Co-reporter:Georg Meisl;Xiaoting Yang;Sara Linse;Tuomas P. J. Knowles
Chemical Science (2010-Present) 2017 vol. 8(Issue 6) pp:4352-4362
Publication Date(Web):2017/05/30
DOI:10.1039/C7SC00215G
The aggregation of the amyloid β peptide (Aβ42), which is linked to Alzheimer's disease, can be altered significantly by modulations of the peptide's intermolecular electrostatic interactions. Variations in sequence and solution conditions have been found to lead to highly variable aggregation behaviour. Here we modulate systematically the electrostatic interactions governing the aggregation kinetics by varying the ionic strength of the solution. We find that changes in the solution ionic strength induce a switch in the reaction pathway, altering the dominant mechanisms of aggregate multiplication. This strategy thereby allows us to continuously sample a large space of different reaction mechanisms and develop a minimal reaction network that unifies the experimental kinetics under a wide range of different conditions. More generally, this universal reaction network connects previously separate systems, such as charge mutants of the Aβ42 peptide, on a continuous mechanistic landscape, providing a unified picture of the aggregation mechanism of Aβ42.
Co-reporter:Patrick Flagmeier;Suman De;David C. Wirthensohn;Steven F. Lee;Cécile Vincke;Serge Muyldermans;Tuomas P. J. Knowles;Sonia Ghi;David Klenerman
Angewandte Chemie International Edition 2017 Volume 56(Issue 27) pp:7750-7754
Publication Date(Web):2017/06/26
DOI:10.1002/anie.201700966
AbstractTo quantify and characterize the potentially toxic protein aggregates associated with neurodegenerative diseases, a high-throughput assay based on measuring the extent of aggregate-induced Ca2+ entry into individual lipid vesicles has been developed. This approach was implemented by tethering vesicles containing a Ca2+ sensitive fluorescent dye to a passivated surface and measuring changes in the fluorescence as a result of membrane disruption using total internal reflection microscopy. Picomolar concentrations of Aβ42 oligomers could be observed to induce Ca2+ influx, which could be inhibited by the addition of a naturally occurring chaperone and a nanobody designed to bind to the Aβ peptide. We show that the assay can be used to study aggregates from other proteins, such as α-synuclein, and to probe the effects of complex biofluids, such as cerebrospinal fluid, and thus has wide applicability.
Co-reporter:Patrick Flagmeier;Suman De;David C. Wirthensohn;Steven F. Lee;Cécile Vincke;Serge Muyldermans;Tuomas P. J. Knowles;Sonia Ghi;David Klenerman
Angewandte Chemie 2017 Volume 129(Issue 27) pp:7858-7862
Publication Date(Web):2017/06/26
DOI:10.1002/ange.201700966
AbstractTo quantify and characterize the potentially toxic protein aggregates associated with neurodegenerative diseases, a high-throughput assay based on measuring the extent of aggregate-induced Ca2+ entry into individual lipid vesicles has been developed. This approach was implemented by tethering vesicles containing a Ca2+ sensitive fluorescent dye to a passivated surface and measuring changes in the fluorescence as a result of membrane disruption using total internal reflection microscopy. Picomolar concentrations of Aβ42 oligomers could be observed to induce Ca2+ influx, which could be inhibited by the addition of a naturally occurring chaperone and a nanobody designed to bind to the Aβ peptide. We show that the assay can be used to study aggregates from other proteins, such as α-synuclein, and to probe the effects of complex biofluids, such as cerebrospinal fluid, and thus has wide applicability.
Co-reporter:Georg Meisl;Luke Rajah;Samuel A. I. Cohen;Manuela Pfammatter;Anđela Šarić;Erik Hellstrand;Alexander K. Buell;Adriano Aguzzi;Sara Linse;Michele Vendruscolo;Tuomas P. J. Knowles
Chemical Science (2010-Present) 2017 vol. 8(Issue 10) pp:7087-7097
Publication Date(Web):2017/09/25
DOI:10.1039/C7SC01965C
The formation of filaments from naturally occurring protein molecules is a process at the core of a range of functional and aberrant biological phenomena, such as the assembly of the cytoskeleton or the appearance of aggregates in Alzheimer's disease. The macroscopic behaviour associated with such processes is remarkably diverse, ranging from simple nucleated growth to highly cooperative processes with a well-defined lagtime. Thus, conventionally, different molecular mechanisms have been used to explain the self-assembly of different proteins. Here we show that this range of behaviour can be quantitatively captured by a single unifying Petri net that describes filamentous growth in terms of aggregate number and aggregate mass concentrations. By considering general features associated with a particular network connectivity, we are able to establish directly the rate-determining steps of the overall aggregation reaction from the system's scaling behaviour. We illustrate the power of this framework on a range of different experimental and simulated aggregating systems. The approach is general and will be applicable to any future extensions of the reaction network of filamentous self-assembly.
Co-reporter:Michele Perni;Martin B. D. Müller;Julius B. Kirkegaard;Roberta Cascella;Patrick Flagmeier;Serene W. Chen;Céline Galvagnion;Cristina Cecchi;Georg Meisl;Pavan K. Challa;Ryan Limbocker;Samuel I. A. Cohen;Fabrizio Chiti;Francesco A. Aprile;Pietro Sormanni;Alexander Maltsev;Nunilo Cremades;Michele Vendruscolo;Ellen A. A. Nollen;Tuomas P. J. Knowles;Adriaan Bax;Michael Zasloff;Gabriella T. Heller
PNAS 2017 Volume 114 (Issue 6 ) pp:E1009-E1017
Publication Date(Web):2017-02-07
DOI:10.1073/pnas.1610586114
The self-assembly of α-synuclein is closely associated with Parkinson’s disease and related syndromes. We show that squalamine,
a natural product with known anticancer and antiviral activity, dramatically affects α-synuclein aggregation in vitro and
in vivo. We elucidate the mechanism of action of squalamine by investigating its interaction with lipid vesicles, which are
known to stimulate nucleation, and find that this compound displaces α-synuclein from the surfaces of such vesicles, thereby
blocking the first steps in its aggregation process. We also show that squalamine almost completely suppresses the toxicity
of α-synuclein oligomers in human neuroblastoma cells by inhibiting their interactions with lipid membranes. We further examine
the effects of squalamine in a Caenorhabditis elegans strain overexpressing α-synuclein, observing a dramatic reduction of α-synuclein aggregation and an almost complete elimination
of muscle paralysis. These findings suggest that squalamine could be a means of therapeutic intervention in Parkinson’s disease
and related conditions.
Co-reporter:Marija Iljina;Liu Hong;Mathew H. Horrocks;Marthe H. Ludtmann
BMC Biology 2017 Volume 15( Issue 1) pp:57
Publication Date(Web):03 July 2017
DOI:10.1186/s12915-017-0390-6
The aggregation of the protein ɑ-synuclein (ɑS) underlies a range of increasingly common neurodegenerative disorders including Parkinson’s disease. One widely explored therapeutic strategy for these conditions is the use of antibodies to target aggregated ɑS, although a detailed molecular-level mechanism of the action of such species remains elusive. Here, we characterize ɑS aggregation in vitro in the presence of two ɑS-specific single-domain antibodies (nanobodies), NbSyn2 and NbSyn87, which bind to the highly accessible C-terminal region of ɑS.We show that both nanobodies inhibit the formation of ɑS fibrils. Furthermore, using single-molecule fluorescence techniques, we demonstrate that nanobody binding promotes a rapid conformational conversion from more stable oligomers to less stable oligomers of ɑS, leading to a dramatic reduction in oligomer-induced cellular toxicity.The results indicate a novel mechanism by which diseases associated with protein aggregation can be inhibited, and suggest that NbSyn2 and NbSyn87 could have significant therapeutic potential.
Co-reporter:Johnny Habchi;Oskar Hansson;Sean Chia;Janet R. Kumita;Samuel I. A. Cohen;Michele Perni;Pavan Kumar Challa;Tuomas P. J. Knowles;Paolo Arosio;Michele Vendruscolo;Minkoo Ahn;Ryan Limbocker;Benedetta Mannini;Sara Linse
PNAS 2017 Volume 114 (Issue 2 ) pp:E200-E208
Publication Date(Web):2017-01-10
DOI:10.1073/pnas.1615613114
The aggregation of the 42-residue form of the amyloid-β peptide (Aβ42) is a pivotal event in Alzheimer’s disease (AD). The
use of chemical kinetics has recently enabled highly accurate quantifications of the effects of small molecules on specific
microscopic steps in Aβ42 aggregation. Here, we exploit this approach to develop a rational drug discovery strategy against
Aβ42 aggregation that uses as a read-out the changes in the nucleation and elongation rate constants caused by candidate small
molecules. We thus identify a pool of compounds that target specific microscopic steps in Aβ42 aggregation. We then test further
these small molecules in human cerebrospinal fluid and in a Caenorhabditis elegans model of AD. Our results show that this strategy represents a powerful approach to identify systematically small molecule
lead compounds, thus offering an appealing opportunity to reduce the attrition problem in drug discovery.
Co-reporter:Farah El-Turk, Francisco N. Newby, Erwin De Genst, Tim Guilliams, Tara Sprules, Anthony Mittermaier, Christopher M. Dobson, and Michele Vendruscolo
Biochemistry 2016 Volume 55(Issue 22) pp:3116-3122
Publication Date(Web):April 20, 2016
DOI:10.1021/acs.biochem.6b00149
α-Synuclein is an intrinsically disordered protein whose aggregation is associated with Parkinson’s disease and other related neurodegenerative disorders. Recently, two single-domain camelid antibodies (nanobodies) were shown to bind α-synuclein with high affinity. Herein, we investigated how these two nanobodies (NbSyn2 and NbSyn87), which are directed to two distinct epitopes within the C-terminal domain of α-synuclein, affect the conformational properties of this protein. Our results suggest that nanobody NbSyn2, which binds to the five C-terminal residues of α-synuclein (residues 136–140), does not disrupt the transient long-range interactions that generate a degree of compaction within the native structural ensemble of α-synuclein. In contrast, the data that we report indicate that NbSyn87, which targets a central region within the C-terminal domain (residues 118–128), has more substantial effects on the fluctuating secondary and tertiary structure of the protein. These results are consistent with the different effects that the two nanobodies have on the aggregation behavior of α-synuclein in vitro. Our findings thus provide new insights into the type of effects that nanobodies can have on the conformational ensemble of α-synuclein.
