Co-reporter:Long Qu;Mingtao Li;Lulu Bian;Qingyang Du
Journal of Solid State Electrochemistry 2017 Volume 21( Issue 12) pp:3659-3673
Publication Date(Web):01 August 2017
DOI:10.1007/s10008-017-3706-0
Strontium-doped Li2FeSiO4/C is prepared by using the sol-gel method with soluble Li, Fe, Si, and Sr sources. X-ray diffraction, scanning electron microscopy, transmission electron microscopy, and X-ray photoelectron spectroscopy measurements are carried out to determine the crystal structures, morphologies, particle sizes, and chemical valence states of the resulting products. Rietveld refinement confirms that strontium-doped Li2FeSiO4 has a monoclinic P21/n structure and that a strontium cation occupies the Fe site in the lattice. The grain size of strontium-doped Li2FeSiO4 is approximately 20 nm, and the nanoparticles are interconnected tightly with amorphous carbon layers. As the cathode material of a lithium-ion battery, strontium-doped Li2FeSiO4/C delivers a high discharge capacity of 181 mAh g−1 at a rate of 0.5 C. The capacity retention after the 100th cycle reaches 76%, which increases by 7 percentage points compared with Li2FeSiO4/C. The cathode exhibits good rate performance, with corresponding discharge capacities of 165, 145, and 119 mAh g−1 for 1, 2, and 5 C rates, respectively. By analyzing the electrochemical impedance spectra, it can be concluded that strontium cation doping helps to increase the Li+ diffusion capability and weakens side reactions between the electrode and electrolyte. In summary, the improvement of the electrochemical performance can be attributed to the strengthened crystal structure stability during charging and discharging after strontium cation doping.
Co-reporter:Jiageng Li, Xueyu Tian, Bolun Yang
Powder Technology 2017 Volume 313(Volume 313) pp:
Publication Date(Web):15 May 2017
DOI:10.1016/j.powtec.2017.03.031
•A bubble-based EMMS model is developed by introducing the pressure-drop balance equation.•A novel solving strategy is proposed to explain the variation of Hd over εg.The dynamic evolutions of the emulsion and bubbles are also revealed by the model solution.•The dynamic evolutions of the emulsion and bubbles are also revealed by the model solution.•Present model can successfully predict the gas-solid hydromechanical behavior in a bubbling fluidized bed.The meso-scale structure has an obvious influence on gas-solid two-phase flow in a bubbling fluidized bed and subsequently affects its heat and mass transfer performance. To accurately simulate the hydromechanical behavior in the bubbling fluidized bed, the influence of the meso-scale structure on gas-solid flow must be reasonably described. In this work, the pressure-drop balance equation is introduced into an original bubble-based energy-minimization multi-scale (EMMS) model to establish a new gas-solid drag model. Compared with previous studies in which the heterogeneous drag force was artificially set at less than the conventional Wen & Yu drag force, a novel solving strategy is proposed to explain this phenomenon. This strategy will give a general method to determine the relationship between heterogeneous and conventional drag forces. Moreover, the dynamic evolution processes of the emulsion phase and bubble phase with the increase of the overall voidage (εg) are clearly revealed by the model solution. Eventually, the present drag model is integrated into a computational fluid dynamics (CFD) solver by user defined functions (UDFs) to simulate the hydromechanical behavior of a bubbling fluidized bed with Geldart A particles. The simulation results give higher accuracy with respect to the reported experimental data compared with those obtained by using the conventional Gidaspow drag model.Download high-res image (341KB)Download full-size image
Co-reporter:Jiageng Li, Bolun Yang
Chemical Engineering Journal 2017 Volume 329(Volume 329) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.cej.2017.05.164
•The local-structure-dependent drag model based on a computational cell is established.•A computational cell is resolved into three sub-systems.•The meso-scale structural parameters and drag force are correlated by implementing the scale resolution.•The grid independence of the new drag model is tested.•The prediction accuracy of the new drag model is significantly improved compared with Gidaspow model.Conventional drag models coupled with a two-fluid model (TFM) are subject to certain restrictions on the predictions of the hydrodynamic behaviors in bubbling fluidized beds with fine particles, owing to the lack of scale resolution and the subsequent neglect of the impact of the meso-scale structure on gas-solid interaction. In this work, a local control volume is resolved into three sub-systems (i.e., the emulsion phase, bubble phase, and interphase) by implementing a designed route of scale resolution. Then, a local-structure-dependent (LSD) drag model based on the energy-minimization multi-scale (EMMS) theory is developed to account for the dependence of the drag force on the meso-scale structure. The LSD drag model is solved using a genetic algorithm and the obtained heterogeneous drag forces are integrated with the TFM to simulate the hydrodynamic behaviors in bubbling fluidized beds for different gas-solid systems. Consequently, the proposed drag model is validated to provide satisfying predictions of the fluidizations of Geldart A, A/B, and B particles. Furthermore, bubble diameters obtained from computational fluid dynamics (CFD) simulations are compared with those obtained from the empirical or semi-empirical correlations. The results indicate that the LSD drag model can capture the gas-solid hydrodynamics in a bubbling fluidized bed.
