg/573000/572913-81-6_b.png)






![SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDINE], 4-(4-METHOXYPHENYL)-](http://img.cochemist.com/ccimg/850200/850175-53-0.png)
![SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDINE], 4-(4-METHOXYPHENYL)-](http://img.cochemist.com/ccimg/850200/850175-53-0_b.png)


![BENZOIC ACID, 3-AMINO-4-[(1-ETHYLPROPYL)AMINO]-, METHYL ESTER](http://img.cochemist.com/ccimg/762300/762295-23-8.png)
![BENZOIC ACID, 3-AMINO-4-[(1-ETHYLPROPYL)AMINO]-, METHYL ESTER](http://img.cochemist.com/ccimg/762300/762295-23-8_b.png)
![SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDINE], 4-PHENYL-](http://img.cochemist.com/ccimg/850200/850175-56-3.png)
![SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDINE], 4-PHENYL-](http://img.cochemist.com/ccimg/850200/850175-56-3_b.png)
![SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDINE], 4-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/850200/850175-55-2.png)
![SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDINE], 4-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/850200/850175-55-2_b.png)
![BENZOIC ACID, 4-[(1-ETHYLPROPYL)AMINO]-3-NITRO-, METHYL ESTER](http://img.cochemist.com/ccimg/762300/762295-20-5.png)
![BENZOIC ACID, 4-[(1-ETHYLPROPYL)AMINO]-3-NITRO-, METHYL ESTER](http://img.cochemist.com/ccimg/762300/762295-20-5_b.png)
![Morpholine,4-[[5-[[1-(1-methylethyl)-4-piperidinyl]oxy]-1H-indol-2-yl]carbonyl]-](http://img.cochemist.com/ccimg/872100/872030-10-9.png)
![Morpholine,4-[[5-[[1-(1-methylethyl)-4-piperidinyl]oxy]-1H-indol-2-yl]carbonyl]-](http://img.cochemist.com/ccimg/872100/872030-10-9_b.png)
![2,4-IMIDAZOLIDINEDIONE, 3-[(4-HYDRAZINOPHENYL)METHYL]- (9CI)](http://img.cochemist.com/ccimg/725300/725268-37-1.png)
![2,4-IMIDAZOLIDINEDIONE, 3-[(4-HYDRAZINOPHENYL)METHYL]- (9CI)](http://img.cochemist.com/ccimg/725300/725268-37-1_b.png)






![N,N-diethyl-4-(5-hydroxyspiro[chromene-2,4'-piperidine]-4-yl)benzamide](http://img.cochemist.com/ccimg/850400/850305-06-5.png)
![N,N-diethyl-4-(5-hydroxyspiro[chromene-2,4'-piperidine]-4-yl)benzamide](http://img.cochemist.com/ccimg/850400/850305-06-5_b.png)

![(4,4-Difluoro-1-piperidinyl){1-isopropyl-5-[(1-isopropyl-4-piperi dinyl)oxy]-1H-indol-2-yl}methanone](http://img.cochemist.com/ccimg/872100/872030-17-6.png)
![(4,4-Difluoro-1-piperidinyl){1-isopropyl-5-[(1-isopropyl-4-piperi dinyl)oxy]-1H-indol-2-yl}methanone](http://img.cochemist.com/ccimg/872100/872030-17-6_b.png)

![4H-CARBAZOL-4-ONE, 1,2,3,9-TETRAHYDRO-9-METHYL-3-[(2-METHYL-1H-IMIDAZOL-1-YL)METHYL]-, (3R)-](http://img.cochemist.com/ccimg/99700/99614-60-5.png)
![4H-CARBAZOL-4-ONE, 1,2,3,9-TETRAHYDRO-9-METHYL-3-[(2-METHYL-1H-IMIDAZOL-1-YL)METHYL]-, (3R)-](http://img.cochemist.com/ccimg/99700/99614-60-5_b.png)
![BENZAMIDE, N,N-DIETHYL-3-SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDIN]-4-YL-](http://img.cochemist.com/ccimg/850200/850175-49-4.png)
![BENZAMIDE, N,N-DIETHYL-3-SPIRO[2H-1-BENZOPYRAN-2,4'-PIPERIDIN]-4-YL-](http://img.cochemist.com/ccimg/850200/850175-49-4_b.png)
![BENZAMIDE, 3-CHLORO-4-METHYL-N-TRICYCLO[3.3.1.13,7]DEC-1-YL-](http://img.cochemist.com/ccimg/461500/461419-63-6.png)
![BENZAMIDE, 3-CHLORO-4-METHYL-N-TRICYCLO[3.3.1.13,7]DEC-1-YL-](http://img.cochemist.com/ccimg/461500/461419-63-6_b.png)
![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0.png)
![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0_b.png)
![(4S,7S)-7-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]-6-oxo-1,2,3,4,7,8,9,10-octahydropyridazino[1,2-a]diazepine-4-carboxylic acid](http://img.cochemist.com/ccimg/88800/88768-40-5.png)
![(4S,7S)-7-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]-6-oxo-1,2,3,4,7,8,9,10-octahydropyridazino[1,2-a]diazepine-4-carboxylic acid](http://img.cochemist.com/ccimg/88800/88768-40-5_b.png)


![5-amino-6-chloro-2-methyl-N-[[1-(2-methylpropyl)piperidin-4-yl]methyl]imidazo[1,2-a]pyridine-8-carboxamide](http://img.cochemist.com/ccimg/519200/519148-48-2.png)
![5-amino-6-chloro-2-methyl-N-[[1-(2-methylpropyl)piperidin-4-yl]methyl]imidazo[1,2-a]pyridine-8-carboxamide](http://img.cochemist.com/ccimg/519200/519148-48-2_b.png)











![N-[4-(4-chlorophenyl)butyl]-N-ethylheptan-1-amine](http://img.cochemist.com/ccimg/72500/72456-59-8.png)
![N-[4-(4-chlorophenyl)butyl]-N-ethylheptan-1-amine](http://img.cochemist.com/ccimg/72500/72456-59-8_b.png)