Co-reporter:Martin B. D. Müller;Ana Rita Costa;Maho Yagi-Utsumi;Priyanka Joshi;Johnny Habchi;Sara Linse;Samuel I. A. Cohen;Tuomas P. J. Knowles;Paolo Arosio;Ellen A. A. Nollen;Michele Vendruscolo;Michele Perni;Sean Chia
Science Advances 2016 Volume 2(Issue 2) pp:e1501244
Publication Date(Web):12 Feb 2016
DOI:10.1126/sciadv.1501244
An approved anticancer drug selectively targets the first step in the molecular cascade resulting in Alzheimer’s disease.
Co-reporter:James W. P. Brown;Céline Galvagnion;Patrick Flagmeier;Myriam M. Ouberai;Emma Sparr;Michele Vendruscolo;Alexander K. Buell
PNAS 2016 Volume 113 (Issue 26 ) pp:7065-7070
Publication Date(Web):2016-06-28
DOI:10.1073/pnas.1601899113
Intracellular α-synuclein deposits, known as Lewy bodies, have been linked to a range of neurodegenerative disorders, including
Parkinson’s disease. α-Synuclein binds to synthetic and biological lipids, and this interaction has been shown to play a crucial
role for both α-synuclein’s native function, including synaptic plasticity, and the initiation of its aggregation. Here, we
describe the interplay between the lipid properties and the lipid binding and aggregation propensity of α-synuclein. In particular,
we have observed that the binding of α-synuclein to model membranes is much stronger when the latter is in the fluid rather
than the gel phase, and that this binding induces a segregation of the lipids into protein-poor and protein-rich populations.
In addition, α-synuclein was found to aggregate at detectable rates only when interacting with membranes composed of the most
soluble lipids investigated here. Overall, our results show that the chemical properties of lipids determine whether or not
the lipids can trigger the aggregation of α-synuclein, thus affecting the balance between functional and aberrant behavior
of the protein.
Co-reporter:Emma V. Yates, Georg Meisl, Tuomas P. J. Knowles, and Christopher M. Dobson
The Journal of Physical Chemistry B 2016 Volume 120(Issue 9) pp:2087-2094
Publication Date(Web):February 11, 2016
DOI:10.1021/acs.jpcb.5b09663
We have explored amyloid formation using poly(amino acid) model systems in which differences in peptide secondary structure and hydrophobicity can be introduced in a controlled manner. We show that an environmentally sensitive fluorescent dye, dapoxyl, is able to identify β-sheet structure and hydrophobic surfaces, structural features likely to be related to toxicity, as a result of changes in its excitation and emission profiles and its relative quantum yield. These results show that dapoxyl is a multidimensional probe of the time dependence of amyloid aggregation, which provides information about the presence and nature of metastable aggregation intermediates that is inaccessible to the conventional probes that rely on changes in quantum yield alone.
Co-reporter:Therese W. Herling;Gonzalo A. Garcia;Wolfgang Grentz;James Dean;Thomas C. T. Michaels;Ulyana Shimanovich;Hongze Gang;Eugene M. Terentjev;Thomas Müller;Batuhan Kav;Tuomas P. J. Knowles
PNAS 2015 Volume 112 (Issue 31 ) pp:9524-9529
Publication Date(Web):2015-08-04
DOI:10.1073/pnas.1417326112
The generation of mechanical forces are central to a wide range of vital biological processes, including the function of the
cytoskeleton. Although the forces emerging from the polymerization of native proteins have been studied in detail, the potential
for force generation by aberrant protein polymerization has not yet been explored. Here, we show that the growth of amyloid
fibrils, archetypical aberrant protein polymers, is capable of unleashing mechanical forces on the piconewton scale for individual
filaments. We apply microfluidic techniques to measure the forces released by amyloid growth for two systems: insulin and
lysozyme. The level of force measured for amyloid growth in both systems is comparable to that observed for actin and tubulin,
systems that have evolved to generate force during their native functions and, unlike amyloid growth, rely on the input of
external energy in the form of nucleotide hydrolysis for maximum force generation. Furthermore, we find that the power density
released from growing amyloid fibrils is comparable to that of high-performance synthetic polymer actuators. These findings
highlight the potential of amyloid structures as active materials and shed light on the criteria for regulation and reversibility
that guide molecular evolution of functional polymers.
Co-reporter:Ulyana Shimanovich, Igor Efimov, Thomas O. Mason, Patrick Flagmeier, Alexander K. Buell, Aharon Gedanken, Sara Linse, Karin S. Åkerfeldt, Christopher M. Dobson, David A. Weitz, and Tuomas P. J. Knowles
ACS Nano 2015 Volume 9(Issue 1) pp:43
Publication Date(Web):December 3, 2014
DOI:10.1021/nn504869d
Nanofibrillar forms of proteins were initially recognized in the context of pathology, but more recently have been discovered in a range of functional roles in nature, including as active catalytic scaffolds and bacterial coatings. Here we show that protein nanofibrils can be used to form the basis of monodisperse microgels and gel shells composed of naturally occurring proteins. We explore the potential of these protein microgels to act as drug carrier agents, and demonstrate the controlled release of four different encapsulated drug-like small molecules, as well as the component proteins themselves. Furthermore, we show that protein nanofibril self-assembly can continue after the initial formation of the microgel particles, and that this process results in active materials with network densities that can be modulated in situ. We demonstrate that these materials are nontoxic to human cells and that they can be used to enhance the efficacy of antibiotics relative to delivery in homogeneous solution. Because of the biocompatibility and biodegradability of natural proteins used in the fabrication of the microgels, as well as their ability to control the release of small molecules and biopolymers, protein nanofibril microgels represent a promising class of functional artificial multiscale materials generated from natural building blocks.Keywords: drug release; lysozyme; microfluidics; microgels; protein nanofibrils;
Co-reporter:Serene W. Chen;Samuel Ness;Francesco A. Aprile;Erwin J. De-Genst;Emma Deas;Myriam Ouberai;Srdja Drakulic;David Klenerman;Rocío Arranz;Tim Guilliams;Carlos Alfonso;Germán Rivas;Andrey Y. Abramov;Nicholas W. Wood;Tuomas P.J. Knowles;Nunilo Cremades;José María Valpuesta;Cintia Roodveldt
PNAS 2015 Volume 112 (Issue 16 ) pp:E1994-E2003
Publication Date(Web):2015-04-21
DOI:10.1073/pnas.1421204112
We describe the isolation and detailed structural characterization of stable toxic oligomers of α-synuclein that have accumulated
during the process of amyloid formation. Our approach has allowed us to identify distinct subgroups of oligomers and to probe
their molecular architectures by using cryo-electron microscopy (cryoEM) image reconstruction techniques. Although the oligomers
exist in a range of sizes, with different extents and nature of β-sheet content and exposed hydrophobicity, they all possess
a hollow cylindrical architecture with similarities to certain types of amyloid fibril, suggesting that the accumulation of
at least some forms of amyloid oligomers is likely to be a consequence of very slow rates of rearrangement of their β-sheet
structures. Our findings reveal the inherent multiplicity of the process of protein misfolding and the key role the β-sheet
geometry acquired in the early stages of the self-assembly process plays in dictating the kinetic stability and the pathological
nature of individual oligomeric species.
Co-reporter:Christopher M. Dobson
Rendiconti Lincei 2015 Volume 26( Issue 3) pp:251-262
Publication Date(Web):2015 September
DOI:10.1007/s12210-015-0453-y
Alzheimer’s disease represents one of the greatest challenges to the social fabric and health care systems of the world since the great plagues of the Middle Ages. At the present time, there are some 40 million sufferers from this disease worldwide, but it is predicted that this number will rise to nearly 150 million by 2050. The predominant reason for this rapidly increasing prevalence is the increase in longevity that has resulted from the tremendous advances in public health and hygiene and in medical and surgical interventions over the last century. This article discusses recent progress in our understanding of the molecular origins of Alzheimer’s disease and emerging ideas for new and rational therapeutic strategies by which to combat its onset and progression.
Co-reporter:Edward P. O’Brien, Prajwal Ciryam, Michele Vendruscolo, and Christopher M. Dobson
Accounts of Chemical Research 2014 Volume 47(Issue 5) pp:1536
Publication Date(Web):May 1, 2014
DOI:10.1021/ar5000117
Protein domains can fold into stable tertiary structures while they are synthesized by the ribosome in a process known as cotranslational folding. If a protein does not fold cotranslationally, however, it has the opportunity to do so post-translationally, that is, after the nascent chain has been fully synthesized and released from the ribosome. The rate at which a ribosome adds an amino acid encoded by a particular codon to the elongating nascent chain can vary significantly and is called the codon translation rate. Recent experiments have illustrated the profound impact that codon translation rates can have on the cotranslational folding process and the acquisition of function by nascent proteins. Synonymous codon mutations in an mRNA molecule change the chemical identity of a codon and its translation rate without changing the sequence of the synthesized protein. This change in codon translation rate can, however, cause a nascent protein to malfunction as a result of cotranslational misfolding. In some situations, such dysfunction can have profound implications; for example, it can alter the substrate specificity of an ABC transporter protein, resulting in patients who are nonresponsive to chemotherapy treatment. Thus, codon translation rates are crucial in coordinating protein folding in a cellular environment and can affect downstream cellular processes that depend on the proper functioning of newly synthesized proteins. As the importance of codon translation rates makes clear, a necessary aspect of fully understanding cotranslational folding lies in considering the kinetics of the process in addition to its thermodynamics.In this Account, we examine the contributions that have been made to elucidating the mechanisms of cotranslational folding by using the theoretical and computational tools of chemical kinetics, molecular simulations, and systems biology. These efforts have extended our ability to understand, model, and predict the influence of codon translation rates on cotranslational protein folding and misfolding. The application of such approaches to this important problem is creating a framework for making quantitative predictions of the impact of synonymous codon substitutions on cotranslational folding that has led to a novel hypothesis regarding the role of fast-translating codons in coordinating cotranslational folding. In addition, it is providing new insights into proteome-wide cotranslational folding behavior and making it possible to identify potential molecular mechanisms by which molecular chaperones can influence such behavior during protein synthesis. As we discuss in this Account, bringing together these theoretical developments with experimental approaches is increasingly helping answer fundamental questions about the nature of nascent protein folding on the ribosome.
Co-reporter:Benedetta Bolognesi, Samuel I. A. Cohen, Pablo Aran Terol, Elin K. Esbjörner, Sofia Giorgetti, Maria F. Mossuto, Antonino Natalello, Ann-Christin Brorsson, Tuomas P. J. Knowles, Christopher M. Dobson, and Leila M. Luheshi
ACS Chemical Biology 2014 Volume 9(Issue 2) pp:378
Publication Date(Web):November 7, 2013
DOI:10.1021/cb400616y
Single point mutations in the Alzheimer’s disease associated Aβ42 peptide are found to alter significantly its neurotoxic properties in vivo and have been associated with early onset forms of this devastating condition. We show that such mutations can induce structural changes in Aβ42 fibrils and are associated with a dramatic switch in the fibril-dependent mechanism by which Aβ42 aggregates. These observations reveal how subtle perturbations to the physicochemical properties of the Aβ peptide, and the structural properties of fibrils that it forms, can have profound effects on the mechanism of its aggregation and pathogenicity.