Co-reporter:Zhiqiang Wu, Wangcai Yang, Xueyu Tian, Bolun Yang
Energy Conversion and Management 2017 Volume 135(Volume 135) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.enconman.2016.12.060
•Influence of three main compounds in spirulina and simulated spirulina on co-pyrolysis behavior was investigated.•Medium chain triglyceride showed positive synergistic effects with higher volatile yield.•Glycine and starch illustrated both positive and negative synergistic effects on char yield.•Non-additivity distribution of activation energy was solved by three model-free methods.Synergistic effects from co-pyrolysis microalgae biomass with low-rank coal were investigated in this work. Model compounds of three main component in microalgae algae (glycine, medium chain triglyceride and starch), spirulina and simulated spirulina were chosen to Shenfu bituminous pyrolysis process. Kinetic parameters were solved through isoconversional method, and scanning electron microscopy with energy dispersive spectroscopy were applied for characterizing the char samples. Results revealed synergistic effects occurred with different forms from co-pyrolysis of microalgae primary compounds and coal. Positive synergistic effects, which were defined as higher volatile yield than calculated value, were found in medium chain triglyceride and coal mixtures at all mass ratio. Whether positive or negative synergistic effects on products yield from glycine or starch blended with coal hinged on the temperature and mixing ratio. Both spirulina and simulated spirulina show optimal performance on volatile yields under 50 wt.% mass ratio. Non-additivity phenomenon was observed on the distribution of average activation energy. Synergistic effects from co-pyrolysis of coal and microalgae biomass may attributes to the integrative action of the three model compounds.
Co-reporter:Lu Wang, Elizabeth G. Mahoney, Shen Zhao, Bolun Yang and Jingguang G. Chen
Chemical Communications 2016 vol. 52(Issue 18) pp:3697-3700
Publication Date(Web):02 Feb 2016
DOI:10.1039/C5CC10439D
Low-loadings of Pt supported over six transition metal carbide (Pt/TMC) powder catalysts were synthesized and evaluated for hydrogen oxidation and evolution reactions in an alkaline electrolyte. The roughness factor of each Pt/TMC catalyst was different, indicating that the carbide supports affect the dispersion of Pt. Furthermore, when normalized by the corresponding roughness factors, all Pt/TMC catalysts were found to have similar intrinsic activities that were comparable to the state-of-the-art commercial Pt/C electrocatalysts.
Co-reporter:Xuedong Jiang, Ning Yang, Bolun Yang
Particuology 2016 Volume 27() pp:95-101
Publication Date(Web):August 2016
DOI:10.1016/j.partic.2015.05.011
•The performances of three drag models were evaluated in the CFD simulation of a riser.•DBS-local drag model gave more reasonable distributions of gas holdup.•The ratio of drag coefficient to bubble diameter is smaller in center and larger near wall.•Liquid velocity distributions were not affected by the drag models.Local hydrodynamics in the riser of an external loop airlift reactor (EL-ALR) are identified and the performances of three drag models are evaluated in computational fluid dynamics simulation. The simulation results show that the Schiller–Naumann drag model underestimated the local gas holdup at lower superficial gas velocity whereas the Tomiyama drag model overestimated that at higher superficial gas velocity. By contrast, the dual-bubble-size (DBS)-local drag model gave more reasonable radial and axial distributions of gas holdup in all cases. The reason is that the DBS-local drag model gave correct values of the lumped parameter, i.e., the ratio of the drag coefficient to bubble diameter, for varying operating conditions and radial positions. This ratio is reasonably expected to decrease with increasing superficial gas velocity and be smaller in the center and larger near the wall. Only the DBS-local drag model correctly reproduced these trends. The radial profiles of the axial velocity of the liquid and gas predicted by the DBS-local model also agreed well with experimental data.
Co-reporter:Lu Wang, Mingtao Li, Zhiyu Huang, Yingming Li, Suitao Qi, Chunhai Yi, Bolun Yang
Journal of Power Sources 2014 Volume 264() pp:282-289
Publication Date(Web):15 October 2014
DOI:10.1016/j.jpowsour.2014.04.104
•The Ni–WC/C catalysts were prepared successfully by a simple impregnation method.•The nickel particles were grown on the WC/C framework in clusters with nanoscale.•Tungsten carbide promotes nickel to more active sites for urea electrooxidation.•Ni–WC/C nanoclusters show superior catalytic activity for urea electrooxidation.A nanocluster Ni–WC/C electrocatalyst is prepared through a sequential impregnation method and is used for the urea electrooxidation in alkaline conditions. The micro-morphology, lattice parameter, composition and surface states of Ni–WC/C particles are determined by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), energy dispersive X-ray (EDX) and X-ray photoelectron spectrometry (XPS) analysis. The electrooxidation activity and stability of the Ni–WC/C catalyst are also investigated by cyclic voltammograms and chronoamperograms. Characterization results indicate that the Ni nanoclusters are uniformly distributed on the WC/C framework, and the Ni–WC/C catalyst shows high electrocatalytic activity and stability for urea electrooxidation. The maximum current density at the Ni–WC/C electrode is almost 700 mA cm−2 mg−1 which is one order of magnitude higher than that at the Ni/C electrode, and the steady current density at the Ni–WC/C electrode is also markedly improved. Furthermore, the ESA values and XPS spectra indicate that the enhanced performance of the Ni–WC/C catalyst could be attributed to the structure effect and electron effect between nickel and tungsten carbide.
Co-reporter:Xingxing Li, Jiageng Li, and Bolun Yang
Industrial & Engineering Chemistry Research 2014 Volume 53(Issue 50) pp:19583-19593
Publication Date(Web):2017-2-22
DOI:10.1021/ie5024063
A cryogenic distillation process for the upgrade of synthetic natural gas (SNG) from methanation of coke oven gas (COG) is designed and controlled using a method of gradually reducing independent variables. Freedom analysis is performed to decide independent variables of the cryogenic distillation column. Based on the equilibrium stage model and Peng–Robinson–Boston–Mathias (PR-BM) thermodynamics method, parameters sensitivity analysis is implemented to obtain the optimal operation conditions using Aspen Plus software. After supplying the physical dimensions and control variables (reflux flow rate and reflux ratio) of the distillation column, three control structures that involve fixed reflux flow rate, fixed reflux ratio and dual-composition controllers are developed to control the cryogenic distillation process. Under the significant disturbances of feed flow rate and feed composition, evaluation results show that the dual-composition control system displays the best effect for maintaining the mole percent of CH4 in column bottoms and gas distillates, which are 99.87% and 0.43%, respectively.