![3(2H)-Pyridazinone,5-chloro-4-[[3-[[2-(3,4-dimethoxyphenyl)ethyl]methylamino]propyl]amino]-](http://img.cochemist.com/ccimg/150900/150800-12-7.png)
![3(2H)-Pyridazinone,5-chloro-4-[[3-[[2-(3,4-dimethoxyphenyl)ethyl]methylamino]propyl]amino]-](http://img.cochemist.com/ccimg/150900/150800-12-7_b.png)
![Acetic acid, [4-[(2-butyl-3-benzofuranyl)carbonyl]-2,6-diiodophenoxy]-](http://img.cochemist.com/ccimg/147100/147030-47-5.png)
![Acetic acid, [4-[(2-butyl-3-benzofuranyl)carbonyl]-2,6-diiodophenoxy]-](http://img.cochemist.com/ccimg/147100/147030-47-5_b.png)
![1,2,4-Triazolo[4,3-a]pyrazine,
7-[[(4R,5S)-5-amino-4-(2,4,5-trifluorophenyl)-1-cyclohexen-1-yl]carbonyl
]-5,6,7,8-tetrahydro-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/913700/913623-69-5.png)
![1,2,4-Triazolo[4,3-a]pyrazine,
7-[[(4R,5S)-5-amino-4-(2,4,5-trifluorophenyl)-1-cyclohexen-1-yl]carbonyl
]-5,6,7,8-tetrahydro-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/913700/913623-69-5_b.png)






![1,8-Naphthyridine-3-carboxylicacid,1-cyclopropyl-6-fluoro-1,4-dihydro-7-[8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-4-oxo-](http://img.cochemist.com/ccimg/219700/219680-11-2.png)
![1,8-Naphthyridine-3-carboxylicacid,1-cyclopropyl-6-fluoro-1,4-dihydro-7-[8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-4-oxo-](http://img.cochemist.com/ccimg/219700/219680-11-2_b.png)
![[4-(2-aminoethoxy)-3,5-diiodophenyl]-(2-butyl-1-benzofuran-3-yl)methanone](http://img.cochemist.com/ccimg/94400/94317-95-0.png)
![[4-(2-aminoethoxy)-3,5-diiodophenyl]-(2-butyl-1-benzofuran-3-yl)methanone](http://img.cochemist.com/ccimg/94400/94317-95-0_b.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)




![1-Piperidinebutanol, a-[4-(1,1-dimethylethyl)phenyl]-4-(hydroxydiphenylmethyl)-,(aS)-](http://img.cochemist.com/ccimg/126600/126588-96-3.png)
![1-Piperidinebutanol, a-[4-(1,1-dimethylethyl)phenyl]-4-(hydroxydiphenylmethyl)-,(aS)-](http://img.cochemist.com/ccimg/126600/126588-96-3_b.png)


![2H-1-Benzopyran-2-one,4-[[1-(1,3-benzodioxol-5-ylmethyl)-4-piperidinyl]amino]-6-chloro-](http://img.cochemist.com/ccimg/638200/638191-35-2.png)
![2H-1-Benzopyran-2-one,4-[[1-(1,3-benzodioxol-5-ylmethyl)-4-piperidinyl]amino]-6-chloro-](http://img.cochemist.com/ccimg/638200/638191-35-2_b.png)
![Pyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione,4-(4-chlorophenyl)-9-hydroxy-](http://img.cochemist.com/ccimg/622900/622855-78-1.png)
![Pyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione,4-(4-chlorophenyl)-9-hydroxy-](http://img.cochemist.com/ccimg/622900/622855-78-1_b.png)
![2-Propanol,1-(9H-carbazol-4-yloxy)-3-[[2-(2-methoxyphenoxy)ethyl]amino]-, (2S)-](http://img.cochemist.com/ccimg/95100/95094-00-1.png)
![2-Propanol,1-(9H-carbazol-4-yloxy)-3-[[2-(2-methoxyphenoxy)ethyl]amino]-, (2S)-](http://img.cochemist.com/ccimg/95100/95094-00-1_b.png)
![Acetic acid,[2-[[(1S,2R,3S,4R)-3-[4-[[(4-cyclohexylbutyl)amino]carbonyl]-2-oxazolyl]-7-oxabicyclo[2.2.1]hept-2-yl]methyl]phenoxy]-](http://img.cochemist.com/ccimg/143500/143443-86-1.png)
![Acetic acid,[2-[[(1S,2R,3S,4R)-3-[4-[[(4-cyclohexylbutyl)amino]carbonyl]-2-oxazolyl]-7-oxabicyclo[2.2.1]hept-2-yl]methyl]phenoxy]-](http://img.cochemist.com/ccimg/143500/143443-86-1_b.png)
![2-Imidazolidinethione,1-[2-[4-[5-chloro-1-(4-fluorophenyl)-1H-indol-3-yl]-1-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/139800/139718-40-4.png)
![2-Imidazolidinethione,1-[2-[4-[5-chloro-1-(4-fluorophenyl)-1H-indol-3-yl]-1-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/139800/139718-40-4_b.png)


![N-[4-(4-chlorophenyl)butyl]-N,N-diethylheptan-1-aminium](http://img.cochemist.com/ccimg/68400/68379-02-2.png)
![N-[4-(4-chlorophenyl)butyl]-N,N-diethylheptan-1-aminium](http://img.cochemist.com/ccimg/68400/68379-02-2_b.png)
![Methanone,(2-butyl-3-benzofuranyl)[4-(2-hydroxyethoxy)-3,5-diiodophenyl]-](http://img.cochemist.com/ccimg/135100/135048-44-1.png)
![Methanone,(2-butyl-3-benzofuranyl)[4-(2-hydroxyethoxy)-3,5-diiodophenyl]-](http://img.cochemist.com/ccimg/135100/135048-44-1_b.png)