Co-reporter:Jane R. Allison, Robert C. Rivers, John C. Christodoulou, Michele Vendruscolo, and Christopher M. Dobson
Biochemistry 2014 Volume 53(Issue 46) pp:
Publication Date(Web):October 28, 2014
DOI:10.1021/bi5009326
α-Synuclein is an intrinsically disordered protein whose aggregation is implicated in Parkinson’s disease. A second member of the synuclein family, β-synuclein, shares significant sequence similarity with α-synuclein but is much more resistant to aggregation. β-Synuclein is missing an 11-residue stretch in the central non-β-amyloid component region that forms the core of α-synuclein amyloid fibrils, yet insertion of these residues into β-synuclein to produce the βSHC construct does not markedly increase the aggregation propensity. To investigate the structural basis of these different behaviors, quantitative nuclear magnetic resonance data, in the form of paramagnetic relaxation enhancement-derived interatomic distances, are combined with molecular dynamics simulations to generate ensembles of structures representative of the solution states of α-synuclein, β-synuclein, and βSHC. Comparison of these ensembles reveals that the differing aggregation propensities of α-synuclein and β-synuclein are associated with differences in the degree of residual structure in the C-terminus coupled to the shorter separation between the N- and C-termini in β-synuclein and βSHC, making protective intramolecular contacts more likely.
Co-reporter:Alexander K. Buell;Tuomas P. J. Knowles;Michele Vendruscolo;Céline Galvagnion;Emma Sparr;Sara Linse;Ricardo Gaspar
PNAS 2014 Volume 111 (Issue 21 ) pp:7671-7676
Publication Date(Web):2014-05-27
DOI:10.1073/pnas.1315346111
The formation of amyloid fibrils by the intrinsically disordered protein α-synuclein is a hallmark of Parkinson disease. To
characterize the microscopic steps in the mechanism of aggregation of this protein we have used in vitro aggregation assays
in the presence of preformed seed fibrils to determine the molecular rate constant of fibril elongation under a range of different
conditions. We show that α-synuclein amyloid fibrils grow by monomer and not oligomer addition and are subject to higher-order
assembly processes that decrease their capacity to grow. We also find that at neutral pH under quiescent conditions homogeneous
primary nucleation and secondary processes, such as fragmentation and surface-assisted nucleation, which can lead to proliferation
of the total number of aggregates, are undetectable. At pH values below 6, however, the rate of secondary nucleation increases
dramatically, leading to a completely different balance between the nucleation and growth of aggregates. Thus, at mildly acidic
pH values, such as those, for example, that are present in some intracellular locations, including endosomes and lysosomes,
multiplication of aggregates is much faster than at normal physiological pH values, largely as a consequence of much more
rapid secondary nucleation. These findings provide new insights into possible mechanisms of α-synuclein aggregation and aggregate
spreading in the context of Parkinson disease.
Co-reporter:Priyanka Narayan ; Kristina A. Ganzinger ; James McColl ; Laura Weimann ; Sarah Meehan ; Seema Qamar ; John A. Carver ̂; Mark R. Wilson ; Peter St. George-Hyslop ; Christopher M. Dobson ;David Klenerman
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1491-1498
Publication Date(Web):January 22, 2013
DOI:10.1021/ja3103567
Oligomers of the 40 and 42 residue amyloid-β peptides (Aβ40 and Aβ42) have been implicated in the neuronal damage and impaired cognitive function associated with Alzheimer’s disease. However, little is known about the specific mechanisms by which these misfolded species induce such detrimental effects on cells. In this work, we use single-molecule imaging techniques to examine the initial interactions between Aβ monomers and oligomers and the membranes of live cells. This highly sensitive method enables the visualization of individual Aβ species on the cell surface and characterization of their oligomerization state, all at biologically relevant, nanomolar concentrations. The results indicate that oligomers preferentially interact with cell membranes, relative to monomers and that the oligomers become immobilized on the cell surface. Additionally, we observe that the interaction of Aβ species with the cell membrane is inhibited by the presence of ATP-independent molecular chaperones. This study demonstrates the power of this methodology for characterizing the interactions between protein aggregates and the membranes of live neuronal cells at physiologically relevant concentrations and opens the door to quantitative studies of the cellular responses to potentially pathogenic oligomers.
Co-reporter:Erwin De Genst, Pak-Ho Chan, Els Pardon, Shang-Te D. Hsu, Janet R. Kumita, John Christodoulou, Linda Menzer, Dimitri Y. Chirgadze, Carol V. Robinson, Serge Muyldermans, André Matagne, Lode Wyns, Christopher M. Dobson, and Mireille Dumoulin
The Journal of Physical Chemistry B 2013 Volume 117(Issue 42) pp:13245-13258
Publication Date(Web):August 6, 2013
DOI:10.1021/jp403425z
We report the effects of the interaction of two camelid antibody fragments, generally called nanobodies, namely cAb-HuL5 and a stabilized and more aggregation-resistant variant cAb-HuL5G obtained by protein engineering, on the properties of two amyloidogenic variants of human lysozyme, I56T and D67H, whose deposition in vital organs including the liver, kidney, and spleen is associated with a familial non-neuropathic systemic amyloidosis. Both NMR spectroscopy and X-ray crystallographic studies reveal that cAb-HuL5 binds to the α-domain, one of the two lobes of the native lysozyme structure. The binding of cAb-HuL5/cAb-HuL5G strongly inhibits fibril formation by the amyloidogenic variants; it does not, however, suppress the locally transient cooperative unfolding transitions, characteristic of these variants, in which the β-domain and the C-helix unfold and which represents key early intermediate species in the formation of amyloid fibrils. Therefore, unlike two other nanobodies previously described, cAb-HuL5/cAb-HuL5G does not inhibit fibril formation via the restoration of the global cooperativity of the native structure of the lysozyme variants to that characteristic of the wild-type protein. Instead, it inhibits a subsequent step in the assembly of the fibrils, involving the unfolding and structural reorganization of the α-domain. These results show that nanobodies can protect against the formation of pathogenic aggregates at different stages in the structural transition of a protein from the soluble native state into amyloid fibrils, illustrating their value as structural probes to study the molecular mechanisms of amyloid fibril formation. Combined with their amenability to protein engineering techniques to improve their stability and solubility, these findings support the suggestion that nanobodies can potentially be developed as therapeutics to combat protein misfolding diseases.
Co-reporter:Marvin J. Bayro;Daniel K. Clare;Anthony W. P. Fitzpatrick;Luchun Wang;Galia T. Debelouchina;Christopher P. Jaroniec;Marc A. Caporini;Vikram S. Bajaj;Vladimir Ladizhansky;Helen R. Mott;Helen R. Saibil;Robert G. Griffin;Tuomas P. J. Knowles;Shirley A. Müller;Alfonso De Simone;Cait E. MacPhee;Michele Vendruscolo;Elena V. Orlova;Christopher A. Waudby
PNAS 2013 Volume 110 (Issue 14 ) pp:5468-5473
Publication Date(Web):2013-04-02
DOI:10.1073/pnas.1219476110
The cross-β amyloid form of peptides and proteins represents an archetypal and widely accessible structure consisting of ordered
arrays of β-sheet filaments. These complex aggregates have remarkable chemical and physical properties, and the conversion
of normally soluble functional forms of proteins into amyloid structures is linked to many debilitating human diseases, including
several common forms of age-related dementia. Despite their importance, however, cross-β amyloid fibrils have proved to be
recalcitrant to detailed structural analysis. By combining structural constraints from a series of experimental techniques
spanning five orders of magnitude in length scale—including magic angle spinning nuclear magnetic resonance spectroscopy,
X-ray fiber diffraction, cryoelectron microscopy, scanning transmission electron microscopy, and atomic force microscopy—we
report the atomic-resolution (0.5 Å) structures of three amyloid polymorphs formed by an 11-residue peptide. These structures
reveal the details of the packing interactions by which the constituent β-strands are assembled hierarchically into protofilaments,
filaments, and mature fibrils.
Co-reporter:Edward P. O’Brien ; John Christodoulou ; Michele Vendruscolo
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:10920-10932
Publication Date(Web):June 8, 2012
DOI:10.1021/ja302305u
The E. coli chaperone trigger factor (TF) interacts directly with nascent polypeptide chains as they emerge from the ribosome exit tunnel. Small protein domains can fold under the cradle created by TF, but the co-translational folding of larger proteins is slowed down by its presence. Because of the great experimental challenges in achieving high spatial and time resolution, it is not yet known whether or not TF alters the folding properties of small proteins and if the reduced rate of folding of larger proteins is the result of kinetic or thermodynamic effects. We show, by molecular simulations employing a coarse-grained model of a series of ribosome nascent-chain complexes, that TF does not alter significantly the co-translational folding process of a small protein G domain but delays that of a large β-galactosidase domain as a result of kinetic trapping of its unfolded ensemble. We demonstrate that this trapping occurs through a combination of three distinct mechanisms: a decrease in the rate of structural rearrangements within the nascent chain, an increase in the effective exit tunnel length due to folding outside the cradle, and entanglement of the nascent chain with TF. We present evidence that this TF-induced trapping represents a trade-off between promoting co-translational folding and sterically shielding the nascent chain from aberrant cytosolic interactions that could lead to its aggregation or degradation.
Co-reporter:Priyanka Narayan, Sarah Meehan, John A. Carver, Mark R. Wilson, Christopher M. Dobson, and David Klenerman
Biochemistry 2012 Volume 51(Issue 46) pp:
Publication Date(Web):October 29, 2012
DOI:10.1021/bi301277k
The aberrant aggregation of the amyloid-β peptide into β-sheet rich, fibrillar structures proceeds via a heterogeneous ensemble of oligomeric intermediates that have been associated with neurotoxicity in Alzheimer’s disease (AD). Of particular interest in this context are the mechanisms by which molecular chaperones, part of the primary biological defenses against protein misfolding, influence Aβ aggregation. We have used single-molecule fluorescence techniques to compare the interactions between distinct aggregation states (monomers, oligomers, and amyloid fibrils) of the AD-associated amyloid-β(1–40) peptide, and two molecular chaperones, both of which are upregulated in the brains of patients with AD and have been found colocalized with Aβ in senile plaques. One of the chaperones, αB-crystallin, is primarily found inside cells, while the other, clusterin, is predominantly located in the extracellular environment. We find that both chaperones bind to misfolded oligomeric species and form long-lived complexes, thereby preventing both their further growth into fibrils and their dissociation. From these studies, we conclude that these chaperones have a common mechanism of action based on sequestering Aβ oligomers. This conclusion suggests that these chaperones, both of which are ATP-independent, are able to inhibit potentially pathogenic Aβ oligomer-associated processes whether they occur in the extracellular or intracellular environment.