Co-reporter:Juan Fan;Chunhai Yi;Xiaorong Lan
Journal of Heterocyclic Chemistry 2014 Volume 51( Issue 4) pp:1058-1062
Publication Date(Web):
DOI:10.1002/jhet.1815
4′,4″(5″) Di-tert-butyldibenzo 18-crown-6 (DTBB18C6) was successfully synthesized by SN2 nucleophilic substitution with 4-tert-butyl catechol as starting material. Effects of cyclization reagents, solvents, and templates were investigated. Reaction process was monitored by the real-time online FTIR to study the actual reaction route. The highest DTBB18C6 yield (above 33%) was obtained by using Cs2CO3 as the template, 2,2′-diethylene glycol ditosylate as the cyclization reagent, and THF as the solvent. From the result of FTIR, four different reaction stages of DTBB18C6 synthesis process were proposed.
Co-reporter:Jun Fan, Chunhai Yi, Xiaorong Lan, and Bolun Yang
Organic Process Research & Development 2013 Volume 17(Issue 3) pp:368-374
Publication Date(Web):February 20, 2013
DOI:10.1021/op3003163
4′,4″(5″)-Di-tert-butyldibenzo-18-crown-6 (DTBB18C6) was synthesized using 4-tert-butyl catechol (TBC) as starting material, 2,2′-diethylene glycol di(p-touenesulfonate) as cyclization reagent, Cs2CO3 as template, and tetrahydrofuran (THF) as solvent. The reaction was carried out in a sealed environment with nitrogen, and Cs2CO3 was supplemented in three steps. Response surface methodology (RSM) was used to investigate the effects of reaction temperature (X1), initial concentration of TBC (X2), reaction time (X3), and Cs2CO3/TBC molar ratio (X4); the experiment method was compared with published methods. From the result of RSM, the operation conditions for maximum yield, such as reaction temperature, reaction time, initial concentration of TBC, and Cs2CO3/TBC molar ratio were found to be 49.15 °C, 72 h, 0.06 mol/L and 3.22, respectively. Under these optimum conditions, DTBB18C6 yield (Y) reached 43.38%.
Co-reporter:Hong Yuan, Xingxing Li, and Bolun Yang
Industrial & Engineering Chemistry Research 2013 Volume 52(Issue 44) pp:15549-15559
Publication Date(Web):2017-2-22
DOI:10.1021/ie401370n
Microwave-absorbing catalyst H3PW12O40·xH2O/C (HPW/C) was prepared for biodiesel synthesis using cottonseed oil and methanol as feed materials. The hot spot temperatures in the catalyst were calculated by considering the absorbed microwave power, temperature rise, and heat transfer between catalyst and liquid phase. The intrinsic kinetics model for heterogeneous transesterification was established on the basis of the Langmuir–Hinshelwood–Hougen–Watson (LHHW) mechanism, and the model parameters were decided by using the kinetics experimental data and the calculated results under the hot spot temperatures. Results showed that the hot spot temperatures of the catalyst were far higher than the temperature of bulk liquid, and the simulated kinetics results agreed well with the experimental data.
Co-reporter:Yulei Guan;Ying Zhang;Chunhai Yi
Chinese Journal of Chemistry 2013 Volume 31( Issue 8) pp:1087-1094
Publication Date(Web):
DOI:10.1002/cjoc.201300204
Abstract
Addition of nitroalkanes into n-alkanes can lower the activation barriers of free-radical production and accelerate the decomposition of n-alkanes at relatively low temperatures. Four initial decomposition mechanisms of the n-butane/nitroethane binary mixture were proposed for the promoting effect and considered theoretically at the B3LYP, BB1K, BMK, MPW1K, and M06-2X levels with MG3S basis set. Energetics above was compared to high-level CBS-QB3 and G4 calculations. Calculated results confirm the feasibility of the four initial decomposition pathways: (I) the CNO2 bond rupture of nitroethane to produce ethyl and ·NO2, (II) HONO elimination from nitroethane followed by decomposition to ·OH and ·NO, (III) rearrangement of nitroethane to ethyl nitrite which further dissociates into CH3CH2O· and ·NO, and (IV) direct hydrogen-abstraction of nitroethane with n-butane.
Co-reporter:Peng Qiu, Lu Wang, Xuedong Jiang, and Bolun Yang
Energy & Fuels 2012 Volume 26(Issue 2) pp:1254
Publication Date(Web):January 2, 2012
DOI:10.1021/ef201732w
A new process that partially integrates reaction and distillation was developed to enhance the transesterification of ethylene carbonate (EC) with ethanol for the production of diethyl carbonate (DEC). Sodium ethoxide was used as a homogeneous catalyst. The top of a three-necked flask reactor was connected to a distillation apparatus equipped with an independent reboiler and θ ring packing to achieve the separation of DEC and ethanol that was returned back to the reactor. The effects of process variables, such as reactant ratio, reflux ratio, reboiler duty, and catalyst concentration, on the DEC production were investigated to obtain the optimum operating conditions. Experimental results indicate that the DEC product can be removed from the reactor and purified by distillation operation during the course of reaction using the proposed process. Under the optimum operating conditions, which are ethanol/EC mole ratio of 4.5, reflux ratio of 1.5, reboiler duty of 124 W, and catalyst concentration of 0.4 wt %, the yield of DEC can reach 91% and the DEC purity can reach 97 wt %.