![Benzenesulfonamide,5-[2-[[2-(2-ethoxyphenoxy)ethyl]amino]propyl]-2-methoxy-](http://img.cochemist.com/ccimg/94700/94666-07-6.png)
![Benzenesulfonamide,5-[2-[[2-(2-ethoxyphenoxy)ethyl]amino]propyl]-2-methoxy-](http://img.cochemist.com/ccimg/94700/94666-07-6_b.png)


![Benzonitrile,4-[3-[ethyl[3-(propylsulfinyl)propyl]amino]-2-hydroxypropoxy]-](/data/chemimg/413200/123955-10-2.png)
![Benzonitrile,4-[3-[ethyl[3-(propylsulfinyl)propyl]amino]-2-hydroxypropoxy]-](/data/chemimg/413200/123955-10-2_b.png)
![Benzamide,4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenyl]ethyl]amino]ethoxy]-](http://img.cochemist.com/ccimg/115300/115256-20-7.png)
![Benzamide,4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenyl]ethyl]amino]ethoxy]-](http://img.cochemist.com/ccimg/115300/115256-20-7_b.png)
![2,4-Quinazolinediamine, 5-[(4-chlorophenyl)thio]-](http://img.cochemist.com/ccimg/169000/168910-48-3.png)
![2,4-Quinazolinediamine, 5-[(4-chlorophenyl)thio]-](http://img.cochemist.com/ccimg/169000/168910-48-3_b.png)
![3-Azabicyclo[3.1.0]hexane, 1-(3,4-dichlorophenyl)-](http://img.cochemist.com/ccimg/66600/66504-40-3.png)
![3-Azabicyclo[3.1.0]hexane, 1-(3,4-dichlorophenyl)-](http://img.cochemist.com/ccimg/66600/66504-40-3_b.png)






![3-(butylamino)-1-[1,3-dichloro-6-(trifluoromethyl)phenanthren-9-yl]propan-1-ol](http://img.cochemist.com/ccimg/69800/69756-48-5.png)
![3-(butylamino)-1-[1,3-dichloro-6-(trifluoromethyl)phenanthren-9-yl]propan-1-ol](http://img.cochemist.com/ccimg/69800/69756-48-5_b.png)
![(4S)-4-ethyl-4-hydroxy-6,12-dihydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,8,14(4H)-trione](http://img.cochemist.com/ccimg/68500/68426-53-9.png)
![(4S)-4-ethyl-4-hydroxy-6,12-dihydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,8,14(4H)-trione](http://img.cochemist.com/ccimg/68500/68426-53-9_b.png)

![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9_b.png)
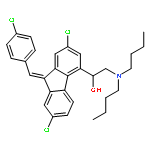
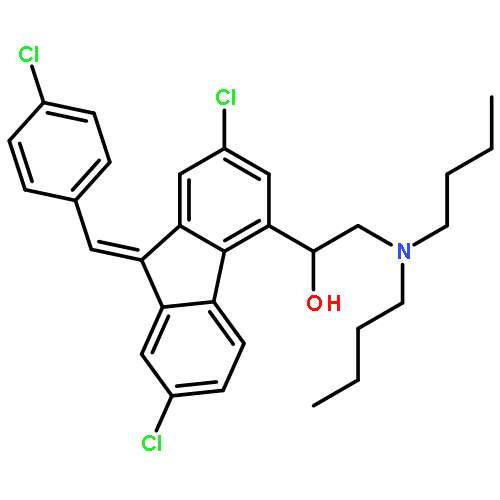
![2-Azabicyclo[2.2.2]octane-3-carboxylic acid, 2-[(2S)-2-[[(1S)-1-(ethoxycarbonyl)-3-phenylpropyl]amino]-1-oxopropyl]-, (3S)-](/data/chemimg/1339900/83059-56-7.png)
![2-Azabicyclo[2.2.2]octane-3-carboxylic acid, 2-[(2S)-2-[[(1S)-1-(ethoxycarbonyl)-3-phenylpropyl]amino]-1-oxopropyl]-, (3S)-](/data/chemimg/1339900/83059-56-7_b.png)


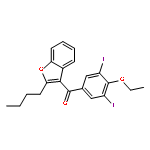
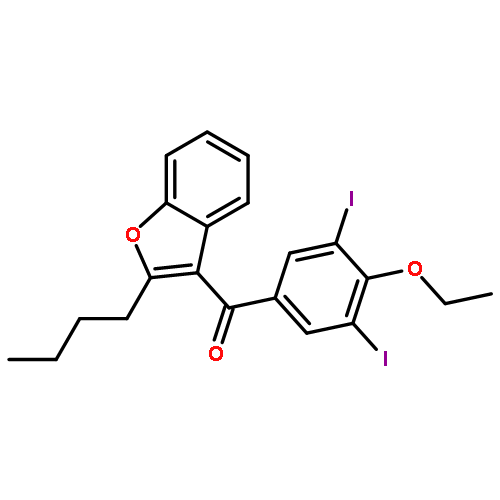
![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, ethyl ester, (1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/600/529-38-4.png)
![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, ethyl ester, (1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/600/529-38-4_b.png)

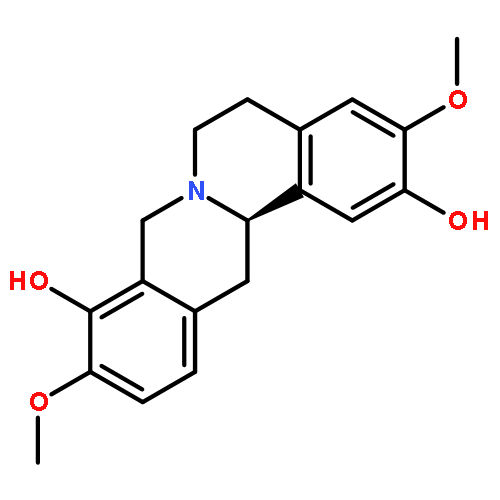
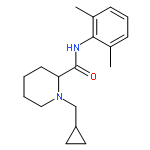
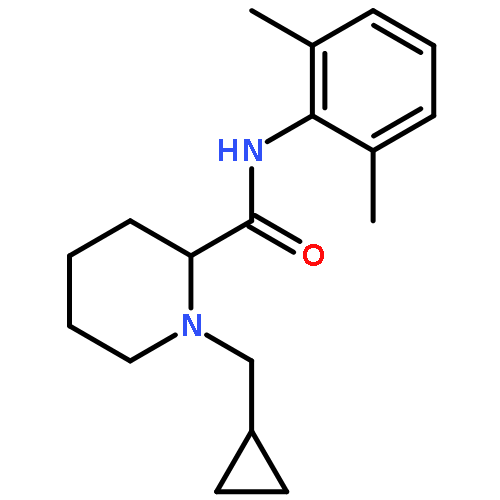