Co-reporter:Alexer K. Buell;Anne Dhulesia;Duncan A. White;Tuomas P. J. Knowles;Mark E. Well
Angewandte Chemie 2012 Volume 124( Issue 21) pp:5339-5344
Publication Date(Web):
DOI:10.1002/ange.201108040
Co-reporter:Alexer K. Buell;Anne Dhulesia;Duncan A. White;Tuomas P. J. Knowles;Mark E. Well
Angewandte Chemie International Edition 2012 Volume 51( Issue 21) pp:5247-5251
Publication Date(Web):
DOI:10.1002/anie.201108040
Co-reporter:Edward P. O’Brien ; John Christodoulou ; Michele Vendruscolo
Journal of the American Chemical Society 2011 Volume 133(Issue 3) pp:513-526
Publication Date(Web):January 4, 2011
DOI:10.1021/ja107863z
Identifying and understanding the differences between protein folding in bulk solution and in the cell is a crucial challenge facing biology. Using Langevin dynamics, we have simulated intact ribosomes containing five different nascent chains arrested at different stages of their synthesis such that each nascent chain can fold and unfold at or near the exit tunnel vestibule. We find that the native state is destabilized close to the ribosome surface due to an increase in unfolded state entropy and a decrease in native state entropy; the former arises because the unfolded ensemble tends to behave as an expanded random coil near the ribosome and a semicompact globule in bulk solution. In addition, the unfolded ensemble of the nascent chain adopts a highly anisotropic shape near the ribosome surface and the cooperativity of the folding-unfolding transition is decreased due to the appearance of partially folded structures that are not populated in bulk solution. The results show, in light of these effects, that with increasing nascent chain length folding rates increase in a linear manner and unfolding rates decrease, with larger and topologically more complex folds being the most highly perturbed by the ribosome. Analysis of folding trajectories, initiated by temperature quench, reveals the transition state ensemble is driven toward compaction and greater native-like structure by interactions with the ribosome surface and exit vestibule. Furthermore, the diversity of folding pathways decreases and the probability increases of initiating folding via the N-terminus on the ribosome. We show that all of these findings are equally applicable to the situation in which protein folding occurs during continuous (non-arrested) translation provided that the time scales of folding and unfolding are much faster than the time scale of monomer addition to the growing nascent chain, which results in a quasi-equilibrium process. These substantial ribosome-induced perturbations to almost all aspects of protein folding indicate that folding scenarios that are distinct from those of bulk solution can occur on the ribosome.
Co-reporter:Alexander K. Buell ; Anne Dhulesia ; Maria F. Mossuto ; Nunilo Cremades ; Janet R. Kumita ; Mireille Dumoulin ; Mark E. Welland ; Tuomas P. J. Knowles ; Xavier Salvatella ▽
Journal of the American Chemical Society 2011 Volume 133(Issue 20) pp:7737-7743
Publication Date(Web):April 29, 2011
DOI:10.1021/ja109620d
The propensity of protein molecules to self-assemble into highly ordered, fibrillar aggregates lies at the heart of understanding many disorders ranging from Alzheimer’s disease to systemic lysozyme amyloidosis. In this paper we use highly accurate kinetic measurements of amyloid fibril growth in combination with spectroscopic tools to quantify the effect of modifications in solution conditions and in the amino acid sequence of human lysozyme on its propensity to form amyloid fibrils under acidic conditions. We elucidate and quantify the correlation between the rate of amyloid growth and the population of nonnative states, and we show that changes in amyloidogenicity are almost entirely due to alterations in the stability of the native state, while other regions of the global free-energy surface remain largely unmodified. These results provide insight into the complex dynamics of a macromolecule on a multidimensional energy landscape and point the way for a better understanding of amyloid diseases.
Co-reporter:Andrew J. Baldwin ; Tuomas P. J. Knowles ; Gian Gaetano Tartaglia ; Anthony W. Fitzpatrick ; Glyn L. Devlin ; Sarah Lucy Shammas ; Christopher A. Waudby ; Maria F. Mossuto ; Sarah Meehan ; Sally L. Gras ; John Christodoulou ; Spencer J. Anthony-Cahill ; Paul D. Barker ; Michele Vendruscolo
Journal of the American Chemical Society 2011 Volume 133(Issue 36) pp:14160-14163
Publication Date(Web):June 8, 2011
DOI:10.1021/ja2017703
An experimental determination of the thermodynamic stabilities of a series of amyloid fibrils reveals that this structural form is likely to be the most stable one that protein molecules can adopt even under physiological conditions. This result challenges the conventional assumption that functional forms of proteins correspond to the global minima in their free energy surfaces and suggests that living systems are conformationally as well as chemically metastable.
Co-reporter:Dr. Gabriele S. Kaminski Schierle;Dr. Carlos W. Bertoncini;Fiona T. S. Chan;Annemieke T. van der Goot;Dr. Stefanie Schwedler;Dr. Jeremy Skepper;Dr. Simon Schlachter;Dr. Tjakko van Ham;Dr. Alessro Esposito;Dr. Janet R. Kumita;Dr. Ellen A. A. Nollen; Christopher M. Dobson;Dr. Clemens F. Kaminski
ChemPhysChem 2011 Volume 12( Issue 3) pp:673-680
Publication Date(Web):
DOI:10.1002/cphc.201000996
Abstract
Misfolding and aggregation of amyloidogenic polypeptides lie at the root of many neurodegenerative diseases. Whilst protein aggregation can be readily studied in vitro by established biophysical techniques, direct observation of the nature and kinetics of aggregation processes taking place in vivo is much more challenging. We describe here, however, a Förster resonance energy transfer sensor that permits the aggregation kinetics of amyloidogenic proteins to be quantified in living systems by exploiting our observation that amyloid assemblies can act as energy acceptors for variants of fluorescent proteins. The observed lifetime reduction can be attributed to fluorescence energy transfer to intrinsic energy states associated with the growing amyloid species. Indeed, for α-synuclein, a protein whose aggregation is linked to Parkinson′s disease, we have used this sensor to follow the kinetics of the self-association reactions taking place in vitro and in vivo and to reveal the nature of the ensuing aggregated species. Experiments were conducted in vitro, in cells in culture and in living Caenorhabditis elegans. For the latter the readout correlates directly with the appearance of a toxic phenotype. The ability to measure the appearance and development of pathogenic amyloid species in a living animal and the ability to relate such data to similar processes observed in vitro provides a powerful new tool in the study of the pathology of the family of misfolding disorders. Our study confirms the importance of the molecular environment in which aggregation reactions take place, highlighting similarities as well as differences between the processes occurring in vitro and in vivo, and their significance for defining the molecular physiology of the diseases with which they are associated.
Co-reporter:Jeremy J. Agresti;Tuomas P. J. Knowles;Ralph A. Sperling;Duncan A. White;Adam R. Abate;Samuel I. A. Cohen;Erwin J. De Genst;David A. Weitz
PNAS 2011 Volume 108 (Issue 36 ) pp:
Publication Date(Web):2011-09-06
DOI:10.1073/pnas.1105555108
The crucial early stages of amyloid growth, in which normally soluble proteins are converted into fibrillar nanostructures,
are challenging to study using conventional techniques yet are critical to the protein aggregation phenomena implicated in
many common pathologies. As with all nucleation and growth phenomena, it is difficult to track individual nuclei in traditional
macroscopic experiments, which probe the overall temporal evolution of the sample, but do not yield detailed information on
the primary nucleation step as they mix independent stochastic events into an ensemble measurement. To overcome this limitation,
we have developed microdroplet assays enabling us to detect single primary nucleation events and to monitor their subsequent
spatial as well as temporal evolution, both of which we find to be determined by secondary nucleation phenomena. By deforming
the droplets to high aspect ratio, we visualize in real-time propagating waves of protein assembly emanating from discrete
primary nucleation sites. We show that, in contrast to classical gelation phenomena, the primary nucleation step is characterized
by a striking dependence on system size, and the filamentous protein self-assembly process involves a highly nonuniform spatial
distribution of aggregates. These findings deviate markedly from the current picture of amyloid growth and uncover a general
driving force, originating from confinement, which, together with biological quality control mechanisms, helps proteins remain
soluble and therefore functional in nature.
Co-reporter:Natàlia Carulla, Min Zhou, Ernest Giralt, Carol V. Robinson and Christopher M. Dobson
Accounts of Chemical Research 2010 Volume 43(Issue 8) pp:1072
Publication Date(Web):June 17, 2010
DOI:10.1021/ar9002784
The aggregation of proteins into amyloid fibrils is a complex and fascinating process associated with debilitating clinical disorders such as Alzheimer’s and Parkinson’s diseases. The process of aggregation involves a series of steps during which many intermediate aggregation states are populated. Recent evidence points to these intermediate states as the toxic moieties primarily responsible for cell damage or cell death, which are critical steps in the origin and progression of these disorders. To understand the molecular basis of these diseases, it is crucial to investigate and define the details of the aggregation process, and to achieve this objective, researchers need the tools to characterize the structure and kinetics of interconversion of the various species present during amyloid fibril formation. Hydrogen−deuterium (HD) exchange experiments are based on solvent accessibilities and provide one means by which this kind of information may be acquired. In this Account, we describe research based on HD exchange processes that is directed toward better understanding the dynamics and structural reorganizations involved in the formation of amyloid fibrils. Amide hydrogens that normally undergo rapid exchange with solvent hydrogens experience much slower exchange when involved in H-bonded structures or when sterically inaccessible to the solvent. The rates of exchange can be monitored by replacing some hydrogens with deuterons. When peptide and protein molecules assemble into amyloid fibrils, the fibrils contain a core region based on repetitive arrays of β-sheets oriented parallel to the fibril axis. HD experiments have been applied extensively to map such structures in different amyloid fibril systems. By an extension of this approach, we have observed that HD exchange can be governed by a mechanism through which molecules making up the fibrils are continuously dissolving and reforming, revealing that amyloid fibrils are not static but dynamic structures. Under such circumstances, the kinetic parameters that define this “recycling” behavior can be determined, and they contain information that could be of significant value in the design of therapeutic strategies directed against amyloid-related diseases. More recently, to gain insights into the variety of intermediates that are thought to be involved in the aggregation process, we have applied a kinetic pulse labeling HD experiment that is able to characterize such species even if they are only transiently populated. Using this approach, we have been able to obtain structural insights into the different aggregates populated during the process of amyloid fibril formation as well as kinetic and mechanistic information on the structural reorganizations that take place during aggregation. HD exchange experiments, when carefully designed, constitute powerful tools for mapping the core structures of amyloid fibrils, for investigating the recycling of fibril components, and for characterizing the various types of structural reorganization that occur during aggregation. Such information is invaluable for understanding and addressing the molecular origins of the increasingly common and highly debilitating diseases associated with protein misfolding and aggregation.