Co-reporter:Yu Liu, Bolun Yang, and Shasha Li
Industrial & Engineering Chemistry Research 2012 Volume 51(Issue 29) pp:9803
Publication Date(Web):June 28, 2012
DOI:10.1021/ie300034x
A new approach that substitutes the original two-step olefinic alkylation of thiophenic sulfur (OATS) technology with a reactive distillation (RD) column was proposed to remove sulfur compounds from fluid catalytic cracking (FCC) gasoline. 3-Methylthiophene (3MT) and isobutylene (IB) were designated as the model compounds for sulfide and olefin, respectively; NKC-9 cation exchange resin mixed with the 2 mm × 2 mm θ ring packing was used as the catalyst bed. The process parameters such as feed composition, molar ratio of 3MT to IB, and reflux ratio were investigated experimentally in the RD column. Simulation for this process was carried out by using the equilibrium stage model—the RADFRAC module of Aspen Plus. The multicomponent vapor–liquid equilibria were predicted by an UNIQUAC method, and the kinetics model that is essential in the RD process was obtained from our previous work. The key design factors (e.g., number of reactive and nonreactive stages, location of feed stage, column pressure, mass ratio of distillate to feed, and catalyst weight) were optimized. The theoretical calculations from the equilibrium stage model matched well with the experimental results. Optimization of the process revealed that applying RD technology into OATS process offers higher alkylation selectivity and better catalytic stability, and the sulfur content in FCC gasoline declined by more than 99%.
Co-reporter:Xiaowei Zhou, Xiaofeng Wu, Bolun Yang, Jianliang Xiao
Journal of Molecular Catalysis A: Chemical 2012 Volume 357() pp:133-140
Publication Date(Web):May 2012
DOI:10.1016/j.molcata.2012.02.002
Asymmetric transfer hydrogenation (ATH) is frequently carried out in the azeotropic mixture of formic acid (F) and triethylamine (T), where the F/T molar ratio is 2.5. This study shows that the F/T ratio affects both the reduction rate and enantioselectivity, with the optimum ratio being 0.2 in the ATH of ketones with the Ru-TsDPEN catalyst. Under such conditions, a range of substrates have been reduced, affording high yields and good to excellent enantioselectivities. In comparison with the common azeotropic F-T system, the reduction is faster. This protocol improves both the classic azeotropic and the aqueous-formate system when using water-insoluble ketones.Graphical abstractHighlights► Asymmetric transfer hydrogenation with ruthenium catalyst. ► Formic acid-triethylamine mixture as reductant and solvent. ► The ratio of formic acid to triethylamine affects the hydrogenation.
Co-reporter:Gangli Zhu, Bolun Yang, Shuyan Wang
International Journal of Hydrogen Energy 2011 Volume 36(Issue 21) pp:13603-13613
Publication Date(Web):October 2011
DOI:10.1016/j.ijhydene.2011.07.112
Dehydrogenation of organic chemical hydrides has been improved by reconstructing the catalyst in the form of hierarchical porous structure nanocatalyst, in which the economical Ni was adopted as catalytic component and nano Al2O3–TiO2 hybrid composite as support. The Al2O3–TiO2 composite was prepared by spontaneous self-assembly of nano Al2O3 and TiO2 aggregates by hydrolysis of tetra-n-butyl-titanate under continuous agitation. The multi-scaled distribution of Al2O3–TiO2 aggregates with hierarchy could be observed in dynamic light scattering spectrometer. The aggregates are comprised of nano-sized γ-Al2O3 and anatase TiO2 crystallites with sizes of about 5 and 7 nm, respectively. The surface modulation by TiO2 could be verified in FTIR Spectra. The migration of Ti species and crystallite growth were hindered by the Al2O3 skeleton and the hierarchical porous structure was sustained during the thermal related process. The multi-scaled distributed pores were confirmed by both TEM analysis and N2 adsorption results. The results of dehydrogenation experiments showed that the hierarchical porous structure nano Ni/Al2O3–TiO2 exhibited superior catalytic performance to Ni/Al2O3 with the optimum conversion of 99.9% at 400 °C, while the catalyst of Ni/Al2O3 exhibited only 16.5% under the same condition.Highlights► Nanocrystallites-forming hierarchical porous Ni/Al2O3–TiO2 catalyst. ► Nano Al2O3 crystallite aggregates as bracing skeleton and TiO2 for modulation. ► Economical Ni catalysts for dehydrogenation of organic chemical hydrides. ► Catalysts exhibited superior catalytic performance to the compared one.
Co-reporter:Tao Chen, Bolun Yang, Shasha Li, Kaile Wang, Xuedong Jiang, Yong Zhang, and Guanwei He
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 19) pp:11043-11048
Publication Date(Web):August 30, 2011
DOI:10.1021/ie201188v
Nickel phosphide catalysts supported on the TiO2–Al2O3 composite oxide were successfully prepared by impregnation of nickel phosphate precursors followed by temperature-programmed reduction. The catalysts were characterized by X-ray diffraction, thermogravimetric analysis, IR spectroscopy, N2 adsorption, and X-ray photoelectron spectroscopy. The catalytic activity was characterized in a fixed-bed reactor for hydrodesulfurization using a model liquid-feed dibenzothiophene, which contained 2000 mg/kg sulfur. Experimental results indicate that the TiO2–Al2O3 composite support effectively prevents the formation of aluminum phosphates due to the strong interaction between phosphorus and γ-Al2O3, and it also shows strong interactions with Ni and P species on the surface, which is the main reason for the enhancement in hydrodesulfurization activity and stability. Among the Ni2P/TiO2–Al2O3 samples tested, the catalyst at a Ni loading of 15 wt % showed the highest activity with a steady-state dibenzothiophene conversion of 99.9% at 613 K, 2.5 MPa, WHSV 3 h–1, and volume ratio of hydrogen/oil of 450.