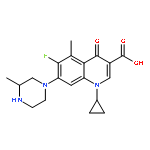
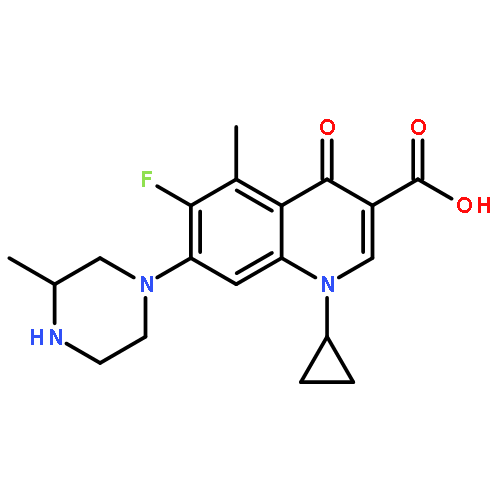

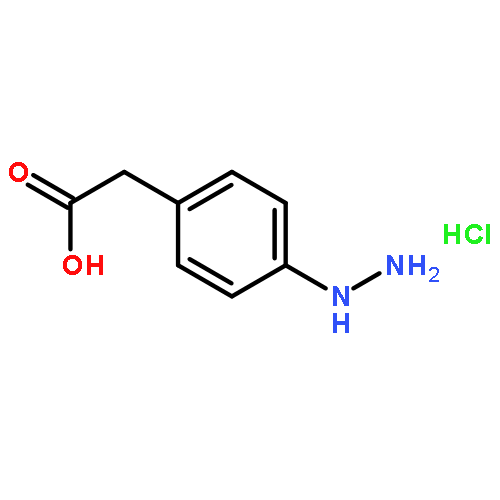
![Benzamide,2-[2-[[2-(4-aminophenyl)ethyl]methylamino]ethoxy]-5-methyl-](http://img.cochemist.com/ccimg/125200/125174-56-3.png)
![Benzamide,2-[2-[[2-(4-aminophenyl)ethyl]methylamino]ethoxy]-5-methyl-](http://img.cochemist.com/ccimg/125200/125174-56-3_b.png)
![Benzamide,5-methyl-2-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenyl]ethyl]amino]ethoxy]-](http://img.cochemist.com/ccimg/125200/125174-73-4.png)
![Benzamide,5-methyl-2-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenyl]ethyl]amino]ethoxy]-](http://img.cochemist.com/ccimg/125200/125174-73-4_b.png)


![Benzeneethanol, b-[2-(dimethylamino)propyl]-a-ethyl-b-phenyl-, 1-acetate](http://img.cochemist.com/ccimg/600/509-74-0.png)
![Benzeneethanol, b-[2-(dimethylamino)propyl]-a-ethyl-b-phenyl-, 1-acetate](http://img.cochemist.com/ccimg/600/509-74-0_b.png)
![Piperazine,1-acetyl-4-[4-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-](http://img.cochemist.com/ccimg/142200/142128-59-4.png)
![Piperazine,1-acetyl-4-[4-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-](http://img.cochemist.com/ccimg/142200/142128-59-4_b.png)
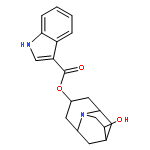
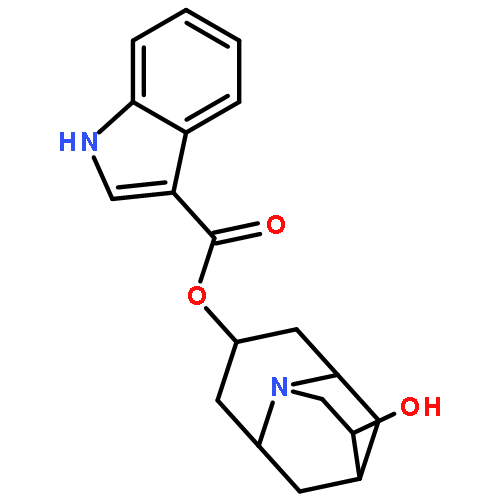


![Benzamide,4-methoxy-N-[2-[2-(1-methyl-2-piperidinyl)ethyl]phenyl]-](http://img.cochemist.com/ccimg/66800/66778-36-7.png)
![Benzamide,4-methoxy-N-[2-[2-(1-methyl-2-piperidinyl)ethyl]phenyl]-](http://img.cochemist.com/ccimg/66800/66778-36-7_b.png)


![Piperazine,1-[bis(4-fluorophenyl)methyl]-4-(3-phenyl-2-propen-1-yl)-](http://img.cochemist.com/ccimg/40300/40218-96-0.png)
![Piperazine,1-[bis(4-fluorophenyl)methyl]-4-(3-phenyl-2-propen-1-yl)-](http://img.cochemist.com/ccimg/40300/40218-96-0_b.png)