Co-reporter:Anne Dhulesia ; Nunilo Cremades ; Janet R. Kumita ; Shang-Te Danny Hsu ; Maria F. Mossuto ; Mireille Dumoulin ; Daniel Nietlispach ; Mikael Akke ; Xavier Salvatella
Journal of the American Chemical Society 2010 Volume 132(Issue 44) pp:15580-15588
Publication Date(Web):October 19, 2010
DOI:10.1021/ja103524m
The partial unfolding of human lysozyme underlies its conversion from the soluble state into amyloid fibrils observed in a fatal hereditary form of systemic amyloidosis. To understand the molecular origins of the disease, it is critical to characterize the structural and physicochemical properties of the amyloidogenic states of the protein. Here we provide a high-resolution view of the unfolding process at low pH for three different lysozyme variants, the wild-type protein and the mutants I56T and I59T, which show variable stabilities and propensities to aggregate in vitro. Using a range of biophysical techniques that includes differential scanning calorimetry and nuclear magnetic resonance spectroscopy, we demonstrate that thermal unfolding under amyloidogenic solution conditions involves a cooperative loss of native tertiary structure, followed by progressive unfolding of a compact, molten globule-like denatured state ensemble as the temperature is increased. The width of the temperature window over which the denatured ensemble progressively unfolds correlates with the relative amyloidogenicity and stability of these variants, and the region of lysozyme that unfolds first maps to that which forms the core of the amyloid fibrils formed under similar conditions. Together, these results present a coherent picture at atomic resolution of the initial events underlying amyloid formation by a globular protein.
Co-reporter:Duncan A. White ; Alexander K. Buell ; Tuomas P. J. Knowles ; Mark E. Welland
Journal of the American Chemical Society 2010 Volume 132(Issue 14) pp:5170-5175
Publication Date(Web):March 24, 2010
DOI:10.1021/ja909997e
The physicochemical parameters of biomolecules are the key determinants of the multitude of processes that govern the normal and aberrant behavior of living systems. A particularly important aspect of such behavior is the role it plays in the self-association of proteins to form organized aggregates such as the amyloid or amyloid-like fibrils that are associated with pathological conditions including Alzheimer’s disease and Type II diabetes. In this study we describe quantitative quartz crystal microbalance measurements of the kinetics of the growth of amyloid fibrils in a range of crowded environments and in conjunction with theoretical predictions demonstrate the existence of general relationships that link the propensities of protein molecules to aggregate with fundamental parameters that describe their specific structures and local environments.
Co-reporter:Edward P. O’Brien ; Shang-Te Danny Hsu ; John Christodoulou ; Michele Vendruscolo
Journal of the American Chemical Society 2010 Volume 132(Issue 47) pp:16928-16937
Publication Date(Web):November 9, 2010
DOI:10.1021/ja106530y
The exit tunnel of the ribosome is commonly considered to be sufficiently narrow that co-translational folding can begin only when specific segments of nascent chains are fully extruded from the tunnel. Here we show, on the basis of molecular simulations and comparison with experiment, that the long-range contacts essential for initiating protein folding can form within a nascent chain when it reaches the last 20 Å of the exit tunnel. We further show that, in this “exit port”, a significant proportion of native and non-native tertiary structure can form without steric overlap with the ribosome itself, and provide a library of structural elements that our simulations predict can form in the exit tunnel and is amenable to experimental testing. Our results show that these elements of folded tertiary structure form only transiently and are at their midpoints of stability at the boundary region between the inside and the outside of the tunnel. These findings provide a framework for interpreting a range of recent experimental studies of ribosome nascent chain complexes and for understanding key aspects of the nature of co-translational folding.
Co-reporter:LisaD. Cabrita ;John Christodoulou
Israel Journal of Chemistry 2010 Volume 50( Issue 1) pp:99-108
Publication Date(Web):
DOI:10.1002/ijch.201000015
Abstract
To maintain optimal protein homeostasis within the cell, a complex interplay exists between factors determining the production, transport, and folding of nascent chains. Here we review the evidence for the structural and dynamical properties of nascent polypeptide chains as they emerge from the ribosome and that enable them to acquire their biologically active, three-dimensional structures. In particular, we focus on the application of NMR spectroscopy, which is now emerging as a powerful technique to probe the behavior and properties of emerging nascent chains.
Co-reporter:Alexander K. Buell, Duncan A. White, Christoph Meier, Mark E. Welland, Tuomas P. J. Knowles and Christopher M. Dobson
The Journal of Physical Chemistry B 2010 Volume 114(Issue 34) pp:10925-10938
Publication Date(Web):August 9, 2010
DOI:10.1021/jp101579n
Chemical control of surface functionality and topography is an essential requirement for many technological purposes. In particular, the covalent attachment of monomeric proteins to surfaces has been the object of intense studies in recent years, for applications as varied as electrochemistry, immuno-sensing, and the production of biocompatible coatings. Little is known, however, about the characteristics and requirements underlying surface attachment of supramolecular protein nanostructures. Amyloid fibrils formed by the self-assembly of peptide and protein molecules represent one important class of such structures. These highly organized β-sheet-rich assemblies are a hallmark of a range of neurodegenerative disorders, including Alzheimer’s disease and type II diabetes, but recent findings suggest that they have much broader significance, potentially representing the global free energy minima of the energy landscapes of proteins and having potential applications in material science. In this paper, we describe strategies for attaching amyloid fibrils formed from different proteins to gold surfaces under different solution conditions. Our methods involve the reaction of sulfur containing small molecules (cystamine and 2-iminothiolane) with the amyloid fibrils, enabling their covalent linkage to gold surfaces. We demonstrate that irreversible attachment using these approaches makes possible quantitative analysis of experiments using biosensor techniques, such as quartz crystal microbalance (QCM) assays that are revolutionizing our understanding of the mechanisms of amyloid growth and the factors that determine its kinetic behavior. Moreover, our results shed light on the nature and relative importance of covalent versus noncovalent forces acting on protein superstructures at metal surfaces.
Co-reporter:Christofer Lendel, Carlos W. Bertoncini, Nunilo Cremades, Christopher A. Waudby, Michele Vendruscolo, Christopher M. Dobson, Dale Schenk, John Christodoulou and Gergely Toth
Biochemistry 2009 Volume 48(Issue 35) pp:
Publication Date(Web):July 31, 2009
DOI:10.1021/bi901285x
Increasing evidence links the misfolding and aberrant self-assembly of proteins with the molecular events that underlie a range of neurodegenerative diseases, yet the mechanistical details of these processes are still poorly understood. The fact that many of these proteins are intrinsically unstructured makes it particularly challenging to develop strategies for discovering small molecule inhibitors of their aggregation. We present here a broad biophysical approach that enables us to characterize the mechanisms of interaction between α-synuclein, a protein whose aggregation is closely connected with Parkinson’s disease, and two small molecules, Congo red and Lacmoid, which inhibit its fibrillization. Both compounds are found to interact with the N-terminal and central regions of the monomeric protein although with different binding mechanisms and affinities. The differences can be attributed to the chemical nature of the compounds as well as their abilities to self-associate. We further show that α-synuclein binding and aggregation inhibition are mediated by small oligomeric species of the compounds that interact with distinct regions of the monomeric protein. These findings provide potential explanations of the nonspecific antiamyloid effect observed for these compounds as well as important mechanistical information for future drug discovery efforts targeting the misfolding and aggregation of intrinsically unstructured proteins.
Co-reporter:Natàlia Carulla;Min Zhou;Muriel Arimon;Margarida Gairí;Ernest Giralt;Carol V. Robinson
PNAS 2009 Volume 106 (Issue 19 ) pp:7828-7833
Publication Date(Web):2009-05-12
DOI:10.1073/pnas.0812227106
Recent experimental evidence points to intermediates populated during the process of amyloid fibril formation as the toxic
moieties primarily responsible for the development of increasingly common disorders such as Alzheimer's disease and type II
diabetes. We describe here the application of a pulse-labeling hydrogen-deuterium (HD) exchange strategy monitored by mass
spectrometry (MS) and NMR spectroscopy (NMR) to characterize the aggregation process of an SH3 domain under 2 different conditions,
both of which ultimately lead to well-defined amyloid fibrils. Under one condition, the intermediates appear to be largely
amorphous in nature, whereas under the other condition protofibrillar species are clearly evident. Under the conditions favoring
amorphous-like intermediates, only species having no protection against HD exchange can be detected in addition to the mature
fibrils that show a high degree of protection. By contrast, under the conditions favoring protofibrillar-like intermediates,
MS reveals that multiple species are present with different degrees of HD exchange protection, indicating that aggregation
occurs initially through relatively disordered species that subsequently evolve to form ordered aggregates that eventually
lead to amyloid fibrils. Further analysis using NMR provides residue-specific information on the structural reorganizations
that take place during aggregation, as well as on the time scales by which they occur.
Co-reporter:Alexer K. Buell;Gian Gaetano Tartaglia Dr.;Neil R. Birkett Dr.;Christopher A. Waudby;Michele Vendruscolo Dr.;Xavier Salvatella Dr.;Mark E. Well ;Tuomas P. J. Knowles Dr.
ChemBioChem 2009 Volume 10( Issue 8) pp:1309-1312
Publication Date(Web):
DOI:10.1002/cbic.200900144
Co-reporter:Pak-Ho Chan, Els Pardon, Linda Menzer, Erwin De Genst, Janet R. Kumita, John Christodoulou, Dirk Saerens, Alain Brans, Fabrice Bouillenne, David B. Archer, Carol V. Robinson, Serge Muyldermans, André Matagne, Christina Redfield, Lode Wyns, Christopher M. Dobson and Mireille Dumoulin
Biochemistry 2008 Volume 47(Issue 42) pp:
Publication Date(Web):September 25, 2008
DOI:10.1021/bi8005797
A single-domain fragment, cAb-HuL22, of a camelid heavy-chain antibody specific for the active site of human lysozyme has been generated, and its effects on the properties of the I56T and D67H amyloidogenic variants of human lysozyme, which are associated with a form of systemic amyloidosis, have been investigated by a wide range of biophysical techniques. Pulse-labeling hydrogen−deuterium exchange experiments monitored by mass spectrometry reveal that binding of the antibody fragment strongly inhibits the locally cooperative unfolding of the I56T and D67H variants and restores their global cooperativity to that characteristic of the wild-type protein. The antibody fragment was, however, not stable enough under the conditions used to explore its ability to perturb the aggregation behavior of the lysozyme amyloidogenic variants. We therefore engineered a more stable version of cAb-HuL22 by adding a disulfide bridge between the two β-sheets in the hydrophobic core of the protein. The binding of this engineered antibody fragment to the amyloidogenic variants of lysozyme inhibited their aggregation into fibrils. These findings support the premise that the reduction in global cooperativity caused by the pathogenic mutations in the lysozyme gene is the determining feature underlying their amyloidogenicity. These observations indicate further that molecular targeting of enzyme active sites, and of protein binding sites in general, is an effective strategy for inhibiting or preventing the aberrant self-assembly process that is often a consequence of protein mutation and the origin of pathogenicity. Moreover, this work further demonstrates the unique properties of camelid single-domain antibody fragments as structural probes for studying the mechanism of aggregation and as potential inhibitors of fibril formation.