Co-reporter:Yu Liu, Bolun Yang, Chunhai Yi, Tao Chen, and Shasha Li
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 16) pp:9609-9616
Publication Date(Web):July 11, 2011
DOI:10.1021/ie200873w
A series of experiments were carried out in a stirred batch reactor at the temperature range of 333–363 K and the pressure of 1.5–2.5 MPa by using NKC-9 ion-exchange resin to investigate the alkylation kinetics of 3-methylthiophene (3MT) with isobutylene (IB). The effect of various operation parameters such as stirring speed, catalyst particle size, temperature, pressure, and sulfur content on the conversion and selectivity of alkylation reaction were studied. Experimental results indicated that the alkylation of 3MT with IB was followed by a parallel reaction of IB oligomerization; higher temperature could improve the alkylation activity while lower temperature was profitable to the reaction selectivity over the NKC-9 catalyst; however, the operating pressure has little effect on the alkylation and oligomerization; much sulfur content could inhibit IB dimerization. The Eley–Rideal (ER) law can be used to explain the reaction mechanism, and the kinetics models that considered the alkylation and oligomerization at the same time were established. Kinetics parameters were solved, and the calculation results agreed well with the experimental results.
Co-reporter:Yulei Guan, Bolun Yang, Suitao Qi, and Chunhai Yi
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 15) pp:9054-9062
Publication Date(Web):June 23, 2011
DOI:10.1021/ie200515g
To elucidate the accelerating effect of 1-nitropropane on the thermal cracking of normal alkanes, ab initio and density functional theory calculations were performed to investigate the elementary reactions involved in the initiated thermal cracking of a 1-nitropropane/n-heptane mixture. The kinetic parameters were evaluated on the basis of standard transition state theory (TST) or variational transition state theory (VTST) with Wigner tunneling correction. The activation energy for the C–N bond rupture of 1-nitropropane to produce primary n-propyl and nitro radicals is calculated to be 234–269 kJ/mol, while the bond dissociation energy of the C–C bond within the n-heptane molecule is predicted to be at least 335 kJ/mol. These calculated results demonstrate that the presence of 1-nitropropane makes the free radical formation become relatively easier compared with single n-heptane cracking. Furthermore, compared with the C–H bond cleavage and 1,3-H intramolecular transfer, the unstable n-propyl radical mainly follows the C–C bond cleavage pathway to produce ethylene and secondary ĊH3 radical. After formation of these free radicals, the H-abstraction of n-heptane with radicals occurs readily with considerably lower activation energy than the radical formation step to initiate the chain reaction. The analysis result indicates that the thermal cracking of n-heptane is accelerated mainly due to the change of the initial step from the C–C bond cleavage of n-heptane to the C–N bond rupture of 1-nitropropane.
Co-reporter:Xiaowei Zhou, Tao Chen, Bolun Yang, Xuedong Jiang, Hailiang Zhang, and Longyan Wang
Energy & Fuels 2011 Volume 25(Issue 6) pp:2427-2437
Publication Date(Web):May 3, 2011
DOI:10.1021/ef200316r
The lumped scheme with consideration of catalyst deactivation was adopted to simulate catalytic cracking of gasoline for predicting the product distribution of secondary reactions. A reactant oriented selective deactivation model was developed using a new strategy of selective deactivation coupled with nonselective independence deactivation. Catalyst deactivation was correlated with the time on stream rather than coke content. Furthermore, a hybrid self-adaptive genetic algorithm (termed AGA/SA), which incorporated evolution strategies and simulated annealing into a genetic algorithm, was developed and applied in parameter estimation of the proposed model. Results suggest that the lumped kinetic scheme incorporating a deactivation function could enhance its fundamentality and the predication accuracy. AGA/SA exhibits the desired improvements such as rapid convergence, high efficiency, strong in hill-climbing, and effective in escaping the local optimum. Good agreement between the predicted results and experimental data indicates that the proposed kinetic model for secondary reactions of fluid catalytic cracking (FCC) gasoline is well established and AGA/SA is reliable.
Co-reporter:Zhao Lei and Bolun Yang , Jianwei Li
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 22) pp:12392-12399
Publication Date(Web):October 11, 2011
DOI:10.1021/ie201715q
There are many methods to simulate coal structure that might be beneficial to coal technologies. However, they often suffer from producing an impractical structure and a large calculation time and could not be applied to most rank coal. Hence, the hybrid inheritance algorithms combined with Elitist strategy (HIA-ES) and Monte Carlo and Elitist Strategy (HIA-MC-ES) are proposed to simulate the coal structure. The reported fragment structures of the coal macromolecule are summarized, and the fragment structure is accurately quantified with an adjacency matrix, an atom identification vector, bonding parameters, a bond characteristic index, and a functional group characteristic parameter, respectively. The humic acid (HA) is used to test the effectiveness of two algorithms since HA has the similar fragment structures with coal. With the use of the method of HIA-ES, the numbers of fragment structures is gained. Then, the parameters of the simulated structures are obtained with the HIA-MC-ES method. Simulated HA and Shenfu coal structures are highly similar with the reported results, indicating that the two novel hybrid inheritance algorithms are effective to simulate a molecular structure for the most rank coal.