![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)
![(2R)-4-[DI(PROPAN-2-YL)AMINO]-2-PHENYL-2-PYRIDIN-2-YLBUTANAMIDE](http://img.cochemist.com/ccimg/74500/74464-83-8.png)
![(2R)-4-[DI(PROPAN-2-YL)AMINO]-2-PHENYL-2-PYRIDIN-2-YLBUTANAMIDE](http://img.cochemist.com/ccimg/74500/74464-83-8_b.png)
![1H-Indazole-3-carboxamide,N-[hexahydro-1-methyl-4-[(3-methylphenyl)methyl]-1H-1,4-diazepin-6-yl]-](http://img.cochemist.com/ccimg/129300/129294-09-3.png)
![1H-Indazole-3-carboxamide,N-[hexahydro-1-methyl-4-[(3-methylphenyl)methyl]-1H-1,4-diazepin-6-yl]-](http://img.cochemist.com/ccimg/129300/129294-09-3_b.png)
![Methanesulfonamide,N-[4-[(1R)-1-hydroxy-2-[(1-methylethyl)amino]ethyl]phenyl]-](http://img.cochemist.com/ccimg/30300/30236-31-8.png)
![Methanesulfonamide,N-[4-[(1R)-1-hydroxy-2-[(1-methylethyl)amino]ethyl]phenyl]-](http://img.cochemist.com/ccimg/30300/30236-31-8_b.png)


![Benzamide,N-[(2R)-2-piperidinylmethyl]-2,5-bis(2,2,2-trifluoroethoxy)-](http://img.cochemist.com/ccimg/99500/99495-90-6.png)
![Benzamide,N-[(2R)-2-piperidinylmethyl]-2,5-bis(2,2,2-trifluoroethoxy)-](http://img.cochemist.com/ccimg/99500/99495-90-6_b.png)
METHANONE](http://img.cochemist.com/ccimg/7200/7187-66-8.png)
METHANONE](http://img.cochemist.com/ccimg/7200/7187-66-8_b.png)


![2-Imidazolidinone,1-[2-[4-[1-(4-fluorophenyl)-1H-indol-3-yl]-1-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/106600/106516-21-6.png)
![2-Imidazolidinone,1-[2-[4-[1-(4-fluorophenyl)-1H-indol-3-yl]-1-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/106600/106516-21-6_b.png)





![Benzenepropanamine, g-[4-(trifluoromethyl)phenoxy]-](http://img.cochemist.com/ccimg/83900/83891-03-6.png)
![Benzenepropanamine, g-[4-(trifluoromethyl)phenoxy]-](http://img.cochemist.com/ccimg/83900/83891-03-6_b.png)
![3,5-Pyridinedicarboxylicacid, 2-[(2-aminoethoxy)methyl]-4-(2-chlorophenyl)-1,4-dihydro-6-methyl-,3-ethyl 5-methyl ester, (4R)-](http://img.cochemist.com/ccimg/103200/103129-81-3.png)
![3,5-Pyridinedicarboxylicacid, 2-[(2-aminoethoxy)methyl]-4-(2-chlorophenyl)-1,4-dihydro-6-methyl-,3-ethyl 5-methyl ester, (4R)-](http://img.cochemist.com/ccimg/103200/103129-81-3_b.png)
![{4-[({4-[(4-BROMO-2-FLUOROPHENYL)AMINO]-6-METHOXY-7-QUINAZOLINYL}<WBR />OXY)METHYL]-1-PIPERIDINYL}METHANOL](http://img.cochemist.com/ccimg/7800/7728-73-6.png)
![{4-[({4-[(4-BROMO-2-FLUOROPHENYL)AMINO]-6-METHOXY-7-QUINAZOLINYL}<WBR />OXY)METHYL]-1-PIPERIDINYL}METHANOL](http://img.cochemist.com/ccimg/7800/7728-73-6_b.png)

![Benzeneethanol,4-chloro-a-[2-(dimethylamino)-1-methylethyl]-a-methyl-](http://img.cochemist.com/ccimg/14900/14860-49-2.png)
![Benzeneethanol,4-chloro-a-[2-(dimethylamino)-1-methylethyl]-a-methyl-](http://img.cochemist.com/ccimg/14900/14860-49-2_b.png)
![2-[[9-(1-Methylethyl)-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-1-butanol](http://img.cochemist.com/ccimg/186700/186692-44-4.png)
![2-[[9-(1-Methylethyl)-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-1-butanol](http://img.cochemist.com/ccimg/186700/186692-44-4_b.png)


![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2,N2-dimethyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/100/91-84-9.png)
![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2,N2-dimethyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/100/91-84-9_b.png)




![1-(4-METHYLPHENYL)-3-AZABICYCLO[3.1.0]HEXANE](http://img.cochemist.com/ccimg/71200/71195-57-8.png)
![1-(4-METHYLPHENYL)-3-AZABICYCLO[3.1.0]HEXANE](http://img.cochemist.com/ccimg/71200/71195-57-8_b.png)
![1-[(4-FLUOROPHENYL)METHYL]-N-PIPERIDIN-4-YLBENZIMIDAZOL-2-AMINE](http://img.cochemist.com/ccimg/76000/75970-99-9.png)
![1-[(4-FLUOROPHENYL)METHYL]-N-PIPERIDIN-4-YLBENZIMIDAZOL-2-AMINE](http://img.cochemist.com/ccimg/76000/75970-99-9_b.png)

![N-ETHYL-N-[4-(4-NITROPHENYL)BUTYL]HEPTAN-1-AMINE](http://img.cochemist.com/ccimg/72500/72456-63-4.png)
![N-ETHYL-N-[4-(4-NITROPHENYL)BUTYL]HEPTAN-1-AMINE](http://img.cochemist.com/ccimg/72500/72456-63-4_b.png)






![Methanone,(4-aminophenyl)[7-(phenylmethyl)-3,7-diazabicyclo[3.3.1]non-3-yl]-](http://img.cochemist.com/ccimg/84000/83991-25-7.png)
![Methanone,(4-aminophenyl)[7-(phenylmethyl)-3,7-diazabicyclo[3.3.1]non-3-yl]-](http://img.cochemist.com/ccimg/84000/83991-25-7_b.png)
![L-Proline,N-[4-(2,3-dihydro-2-benzofuranyl)-1-(ethoxycarbonyl)butyl]-L-alanyl-](http://img.cochemist.com/ccimg/84800/84768-09-2.png)
![L-Proline,N-[4-(2,3-dihydro-2-benzofuranyl)-1-(ethoxycarbonyl)butyl]-L-alanyl-](http://img.cochemist.com/ccimg/84800/84768-09-2_b.png)