Co-reporter:Angel Orte;Neil R. Birkett;Richard W. Clarke;Glyn L. Devlin;David Klenerman
PNAS 2008 Volume 105 (Issue 38 ) pp:14424-14429
Publication Date(Web):2008-09-23
DOI:10.1073/pnas.0803086105
A key issue in understanding the pathogenic conditions associated with the aberrant aggregation of misfolded proteins is the
identification and characterization of species formed during the aggregation process. Probing the nature of such species has,
however, proved to be extremely challenging to conventional techniques because of their transient and heterogeneous character.
We describe here the application of a two-color single-molecule fluorescence technique to examine the assembly of oligomeric
species formed during the aggregation of the SH3 domain of PI3 kinase. The single-molecule experiments show that the species
formed at the stage of the reaction where aggregates have previously been found to be maximally cytotoxic are a heterogeneous
ensemble of oligomers with a median size of 38 ± 10 molecules. This number is remarkably similar to estimates from bulk measurements
of the critical size of species observed to seed ordered fibril formation and of the most infective form of prion particles.
Moreover, although the size distribution of the SH3 oligomers remains virtually constant as the time of aggregation increases,
their stability increases substantially. These findings together provide direct evidence for a general mechanism of amyloid
aggregation in which the stable cross-β structure emerges via internal reorganization of disordered oligomers formed during
the lag phase of the self-assembly reaction.
Co-reporter:AndrewJ. Baldwin;SpencerJ. Anthony-Cahill;TuomasP.J. Knowles;Guy Lippens;John Christodoulou;PaulD. Barker;ChristopherM. Dobson
Angewandte Chemie 2008 Volume 120( Issue 18) pp:3433-3435
Publication Date(Web):
DOI:10.1002/ange.200703915
Co-reporter:AndrewJ. Baldwin;SpencerJ. Anthony-Cahill;TuomasP.J. Knowles;Guy Lippens;John Christodoulou;PaulD. Barker;ChristopherM. Dobson
Angewandte Chemie International Edition 2008 Volume 47( Issue 18) pp:3385-3387
Publication Date(Web):
DOI:10.1002/anie.200703915
Co-reporter:Tuomas P. J. Knowles;Wenmiao Shu;Sarah Meehan;Glyn L. Devlin;Mark E. Welland;Stefan Auer
PNAS 2007 Volume 104 (Issue 24 ) pp:10016-10021
Publication Date(Web):2007-06-12
DOI:10.1073/pnas.0610659104
Aggregation of proteins and peptides is a widespread and much-studied problem, with serious implications in contexts ranging
from biotechnology to human disease. An understanding of the proliferation of such aggregates under specific conditions requires
a quantitative knowledge of the kinetics and thermodynamics of their formation; measurements that to date have remained elusive.
Here, we show that precise determination of the growth rates of ordered protein aggregates such as amyloid fibrils can be
achieved through real–time monitoring, using a quartz crystal oscillator, of the changes in the numbers of molecules in the
fibrils from variations in their masses. We show further that this approach allows the effect of other molecular species on
fibril growth to be characterized quantitatively. This method is widely applicable, and we illustrate its power by exploring
the free-energy landscape associated with the conversion of the protein insulin to its amyloid form and elucidate the role
of a chemical chaperone and a small heat shock protein in inhibiting the aggregation reaction.
Co-reporter:Tuomas P. Knowles;Anthony W. Fitzpatrick;Helen R. Mott;Michele Vendruscolo;Mark E. Welland;Sarah Meehan
Science 2007 Volume 318(Issue 5858) pp:
Publication Date(Web):
DOI:10.1126/science.1150057
Abstract
Protein molecules have the ability to form a rich variety of natural and artificial structures and materials. We show that amyloid fibrils, ordered supramolecular nanostructures that are self-assembled from a wide range of polypeptide molecules, have rigidities varying over four orders of magnitude, and constitute a class of high-performance biomaterials. We elucidate the molecular origin of fibril material properties and show that the major contribution to their rigidity stems from a generic interbackbone hydrogen-bonding network that is modulated by variable side-chain interactions.
Co-reporter:Jeffrey F. Smith;Tuomas P. J. Knowles;Cait E. MacPhee;Mark E. Welland
PNAS 2006 Volume 103 (Issue 43 ) pp:15806-15811
Publication Date(Web):2006-10-24
DOI:10.1073/pnas.0604035103
We report the detailed mechanical characterization of individual amyloid fibrils by atomic force microscopy and spectroscopy.
These self-assembling materials, formed here from the protein insulin, were shown to have a strength of 0.6 ± 0.4 GPa, comparable
to that of steel (0.6–1.8 GPa), and a mechanical stiffness, as measured by Young's modulus, of 3.3 ± 0.4 GPa, comparable to
that of silk (1–10 GPa). The values of these parameters reveal that the fibrils possess properties that make these structures
highly attractive for future technological applications. In addition, analysis of the solution-state growth kinetics indicated
a breakage rate constant of 1.7 ± 1.3 × 10−8 s−1, which reveals that a fibril 10 μm in length breaks spontaneously on average every 47 min, suggesting that internal fracturing
is likely to be of fundamental importance in the proliferation of amyloid fibrils and therefore for understanding the progression
of their associated pathogenic disorders.
Co-reporter:Kresten Lindorff-Larsen,
Robert B. Best,
Mark A. DePristo,
Christopher M. Dobson
and
Michele Vendruscolo
Nature 2005 433(7022) pp:128
Publication Date(Web):
DOI:10.1038/nature03199
Co-reporter:Caroline F. Wright, Sarah A. Teichmann, Jane Clarke
and Christopher M. Dobson
Nature 2005 438(7069) pp:878
Publication Date(Web):
DOI:10.1038/nature04195
Co-reporter:Natàlia Carulla,
Gemma L. Caddy,
Damien R. Hall,
Jesús Zurdo,
Margarida Gairí,
Miguel Feliz,
Ernest Giralt,
Carol V. Robinson
&
Christopher M. Dobson
Nature 2005 436(7050) pp:554
Publication Date(Web):
DOI:10.1038/nature03986
Co-reporter:Michele Vendruscolo
PNAS 2005 102 (16 ) pp:5641-5642
Publication Date(Web):2005-04-19
DOI:10.1073/pnas.0500274102
Co-reporter:
Nature Structural and Molecular Biology 2004 11(5) pp:443-449
Publication Date(Web):18 April 2004
DOI:10.1038/nsmb765
We present a structural analysis of the folding transition states of three SH3 domains. Our results reveal that the secondary structure is not yet fully formed at this stage of folding and that the solvent is only partially excluded from the interior of the protein. Comparison of the members of the transition state ensemble with a database of native folds shows that, despite substantial local variability, the transition state structures can all be classified as having the topology characteristic of an SH3 domain. Our results suggest a mechanism for folding in which the formation of a network of interactions among a subset of hydrophobic residues ensures that the native topology is generated. Such a mechanism enables high fidelity in folding while minimizing the need to establish a large number of specific interactions in the conformational search.
Co-reporter:John Christodoulou;Göran Larsson;Paola Fucini;Sean R. Connell;Thelma A. Pertinhez;Charlotte L. Hanson;Knud H. Nierhaus;Christina Redfield;Jürgen Schleucher;Carol V. Robinson
PNAS 2004 Volume 101 (Issue 30 ) pp:10949-10954
Publication Date(Web):2004-07-27
DOI:10.1073/pnas.0400928101
15N-1H NMR spectroscopy has been used to probe the dynamic properties of uniformly 15N labeled Escherichia coli ribosomes. Despite the high molecular weight of the complex (≈2.3 MDa), [1H-15N] heteronuclear single-quantum correlation spectra contain ≈100 well resolved resonances, the majority of which arise from
two of the four C-terminal domains of the stalk proteins, L7/L12. Heteronuclear pulse-field gradient NMR experiments show
that the resonances arise from species with a translational diffusion constant consistent with that of the intact ribosome.
Longitudinal relaxation time (T
1) and T
1ρ
15N-spin relaxation measurements show that the observable domains tumble anisotropically, with an apparent rotational correlation
time significantly longer than that expected for a free L7/L12 domain but much shorter than expected for a protein rigidly
incorporated within the ribosomal particle. The relaxation data allow the ribosomally bound C-terminal domains to be oriented
relative to the rotational diffusion tensor. Binding of elongation factor G to the ribosome results in the disappearance of
the resonances of the L7/L12 domains, indicating a dramatic reduction in their mobility. This result is in agreement with
cryoelectron microscopy studies showing that the ribosomal stalk assumes a single rigid orientation upon elongation factor
G binding. As well as providing information about the dynamical properties of L7/L12, these results demonstrate the utility
of heteronuclear NMR in the study of mobile regions of large biological complexes and form the basis for further NMR studies
of functional ribosomal complexes in the context of protein synthesis.