Co-reporter:Weiqin Zhao, Bolun Yang, Chunhai Yi, Zhao Lei, and Jie Xu
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 24) pp:12399-12404
Publication Date(Web):November 18, 2010
DOI:10.1021/ie101461g
A carbon-based solid acid catalyst was prepared by sulfonation of partially carbonized peanut shell, and characterized by SEM, EDS, BET analysis, FTIR spectroscopy, NH3 TPD, and TGA. The analytic results indicate that sulfonated peanut shell catalyst has an amorphous porous structure with a high acid capacity and good thermal stability and exhibits better catalytic activity for the glycerol etherification reaction than cation-exchange resin. With a molar ratio of isobutylene to glycerol of 4:1, a catalyst-to-glycerol mass ratio of 6 wt %, a reaction temperature of 343 K, and a reaction time of 2 h, glycerol was completely transformed into a mixture of glycerol ethers including mono-tert-butylglycerols (MTBGs), di-tert-butylglycerols (DTBGs), and tri-tert-butylglycerol (TTBG), and the selectivity toward the sum of the desired DTBGs and TTBG of 92.1% was obtained. Moreover, excellent reusability of the catalyst was also confirmed by repeated experiments.
Co-reporter:Xiaowei Zhou;Chunhai Yi;Jun Yuan ;Longyan Wang
Journal of Chemometrics 2010 Volume 24( Issue 9) pp:574-583
Publication Date(Web):
DOI:10.1002/cem.1317
Abstract
Levenberg–Marquardt (LM) algorithm was adopted to optimize the multiple parameters of the support vector machines (SVM) model to overcome the difficulty in selecting the parameters of SVM and to fit relational expression of high nonlinearity. Strategy of dividing the training data into working data to train SVM and the testing data so as to avoid over-fitting was performed. Comparison of the proposed LM/SVM method with three reported hybridized SVM approaches (GA/SVM, SM/SVM and SQP/SVM) was also carried out. The new method was applied in modelling for the prediction of propylene by secondary reactions of FCC gasoline. Best performance of LM/SVM employing polynomial kernel was demonstrated. Good agreement between predicted results and experimental data suggests that the LM/SVM method is successfully developed and the SVM model for increasing propylene is well established. Finally, sequential quadratic programming (SQP) algorithm was employed to optimize the operation conditions of FCC gasoline secondary reaction for maximizing the propylene yield. The obtained optimization conditions are consistent with experimental data and reported results, indicating that the optimization results are reliable. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Miaoli Hao, Bolun Yang, Haiguo Wang, Gong Liu and Suitao Qi, Jianming Yang, Chunying Li and Jian Lv
The Journal of Physical Chemistry A 2010 Volume 114(Issue 11) pp:3811-3817
Publication Date(Web):September 29, 2009
DOI:10.1021/jp9060363
To investigate the kinetics behaviors of dicyclopentadiene hydrogenation, a series of experiments were performed at different temperatures (323−353 K) under varying hydrogen pressure (0.5−1.5 MPa) with a range of Pd/C catalyst loading (0.25−1.00 wt %) using ethanol as solvent in a batch reactor. The time dependent concentration variations for each component were traced under the conditions of removing both the internal and external diffusion effects. The Langmuir−Hinshelwood mechanism was proposed with the consideration of the noncompetitive adsorption between the organic species with hydrogen, and the surface reaction was the rate-determining step. The kinetic equations for the sequence reaction were derived on the basis of the analysis of mechanisms, and the model parameters were determined by fitting the experimental data in differential temperature using the method of Runge−Kutta. The reaction activation energies for the first and second steps are 3.19 and 31.69 kJ·mol−1, respectively, and the reliability of the model was verified by these experimental results to change hydrogen pressure, reactant concentration and catalyst loading. The simulation results agreed well with the experimental data.
Co-reporter:Peng Qiu;Chunhai Yi;Suitao Qi
Catalysis Letters 2010 Volume 137( Issue 3-4) pp:232-238
Publication Date(Web):2010 July
DOI:10.1007/s10562-010-0354-8
KF/γ-Al2O3 catalysts were prepared by impregnation method and investigated for the transesterification of ethylene carbonate (EC) with ethanol to synthesize diethyl carbonate (DEC). The KF/γ-Al2O3 catalysts were characterized by nitrogen physisorption, XRD and FT-IR techniques, and three new species: K3AlF6, KOH and K2CO3 were found on the catalysts. Experimental results indicate that KOH and K2CO3 are the major active species and K3AlF6 is inactive for DEC synthesis. Increasing the KF loading favors the formation of K2CO3 and consequently enhances the activity of the KF/γ-Al2O3 catalysts. However, when KF loading exceeded 50 mmol/g, the activity of the KF/γ-Al2O3 catalysts decreased. This may be due to the presence of intact KF on the catalyst, which may dilute the content of active species in the catalyst and cover the active species. The KF/γ-Al2O3 (50 mmol/g) catalyst exhibits the best catalytic performance. With this catalyst, a 72 mol% yield of DEC (based on EC) was obtained at 298 K.