![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(1-piperazinyl)-](http://img.cochemist.com/ccimg/6200/6104-71-8.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(1-piperazinyl)-](http://img.cochemist.com/ccimg/6200/6104-71-8_b.png)
![Phenol,4-[2-[4-[[1-[(4-fluorophenyl)methyl]-1H-benzimidazol-2-yl]amino]-1-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/73800/73736-50-2.png)
![Phenol,4-[2-[4-[[1-[(4-fluorophenyl)methyl]-1H-benzimidazol-2-yl]amino]-1-piperidinyl]ethyl]-](http://img.cochemist.com/ccimg/73800/73736-50-2_b.png)






![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5.png)
![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5_b.png)







![2(1H)-Quinolinone,6-[3-[[(3,4-dimethoxyphenyl)methyl]amino]-2-hydroxypropoxy]-](http://img.cochemist.com/ccimg/143400/143343-83-3.png)
![2(1H)-Quinolinone,6-[3-[[(3,4-dimethoxyphenyl)methyl]amino]-2-hydroxypropoxy]-](http://img.cochemist.com/ccimg/143400/143343-83-3_b.png)
![N-methyl-N-{2-[methyl(1-methyl-1H-benzimidazol-2-yl)amino]ethyl}-4-[(methylsulfonyl)amino]benzenesulfonamide](http://img.cochemist.com/ccimg/138500/138490-53-6.png)
![N-methyl-N-{2-[methyl(1-methyl-1H-benzimidazol-2-yl)amino]ethyl}-4-[(methylsulfonyl)amino]benzenesulfonamide](http://img.cochemist.com/ccimg/138500/138490-53-6_b.png)
![(3S,5S,8R,9S,10S,13S,14S,17S)-17-[(1R)-1-HYDROXYETHYL]-10,13-DIMETHYL-2,3,4,5,6,7,8,9,11,12,14,15,16,17-TETRADECAHYDRO-1H-CYCLOPENTA[A]PHENANTHREN-3-OL](http://img.cochemist.com/ccimg/6100/6018-40-2.png)
![(3S,5S,8R,9S,10S,13S,14S,17S)-17-[(1R)-1-HYDROXYETHYL]-10,13-DIMETHYL-2,3,4,5,6,7,8,9,11,12,14,15,16,17-TETRADECAHYDRO-1H-CYCLOPENTA[A]PHENANTHREN-3-OL](http://img.cochemist.com/ccimg/6100/6018-40-2_b.png)


![1-(3-(4-Benzhydrylpiperazin-1-yl)propyl)-1H-benzo[d]imidazol-2(3H)-one](http://img.cochemist.com/ccimg/60700/60607-34-3.png)
![1-(3-(4-Benzhydrylpiperazin-1-yl)propyl)-1H-benzo[d]imidazol-2(3H)-one](http://img.cochemist.com/ccimg/60700/60607-34-3_b.png)



![Benzeneethanol, a-ethyl-b-[2-(methylamino)propyl]-b-phenyl-, 1-acetate](http://img.cochemist.com/ccimg/1500/1477-39-0.png)
![Benzeneethanol, a-ethyl-b-[2-(methylamino)propyl]-b-phenyl-, 1-acetate](http://img.cochemist.com/ccimg/1500/1477-39-0_b.png)


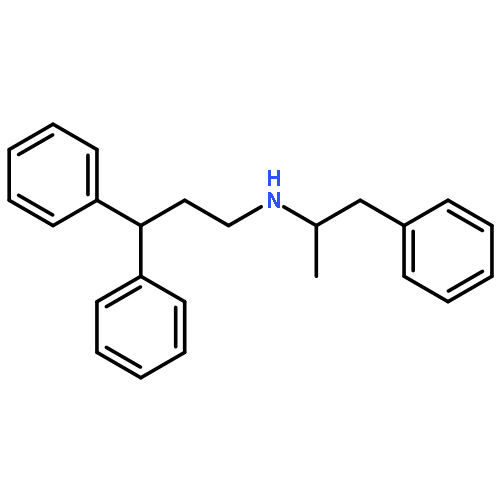
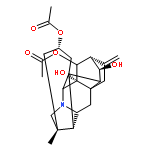
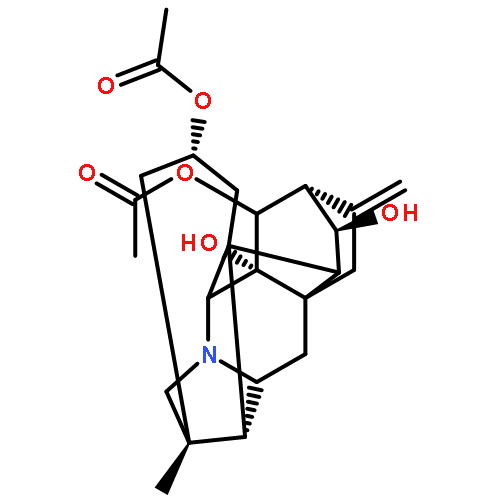
![6-(Trifluoromethyl)-3,4-dihydro-2H-benzo[e][1,2,4]thiadiazine-7-sulfonamide 1,1-dioxide](http://img.cochemist.com/ccimg/200/135-09-1.png)
![6-(Trifluoromethyl)-3,4-dihydro-2H-benzo[e][1,2,4]thiadiazine-7-sulfonamide 1,1-dioxide](http://img.cochemist.com/ccimg/200/135-09-1_b.png)


![Benzeneethanol, b-[(2S)-2-(dimethylamino)propyl]-a-ethyl-b-phenyl-, 1-acetate, (aS)-](http://img.cochemist.com/ccimg/1500/1477-40-3.png)
![Benzeneethanol, b-[(2S)-2-(dimethylamino)propyl]-a-ethyl-b-phenyl-, 1-acetate, (aS)-](http://img.cochemist.com/ccimg/1500/1477-40-3_b.png)
![1,3-BIS{[BIS(2-CHLOROETHYL)AMINO]METHYL}URE](http://img.cochemist.com/ccimg/3400/3376-94-1.png)
![1,3-BIS{[BIS(2-CHLOROETHYL)AMINO]METHYL}URE](http://img.cochemist.com/ccimg/3400/3376-94-1_b.png)