Co-reporter:Dmitry M. Korzhnev,
Xavier Salvatella,
Michele Vendruscolo,
Ariel A. Di Nardo,
Alan R. Davidson,
Christopher M. Dobson
and
Lewis E. Kay
Nature 2004 430(6999) pp:586
Publication Date(Web):
DOI:10.1038/nature02655
Co-reporter:Mireille Dumoulin,
Alexander M. Last,
Aline Desmyter,
Klaas Decanniere,
Denis Canet,
Göran Larsson,
Andrew Spencer,
David B. Archer,
Jurgen Sasse,
Serge Muyldermans,
Lode Wyns,
Christina Redfield,
André Matagne,
Carol V. Robinson
and
Christopher M. Dobson
Nature 2003 424(6950) pp:783
Publication Date(Web):
DOI:10.1038/nature01870
Co-reporter:Fabrizio Chiti,
Massimo Stefani,
Niccolò Taddei,
Giampietro Ramponi
and
Christopher M. Dobson
Nature 2003 424(6950) pp:805
Publication Date(Web):
DOI:10.1038/nature01891
Co-reporter:
Nature Structural and Molecular Biology 2002 9(4) pp:308 - 315
Publication Date(Web):11 March 2002
DOI:10.1038/nsb768
Co-reporter:Maria F. Mossuto, Anne Dhulesia, Glyn Devlin, Erica Frare, ... Xavier Salvatella
Journal of Molecular Biology (8 October 2010) Volume 402(Issue 5) pp:783-796
Publication Date(Web):8 October 2010
DOI:10.1016/j.jmb.2010.07.005
Identifying the cause of the cytotoxicity of species populated during amyloid formation is crucial to understand the molecular basis of protein deposition diseases. We have examined different types of aggregates formed by lysozyme, a protein found as fibrillar deposits in patients with familial systemic amyloidosis, by infrared spectroscopy, transmission electron microscopy, and depolymerization experiments, and analyzed how they affect cell viability. We have characterized two types of human lysozyme amyloid structures formed in vitro that differ in morphology, molecular structure, stability, and size of the cross-β core. Of particular interest is that the fibrils with a smaller core generate a significant cytotoxic effect. These findings indicate that protein aggregation can give rise to species with different degree of cytotoxicity due to intrinsic differences in their physicochemical properties.
Co-reporter:Francesco Bemporad, Alfonso De Simone, Fabrizio Chiti, Christopher M. Dobson
Biophysical Journal (6 June 2012) Volume 102(Issue 11) pp:
Publication Date(Web):6 June 2012
DOI:10.1016/j.bpj.2012.03.057
Folded proteins can access aggregation-prone states without the need for transitions that cross the energy barriers for unfolding. In this study we characterized the initial steps of aggregation from a native-like state of the acylphosphatase from Sulfolobus solfataricus (Sso AcP). Using computer simulations restrained by experimental hydrogen/deuterium (H/D) exchange data, we provide direct evidence that under aggregation-promoting conditions Sso AcP populates a conformational ensemble in which native-like structure is retained throughout the sequence in the absence of local unfolding (N∗), although the protein exhibits an increase in hydrodynamic radius and dynamics. This transition leads an edge strand to experience an increased affinity for a specific unfolded segment of the protein. Direct measurements by means of H/D exchange rates, isothermal titration calorimetry, and intermolecular relaxation enhancements show that after formation of N∗, an intermolecular interaction with an antiparallel arrangement is established between the edge strand and the unfolded segment of the protein. However, under conditions that favor the fully native state of Sso AcP, such an interaction is not established. Thus, these results reveal a novel (to our knowledge) self-assembly mechanism for a folded protein that is based on the increased flexibility of highly aggregation-prone segments in the absence of local unfolding.
Co-reporter:Daizo Hamada, Toshiki Tanaka, Gian Gaetano Tartaglia, Amol Pawar, ... Christopher M. Dobson
Journal of Molecular Biology (27 February 2009) Volume 386(Issue 3) pp:878-890
Publication Date(Web):27 February 2009
DOI:10.1016/j.jmb.2008.12.038
We show that a series of peptides corresponding to individual β-strands in native β-lactoglobulin readily form amyloid aggregates and that such aggregates are capable of seeding fibril formation by a full-length form of β-lactoglobulin in which the disulfide bonds are reduced. By contrast, preformed fibrils corresponding to only one of the β-strands that we considered, βA, were found to promote fibril formation by a full-length form of β-lactoglobulin in which the disulfide bonds are intact. These results indicate that regions of high intrinsic aggregation propensity do not give rise to aggregation unless at least partial unfolding takes place. Furthermore, we found that the high aggregation propensity of one of the edge strands, βI, promotes dimerisation of the native structure rather than misfolding and aggregation since the structure of βI is stabilised by the presence of a disulfide bond. These findings demonstrate that the interactions that promote folding and native-state oligomerisation can also result in high intrinsic amyloidogenicity. However, we show that the presence of the remainder of the sequence dramatically reduces the net overall aggregation propensity by negative design principles that we suggest are very common in biological systems as a result of evolutionary processes.
Co-reporter:Janet R. Kumita, Stephen Poon, Gemma L. Caddy, Christine L. Hagan, ... Christopher M. Dobson
Journal of Molecular Biology (25 May 2007) Volume 369(Issue 1) pp:157-167
Publication Date(Web):25 May 2007
DOI:10.1016/j.jmb.2007.02.095
We have studied the effects of the extracellular molecular chaperone, clusterin, on the in vitro aggregation of mutational variants of human lysozyme, including one associated with familial amyloid disease. The aggregation of the amyloidogenic variant I56T is inhibited significantly at clusterin to lysozyme ratios as low as 1:80 (i.e. one clusterin molecule per 80 lysozyme molecules). Experiments indicate that under the conditions where inhibition of aggregation occurs, clusterin does not bind detectably to the native or fibrillar states of lysozyme, or to the monomeric transient intermediate known to be a key species in the aggregation reaction. Rather, it seems to interact with oligomeric species that are present at low concentrations during the lag (nucleation) phase of the aggregation reaction. This behavior suggests that clusterin, and perhaps other extracellular chaperones, could have a key role in curtailing the potentially pathogenic effects of the misfolding and aggregation of proteins that, like lysozyme, are secreted into the extracellular environment.
Co-reporter:Tim Guilliams, Farah El-Turk, Alexander K. Buell, Elizabeth M. O'Day, ... Erwin De Genst
Journal of Molecular Biology (24 July 2013) Volume 425(Issue 14) pp:2397-2411
Publication Date(Web):24 July 2013
DOI:10.1016/j.jmb.2013.01.040
► NbSyn2 and NbSyn87 bind, respectively, to residues 137–140 and ca residues 118–131 of αSyn. ► The nanobodies bind and distinguish between αSyn fibrils at different maturation stages. ► The accessibility of the C-terminal region of αSyn changes upon fibril maturation. ► Conformation-sensitive nanobodies can study time-dependent aspects of fibril maturation.Nanobodies are single-domain fragments of camelid antibodies that are emerging as versatile tools in biotechnology. We describe here the interactions of a specific nanobody, NbSyn87, with the monomeric and fibrillar forms of α-synuclein (αSyn), a 140-residue protein whose aggregation is associated with Parkinson's disease. We have characterized these interactions using a range of biophysical techniques, including nuclear magnetic resonance and circular dichroism spectroscopy, isothermal titration calorimetry and quartz crystal microbalance measurements. In addition, we have compared the results with those that we have reported previously for a different nanobody, NbSyn2, also raised against monomeric αSyn. This comparison indicates that NbSyn87 and NbSyn2 bind with nanomolar affinity to distinctive epitopes within the C-terminal domain of soluble αSyn, comprising approximately amino acids 118–131 and 137–140, respectively. The calorimetric and quartz crystal microbalance data indicate that the epitopes of both nanobodies are still accessible when αSyn converts into its fibrillar structure. The apparent affinities and other thermodynamic parameters defining the binding between the nanobody and the fibrils, however, vary significantly with the length of time that the process of fibril formation has been allowed to progress and with the conditions under which formation occurs, indicating that the environment of the C-terminal domain of αSyn changes as fibril assembly takes place. These results demonstrate that nanobodies are able to target forms of potentially pathogenic aggregates that differ from each other in relatively minor details of their structure, such as those associated with fibril maturation.Download high-res image (211KB)Download full-size image
Co-reporter:Leila M. Luheshi, Christopher M. Dobson
FEBS Letters (20 August 2009) Volume 583(Issue 16) pp:2581-2586
Publication Date(Web):20 August 2009
DOI:10.1016/j.febslet.2009.06.030
Protein misfolding and aggregation are pathognomic for a number of the most common age-related degenerative diseases. Great progress has been made in studying protein aggregation in the test tube and also in replicating protein aggregation in vertebrate animal models of these diseases. However, we argue here that the development and effective integration of emerging techniques such as the methods of nanoscience and the use of invertebrate models are now providing powerful new opportunities to advance our current understanding of the fundamental origins of these disorders.
Co-reporter:Alexandra E. Porter, Tuomas P.J. Knowles, Karin Muller, Sarah Meehan, ... Christopher M. Dobson
Journal of Molecular Biology (2 October 2009) Volume 392(Issue 4) pp:868-871
Publication Date(Web):2 October 2009
DOI:10.1016/j.jmb.2009.07.061
The process of aggregation leading to amyloid formation by peptides and proteins is associated with diseases ranging from systemic amyloidoses to neurodegenerative disorders such as Alzheimer's disease. A key question in understanding the link between amyloid formation and its pathological consequences is the ultrastructural localisation and morphological form of amyloid species within the cellular environment. The acquisition of such information has proven to be challenging, but we report here a novel approach that enables amyloid fibrils to be visualised directly within a cell. First, fibrils are assembled from selenium analogues of the sulfur-containing cysteine peptides, and then, atomic number contrast transmission electron microscopy is used to detect the selenium doped species selectively within the carbon-rich background of the cell. We demonstrate the power of this approach by imaging human monocyte-derived macrophage cells that have been exposed to fibrils from an amyloidogenic fragment of the disease-associated protein transthyretin. The ready incorporation of seleno-cysteine and methionine instead of their natural sulfur-containing analogues, a feature that is already commonly used in X-ray diffraction studies of proteins, suggests that this method can be used as a general strategy to image specific peptides and proteins within the cellular environment using electron microscopy.
Co-reporter:Erwin J. De Genst, Tim Guilliams, Joke Wellens, Elizabeth M. O'Day, ... Christopher M. Dobson
Journal of Molecular Biology (17 September 2010) Volume 402(Issue 2) pp:326-343
Publication Date(Web):17 September 2010
DOI:10.1016/j.jmb.2010.07.001
The aggregation of the intrinsically disordered protein α-synuclein to form fibrillar amyloid structures is intimately associated with a variety of neurological disorders, most notably Parkinson's disease. The molecular mechanism of α-synuclein aggregation and toxicity is not yet understood in any detail, not least because of the paucity of structural probes through which to study the behavior of such a disordered system. Here, we describe an investigation involving a single-domain camelid antibody, NbSyn2, selected by phage display techniques to bind to α-synuclein, including the exploration of its effects on the in vitro aggregation of the protein under a variety of conditions. We show using isothermal calorimetric methods that NbSyn2 binds specifically to monomeric α-synuclein with nanomolar affinity and by means of NMR spectroscopy that it interacts with the four C-terminal residues of the protein. This latter finding is confirmed by the determination of a crystal structure of NbSyn2 bound to a peptide encompassing the nine C-terminal residues of α-synuclein. The NbSyn2:α-synuclein interaction is mediated mainly by side-chain interactions while water molecules cross-link the main-chain atoms of α-synuclein to atoms of NbSyn2, a feature we believe could be important in intrinsically disordered protein interactions more generally. The aggregation behavior of α-synuclein at physiological pH, including the morphology of the resulting fibrillar structures, is remarkably unaffected by the presence of NbSyn2 and indeed we show that NbSyn2 binds strongly to the aggregated as well as to the soluble forms of α-synuclein. These results give strong support to the conjecture that the C-terminal region of the protein is not directly involved in the mechanism of aggregation and suggest that binding of NbSyn2 could be a useful probe for the identification of α-synuclein aggregation in vitro and possibly in vivo.