Co-reporter:H. Yuan, B. L. Yang and G. L. Zhu
Energy & Fuels 2009 Volume 23(Issue 1) pp:548
Publication Date(Web):December 9, 2008
DOI:10.1021/ef800577j
To enhance the synthesis process for biodiesel (fatty acid methyl ester, FAME), microwave absorption solid acid catalysts were used for transesterification under microwave radiation. The H2SO4/C catalyst was prepared by an impregnation method. The surface areas of active carbon and H2SO4/C were measured by the BET method. Synthesis reactions were carried out under different conditions using castor oil and methanol as the feedstock. The amounts of FAME in the product were analyzed by high-performance liquid chromatography. Experimental results showed that the microwave radiation exhibited a notable enhanced effect for transesterification by using the microwave absorption solid acid catalysts (H2SO4/C) compared with that of the conventional heating method. When the transesterification was carried out at 338 K, with 12:1 molar ratio of methanol to castor oil, 50 wt % loading amounts of H2SO4 and 5 wt % of catalyst to castor oil, after 60 min the yield of FAME 94 wt % was obtained. This method can also be used in the case of castor oil with high free fatty acid.
Co-reporter:Kaile Wang, Bolun Yang, Yu Liu and Chunhai Yi
Energy & Fuels 2009 Volume 23(Issue 9) pp:4209-4214
Publication Date(Web):July 23, 2009
DOI:10.1021/ef9002523
Composite TiO2−Al2O3 supports were prepared by the sol−gel technique using tetra-n-butyl-titanate and γ-Al2O3 as raw materials, and the Ni2P/TiO2−Al2O3 catalysts were obtained by incipient wetness impregnation of aqueous metal phosphate precursors, followed by temperature-programmed reduction of inflowing H2. The supports and catalysts were characterized by X-ray diffraction, infrared spectroscopy, transmission electron microscopy, and N2 adsorption. The hydrodesulfurization (HDS) activity was examined in a fixed-bed reactor. Experimental results indicate that the composite support can effectively prevent the formation of aluminum phosphates based on the strong interaction between P and γ-Al2O3, overcome the disadvantage of low surface titania, and improve metal support interaction, which significantly increase catalyst activity and selectivity. The Ni2P/TiO2−Al2O3 catalyst exhibits good activity for the HDS of 3-methylthiophene (3-MT), used as a model compound. The activity and stability of the Ni2P/TiO2−Al2O3 catalyst are affected by the phosphorus content, both achieving a maximum with an initial Ni/P molar ratio of 1:2. The reaction temperature and the weight hourly space velocity (WHSV) show significant influence, but the reaction pressure and the volume ratio of hydrogen/oil have little effect on the HDS performance of the Ni2P/TiO2−Al2O3 catalyst. The conversion of 3-MT is close to 100% when the reaction temperature reaches 603 K, reaction pressure reaches 2.0 MPa, WHSV reaches 1.275 h−1, and volume ratio of hydrogen/oil reaches 400.
Co-reporter:Hong Yuan;Jiming Yang
Journal of the American Oil Chemists' Society 2009 Volume 86( Issue 4) pp:375-382
Publication Date(Web):2009 April
DOI:10.1007/s11746-009-1354-y
A new topological index for individual components based on the information of molar mass, bond length and bond energy was established to reflect the molecular structure of fatty acid methyl esters (FAME). Combined with the modified Grunberg–Nissan or Hind equation, the two series mixture topological index values of the biodiesels (mixture of FAME) were calculated, respectively. Some basic properties such as the density, viscosity, flash point (FP), high heating value (HHV) for biodiesel were correlated with these mixture topological indexes to find the relationship of structure and properties. The results show that the topological index can reflect the information of the molecular structure for FAME, such as the size of molecule, unsaturated bond, intensity of bond and branch degree. The modified Grunberg–Nissan equation has a higher precision of predicting for the properties of biodiesel studied than the Hind equation.
Co-reporter:Bolun Yang, Xiaowei Zhou, Chun Chen, Jun Yuan and Longyan Wang
Industrial & Engineering Chemistry Research 2008 Volume 47(Issue 14) pp:4648-4657
Publication Date(Web):June 19, 2008
DOI:10.1021/ie800023x
To overcome the drawback of the traditional lumping method, a new method constructed from structure oriented lumping (SOL) combined with Monte Carlo (MC) to simulate the secondary reactions process of fluid catalytic cracking (FCC) gasoline was developed. The SOL method was applied to represent the feedstocks and products configuration framework; then, more than 60 item reaction rules were established to produce all the reaction networks for the secondary reactions of FCC gasoline. By integral calculating each molecule reaction probability using the MC method, the product distribution thus can be obtained. Three samples of catalytic cracking gasoline, taken from the industrial FCC units of China have been used as feedstock of the simulation to check the validity of the proposed method. Some integral properties, such as average molecular weight, element compositions, and hydrocarbon compositions were predicted. The yield of upgraded gasoline, dry gas, liquefied petroleum gas (LPG), light cycle oil (LCO), heavy oil, coke, and olefin changes with extent of reaction were also simulated with this method and the predicted results agreed well with the experimental data.
Co-reporter:Jianjun Sun, Bolun Yang, Hongye Lin, Xiaoping Wang, Dongpeng Wang
Journal of Organometallic Chemistry 2005 Volume 690(Issue 5) pp:1300-1305
Publication Date(Web):1 March 2005
DOI:10.1016/j.jorganchem.2004.11.039
A new process for synthesis of 1,1,3,3-tetrabutyl-1-methoxy-3-isocyanatodistannoxane (TBMI) from dibutyltin oxide (DSnO), methyl carbamate and methanol was proposed. The structure of the TBMI was confirmed by UV–Vis, elemental analyses, FTIR and HPLC. The effects of various conditions, such as reaction temperature, pressure, reaction time, molar ratio of the reactants and the stirring speed were investigated in the terms of TBMI yield. The experimental results indicated that the optimal reaction conditions were the molar ratio of methyl carbamate:DSnO of 2:1, the reaction time of 3 h, the reaction temperature of 433 K, the methanol:DSnO molar ratio of 2:1, the initial pressure of 0.4 MPa and the stirring speed of 1000 rpm, respectively. TBMI yield of 96.9% was obtained in the optimal reaction conditions. This process shows some advantages like easy to operate, higher yield, shorter reaction time and lower cost.A new process for synthesis of 1,1,3,3-tetrabutyl-1-methoxy-3-isocyanatodistannoxane (TBMI) from dibutyltin oxide (DSnO), methyl carbamate and methanol was proposed. The structure of the TBMI was confirmed by UV–Vis, elemental analyses, FTIR and HPLC. The effects of various conditions, such as reaction temperature, pressure, reaction time, molar ratio of the reactants and the stirring speed were investigated in the terms of TBMI yield.