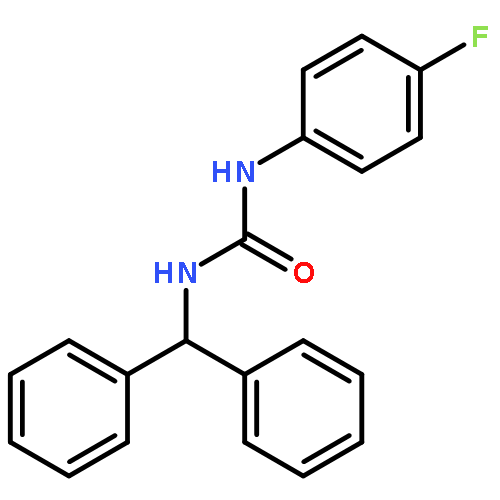












![Dibenz[b,f][1,4]oxazepine](http://img.cochemist.com/ccimg/300/257-07-8.png)
![Dibenz[b,f][1,4]oxazepine](http://img.cochemist.com/ccimg/300/257-07-8_b.png)

![Benzamide,4-amino-N-(1R,4S,5R)-1-azabicyclo[3.3.1]non-4-yl-5-chloro-2-methoxy-, rel-](http://img.cochemist.com/ccimg/112800/112727-80-7.png)
![Benzamide,4-amino-N-(1R,4S,5R)-1-azabicyclo[3.3.1]non-4-yl-5-chloro-2-methoxy-, rel-](http://img.cochemist.com/ccimg/112800/112727-80-7_b.png)
![1H-Indazole-3-carboxamide,N-[(7-endo)-3,9-dimethyl-3,9-diazabicyclo[3.3.1]non-7-yl]-](http://img.cochemist.com/ccimg/141600/141549-75-9.png)
![1H-Indazole-3-carboxamide,N-[(7-endo)-3,9-dimethyl-3,9-diazabicyclo[3.3.1]non-7-yl]-](http://img.cochemist.com/ccimg/141600/141549-75-9_b.png)




![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-, (2S)-](/data/chemimg/55200/4199-09-1.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-, (2S)-](/data/chemimg/55200/4199-09-1_b.png)
![[1,1'-Biphenyl]-2-carboxamide,2'-[[[(4-methoxyphenyl)acetyl]amino]methyl]-N-[2-(3-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/498600/498577-53-0.png)
![[1,1'-Biphenyl]-2-carboxamide,2'-[[[(4-methoxyphenyl)acetyl]amino]methyl]-N-[2-(3-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/498600/498577-53-0_b.png)
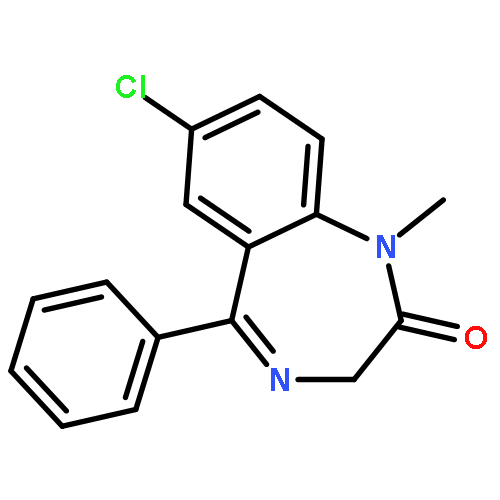


![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7.png)
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7_b.png)
![Benzamide,N-(3,4-dimethoxyphenyl)-N-[3-[[2-(3,4-dimethoxyphenyl)ethyl]methylamino]propyl]-4-nitro-,hydrochloride (1:1)](http://img.cochemist.com/ccimg/113300/113241-47-7.png)
![Benzamide,N-(3,4-dimethoxyphenyl)-N-[3-[[2-(3,4-dimethoxyphenyl)ethyl]methylamino]propyl]-4-nitro-,hydrochloride (1:1)](http://img.cochemist.com/ccimg/113300/113241-47-7_b.png)


![5-chloro-3-[1-((imidazolidin-2-one-1-yl)-ethyl)-1,2,5,6-tetrahydropyridin-4-yl]-1-(4-fluorophenyl)-indole](http://img.cochemist.com/ccimg/106600/106516-54-5.png)
![5-chloro-3-[1-((imidazolidin-2-one-1-yl)-ethyl)-1,2,5,6-tetrahydropyridin-4-yl]-1-(4-fluorophenyl)-indole](http://img.cochemist.com/ccimg/106600/106516-54-5_b.png)

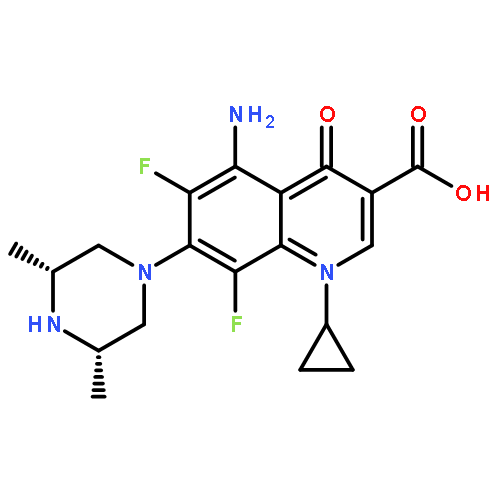




![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7.png)
![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7_b.png)


![3-Quinolinecarboxylicacid,1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-4-oxo-](http://img.cochemist.com/ccimg/354900/354812-41-2.png)
![3-Quinolinecarboxylicacid,1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-4-oxo-](http://img.cochemist.com/ccimg/354900/354812-41-2_b.png)