Co-reporter:Sarah Meehan, Tuomas P.J. Knowles, Andrew J. Baldwin, Jeffrey F. Smith, ... John A. Carver
Journal of Molecular Biology (14 September 2007) Volume 372(Issue 2) pp:470-484
Publication Date(Web):14 September 2007
DOI:10.1016/j.jmb.2007.06.060
αB-Crystallin is a ubiquitous small heat-shock protein (sHsp) renowned for its chaperone ability to prevent target protein aggregation. It is stress-inducible and its up-regulation is associated with a number of disorders, including those linked to the deposition of misfolded proteins, such as Alzheimer's and Parkinson's diseases. We have characterised the formation of amyloid fibrils by human αB-crystallin in detail, and also that of αA-crystallin and the disease-related mutant R120G αB-crystallin. We find that the last 12 amino acid residues of the C-terminal region of αB-crystallin are predicted from their physico-chemical properties to have a very low propensity to aggregate. 1H NMR spectroscopy reveals that this hydrophilic C-terminal region is flexible both in its solution state and in amyloid fibrils, where it protrudes from the fibrillar core. We demonstrate, in addition, that the equilibrium between different protofilament assemblies can be manipulated and controlled in vitro to select for particular αB-crystallin amyloid morphologies. Overall, this study suggests that there could be a fine balance in vivo between the native functional sHsp state and the formation of amyloid fibrils.
Co-reporter:Martino Calamai, Gian Gaetano Tartaglia, Michele Vendruscolo, Fabrizio Chiti, Christopher M. Dobson
Journal of Molecular Biology (10 April 2009) Volume 387(Issue 4) pp:965-974
Publication Date(Web):10 April 2009
DOI:10.1016/j.jmb.2008.09.003
We have performed an extensive mutational analysis of aggregation and disaggregation of amyloid-like protofibrils of human muscle acylphosphatase. Our findings indicate that the regions that promote aggregation in 25% (v/v) 2,2,2 trifluoroethanol (TFE) are different from those that promote disaggregation under milder conditions (5% TFE). Significant changes in the rate of disaggregation of protofibrils in 5% TFE result not only from mutations situated in the regions of the sequence that play a key role in the mechanism of aggregation in 25% TFE, but also from mutations located in other regions. In order to rationalise these results, we have used a modified version of the Zyggregator aggregation propensity prediction algorithm to take into account structural rearrangements of the protofibrils that may be induced by changes in solution conditions. Our results suggest that a wider range of residues contributes to the stability of the aggregates in addition to those that play an important kinetic role in the aggregation process. The mutational approach described here is capable of providing residue-specific information on the structure and dynamics of amyloid protofibrils under conditions close to physiological and should be widely applicable to other systems.
Co-reporter:Georg Meisl, Xiaoting Yang, Christopher M. Dobson, Sara Linse and Tuomas P. J. Knowles
Chemical Science (2010-Present) 2017 - vol. 8(Issue 6) pp:NaN4362-4362
Publication Date(Web):2017/04/26
DOI:10.1039/C7SC00215G
The aggregation of the amyloid β peptide (Aβ42), which is linked to Alzheimer's disease, can be altered significantly by modulations of the peptide's intermolecular electrostatic interactions. Variations in sequence and solution conditions have been found to lead to highly variable aggregation behaviour. Here we modulate systematically the electrostatic interactions governing the aggregation kinetics by varying the ionic strength of the solution. We find that changes in the solution ionic strength induce a switch in the reaction pathway, altering the dominant mechanisms of aggregate multiplication. This strategy thereby allows us to continuously sample a large space of different reaction mechanisms and develop a minimal reaction network that unifies the experimental kinetics under a wide range of different conditions. More generally, this universal reaction network connects previously separate systems, such as charge mutants of the Aβ42 peptide, on a continuous mechanistic landscape, providing a unified picture of the aggregation mechanism of Aβ42.

![3-[4-hydroxy-3-(5,5,8,8-tetramethyl-3-pentoxy-6,7-dihydronaphthalen-2-yl)phenyl]prop-2-enoic acid](http://img.cochemist.com/ccimg/847300/847239-17-2.png)
![3-[4-hydroxy-3-(5,5,8,8-tetramethyl-3-pentoxy-6,7-dihydronaphthalen-2-yl)phenyl]prop-2-enoic acid](http://img.cochemist.com/ccimg/847300/847239-17-2_b.png)

![5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methyl-1,2-oxazole](http://img.cochemist.com/ccimg/98100/98033-68-2.png)
![5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methyl-1,2-oxazole](http://img.cochemist.com/ccimg/98100/98033-68-2_b.png)













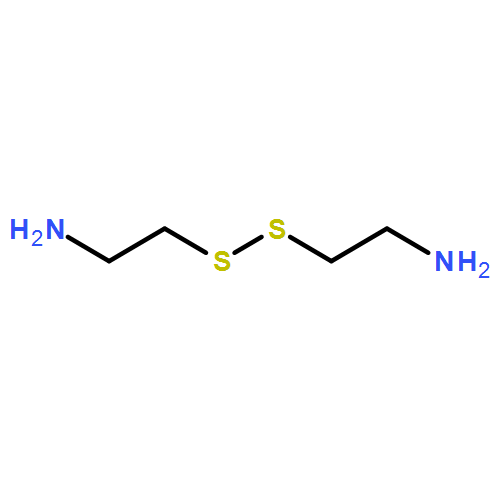
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-1-methyl-2-oxopropyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/71800/71732-53-1.png)
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-1-methyl-2-oxopropyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/71800/71732-53-1_b.png)




![Poly[imino[(1S)-1-(3-amino-3-oxopropyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/69900/69864-43-3.png)
![Poly[imino[(1S)-1-(3-amino-3-oxopropyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/69900/69864-43-3_b.png)
![4,6,10-Trioxa-5-phosphatetracosanoicacid, 2-amino-5-hydroxy-11-oxo-8-[(1-oxotetradecyl)oxy]-, 5-oxide, (2S,8R)-](http://img.cochemist.com/ccimg/64100/64023-32-1.png)
![4,6,10-Trioxa-5-phosphatetracosanoicacid, 2-amino-5-hydroxy-11-oxo-8-[(1-oxotetradecyl)oxy]-, 5-oxide, (2S,8R)-](http://img.cochemist.com/ccimg/64100/64023-32-1_b.png)
![Cholestane-7,24-diol,3-[[3-[(4-aminobutyl)amino]propyl]amino]-, 24-(hydrogen sulfate), (3b,5a,7a,24R)-](http://img.cochemist.com/ccimg/148800/148717-90-2.png)
![Cholestane-7,24-diol,3-[[3-[(4-aminobutyl)amino]propyl]amino]-, 24-(hydrogen sulfate), (3b,5a,7a,24R)-](http://img.cochemist.com/ccimg/148800/148717-90-2_b.png)
![Benzoic acid,4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b][1,4]diazepin-13-yl)-](http://img.cochemist.com/ccimg/155900/155877-83-1.png)
![Benzoic acid,4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b][1,4]diazepin-13-yl)-](http://img.cochemist.com/ccimg/155900/155877-83-1_b.png)











![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
![Benzoic acid,4-[(1E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/71500/71441-28-6.png)
![Benzoic acid,4-[(1E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/71500/71441-28-6_b.png)
![4-[2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dioxolan-2-yl]benzoic acid](http://img.cochemist.com/ccimg/146700/146670-40-8.png)
![4-[2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dioxolan-2-yl]benzoic acid](http://img.cochemist.com/ccimg/146700/146670-40-8_b.png)
![[benzene-1,4-diylbis(methanediylpyridin-1-yl-4-ylidene)]bis(N-oxomethanaminium) dibromide](http://img.cochemist.com/ccimg/4700/4605-75-8.png)
![[benzene-1,4-diylbis(methanediylpyridin-1-yl-4-ylidene)]bis(N-oxomethanaminium) dibromide](http://img.cochemist.com/ccimg/4700/4605-75-8_b.png)


![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)



![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)



![4-[6-(1-adamantyl)-7-hydroxy-2-naphthyl]benzoic acid](http://img.cochemist.com/ccimg/107500/107430-66-0.png)
![4-[6-(1-adamantyl)-7-hydroxy-2-naphthyl]benzoic acid](http://img.cochemist.com/ccimg/107500/107430-66-0_b.png)
![Benzoic acid,4-[(1E)-3-[3,5-bis(1,1-dimethylethyl)phenyl]-3-oxo-1-propen-1-yl]-](http://img.cochemist.com/ccimg/110400/110368-33-7.png)
![Benzoic acid,4-[(1E)-3-[3,5-bis(1,1-dimethylethyl)phenyl]-3-oxo-1-propen-1-yl]-](http://img.cochemist.com/ccimg/110400/110368-33-7_b.png)


![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)




![Sodium;(2s)-2-azaniumyl-3-[[(2r)-2,3-di(hexadecanoyloxy)propoxy]-oxidophosphoryl]oxypropanoate](http://img.cochemist.com/ccimg/145900/145849-32-7.png)
![Sodium;(2s)-2-azaniumyl-3-[[(2r)-2,3-di(hexadecanoyloxy)propoxy]-oxidophosphoryl]oxypropanoate](http://img.cochemist.com/ccimg/145900/145849-32-7_b.png)
























![2-(4-{3-[2-(TRIFLUOROMETHYL)-10H-PHENOTHIAZIN-10-YL]PROPYL}-1-PIP<WBR />ERAZINYL)ETHYL HEPTANOATE](http://img.cochemist.com/ccimg/215100/215030-90-3.png)
![2-(4-{3-[2-(TRIFLUOROMETHYL)-10H-PHENOTHIAZIN-10-YL]PROPYL}-1-PIP<WBR />ERAZINYL)ETHYL HEPTANOATE](http://img.cochemist.com/ccimg/215100/215030-90-3_b.png)