![10H-Thieno[2,3-b][1,5]benzodiazepine-2-methanol,4-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/174800/174756-45-7.png)
![10H-Thieno[2,3-b][1,5]benzodiazepine-2-methanol,4-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/174800/174756-45-7_b.png)
![N-[4-[[1-[2-(6-METHYL-2-PYRIDINYL)ETHYL]-4-PIPERIDINYL]CARBONYL]PHENYL]METHANESULFONAMIDE DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/113600/113558-89-7.png)
![N-[4-[[1-[2-(6-METHYL-2-PYRIDINYL)ETHYL]-4-PIPERIDINYL]CARBONYL]PHENYL]METHANESULFONAMIDE DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/113600/113558-89-7_b.png)
![2-Methyl-4-(1-piperazinyl)-10H-thienol[2,3-b][1,5]benzodiazepine](http://img.cochemist.com/ccimg/161700/161696-76-0.png)
![2-Methyl-4-(1-piperazinyl)-10H-thienol[2,3-b][1,5]benzodiazepine](http://img.cochemist.com/ccimg/161700/161696-76-0_b.png)














![1-Piperazineacetamide,4-[4,4-bis(4-fluorophenyl)butyl]-N-(2,6-dimethylphenyl)-](http://img.cochemist.com/ccimg/3500/3416-26-0.png)
![1-Piperazineacetamide,4-[4,4-bis(4-fluorophenyl)butyl]-N-(2,6-dimethylphenyl)-](http://img.cochemist.com/ccimg/3500/3416-26-0_b.png)
![10H-Phenothiazine-2-carbonitrile,10-[3-(dimethylamino)-2-methylpropyl]-](http://img.cochemist.com/ccimg/3600/3546-03-0.png)
![10H-Phenothiazine-2-carbonitrile,10-[3-(dimethylamino)-2-methylpropyl]-](http://img.cochemist.com/ccimg/3600/3546-03-0_b.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7_b.png)


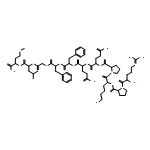
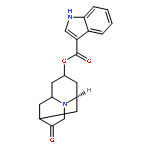
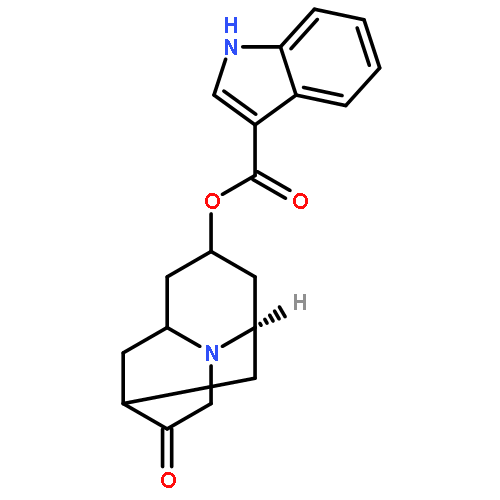
![5-[(2R)-2-[2-(2-ETHOXYPHENOXY)ETHYLAMINO]PROPYL]-2-METHOXYBENZENESULFONAMIDE](/data/chemimg/307000/106133-20-4.png)
![5-[(2R)-2-[2-(2-ETHOXYPHENOXY)ETHYLAMINO]PROPYL]-2-METHOXYBENZENESULFONAMIDE](/data/chemimg/307000/106133-20-4_b.png)








![6-Fluoro-1-methyl-7-(4-((5-methyl-2-oxo-1,3-dioxol-4-yl)methyl)piperazin-1-yl)-4-oxo-1,4-dihydro-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid](http://img.cochemist.com/ccimg/123500/123447-62-1.png)
![6-Fluoro-1-methyl-7-(4-((5-methyl-2-oxo-1,3-dioxol-4-yl)methyl)piperazin-1-yl)-4-oxo-1,4-dihydro-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid](http://img.cochemist.com/ccimg/123500/123447-62-1_b.png)


![(S)-N-Methyl-gamma-[4-(trifluoromethyl)phenoxy]benzenepropanamine](http://img.cochemist.com/ccimg/100600/100568-02-3.png)
![(S)-N-Methyl-gamma-[4-(trifluoromethyl)phenoxy]benzenepropanamine](http://img.cochemist.com/ccimg/100600/100568-02-3_b.png)








![Acetic acid, methoxy-,(1S,2S)-2-[2-[[3-(1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-(1-methylethyl)-2-naphthalenylester](http://img.cochemist.com/ccimg/116700/116644-53-2.png)
![Acetic acid, methoxy-,(1S,2S)-2-[2-[[3-(1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-(1-methylethyl)-2-naphthalenylester](http://img.cochemist.com/ccimg/116700/116644-53-2_b.png)
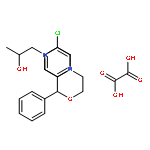





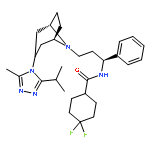


![4H-Dibenzo[de,g]quinoline-10,11-diol,5,6,6a,7-tetrahydro-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/100/58-00-4.png)
![4H-Dibenzo[de,g]quinoline-10,11-diol,5,6,6a,7-tetrahydro-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/100/58-00-4_b.png)




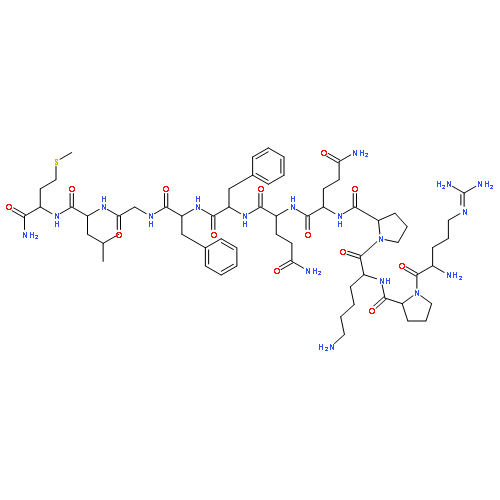
![[pyr1]-apelin-13](http://img.cochemist.com/ccimg/217100/217082-60-5.png)
![[pyr1]-apelin-13](http://img.cochemist.com/ccimg/217100/217082-60-5_b.png)