Co-reporter:Yongjun He, Bolun Yang, Guangxu Cheng
Materials Letters 2003 Volume 57(13–14) pp:1880-1884
Publication Date(Web):April 2003
DOI:10.1016/S0167-577X(02)01093-5
CeO2 nanoparticles with good monodispersity and narrow size distribution were synthesized by the combined method of homogenous precipitation with microemulsion. The affect factors of preparing conditions were investigated. The products were characterized by XRD, TEM, FTIR, DTA and TGA. It was found that the average size of CeO2 nanoparticles could be tailored by adjusting the precipitation temperature, calcination temperature, and the sort of surfactants or the relative concentration of reactants.
Co-reporter:Xingxing Li, Bolun Yang, Yong Zhang
Journal of Process Control (November 2013) Volume 23(Issue 10) pp:1360-1370
Publication Date(Web):1 November 2013
DOI:10.1016/j.jprocont.2013.09.003
•Nonlinear and linear characteristics of the methanation reactor are analyzed.•Effects of mass, heat recycle and split ratio on reactor stability are revealed.•Linear model implies that the integral element is necessary for reactor control.•The High Selector module can maintain the real peak temperature well.Both nonlinear and linear dynamic characteristics of the low temperature methanation reactor with recycle loop are analyzed for the process safe operation and effective control. Aspen Dynamics tool is adopted to get transient behaviors of the reactor under step changes of the inlet temperature and CO mole fraction. A double-input-multi-output linearized system based on the nonlinear dynamic model is developed for further insight into the process stability, response rapidity and controllability. Similar responses are obtained in the nonlinear and linear models for commendable mutual authenticating. The presence of material recycle because of the unconverted reactants, together with the thermal recycle for energy saving, renders a more sensitive stability of the system, where thermal feedback has the major effect, but the existing of mass feedback may weaken this effect when considerable heat is recycled. Moreover, the transfer function of the linear model indicates that the integral term is necessary for the reactor variables control, and the proposed control strategy using a High Selector module in Aspen Dynamics can successfully maintain the hot spot temperature within the reactor by considering the migration of the peak temperature under the two disturbances.
Co-reporter:Xingxing Li, Jiageng Li, Bolun Yang, Yong Zhang
Chinese Journal of Chemical Engineering (February 2015) Volume 23(Issue 2) pp:389-397
Publication Date(Web):1 February 2015
DOI:10.1016/j.cjche.2014.11.007
A double-input–multi-output linearized system is developed using the state-space method for dynamic analysis of methanation process of coke oven gas. The stability of reactor alone and reactor with feed-effluent heat exchanger is compared through the dominant poles of the system transfer functions. With single or double disturbance of temperature and CO concentration at the reactor inlet, typical dynamic behavior in the reactor, including fast concentration response, slow temperature response and inverse response, is revealed for further understanding of the counteraction and synergy effects caused by simultaneous variation of concentration and temperature. Analysis results show that the stability of the reactor loop is more sensitive than that of reactor alone due to the positive heat feedback. Remarkably, with the decrease of heat exchange efficiency, the reactor system may display limit cycle behavior for a pair of complex conjugate poles across the imaginary axis.The stability of the methanation reactor with feed-effluent heat exchanger is more sensitive than that of reactor alone. Remarkably, with the decrease of the heat exchange efficiency, temperature and concentration oscillations have been confirmed because of the presence of inverse temperature response through the reactor combined with the positive feedback caused by the feed-effluent heat exchanger.Download full-size image
Co-reporter:Lu Wang, Elizabeth G. Mahoney, Shen Zhao, Bolun Yang and Jingguang G. Chen
Chemical Communications 2016 - vol. 52(Issue 18) pp:NaN3700-3700
Publication Date(Web):2016/02/02
DOI:10.1039/C5CC10439D
Low-loadings of Pt supported over six transition metal carbide (Pt/TMC) powder catalysts were synthesized and evaluated for hydrogen oxidation and evolution reactions in an alkaline electrolyte. The roughness factor of each Pt/TMC catalyst was different, indicating that the carbide supports affect the dispersion of Pt. Furthermore, when normalized by the corresponding roughness factors, all Pt/TMC catalysts were found to have similar intrinsic activities that were comparable to the state-of-the-art commercial Pt/C electrocatalysts.

![Benzenemethanaminium, 4-ethenyl-N,N,N-trimethyl-, salt with 1,1,1-trifluoro-N-[(trifluoromethyl)sulfonyl]methanesulfonamide (1:1), homopolymer](/data/chemimg/3779900/876048-28-1.png)
![Benzenemethanaminium, 4-ethenyl-N,N,N-trimethyl-, salt with 1,1,1-trifluoro-N-[(trifluoromethyl)sulfonyl]methanesulfonamide (1:1), homopolymer](/data/chemimg/3779900/876048-28-1_b.png)





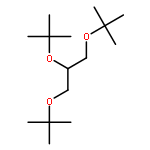
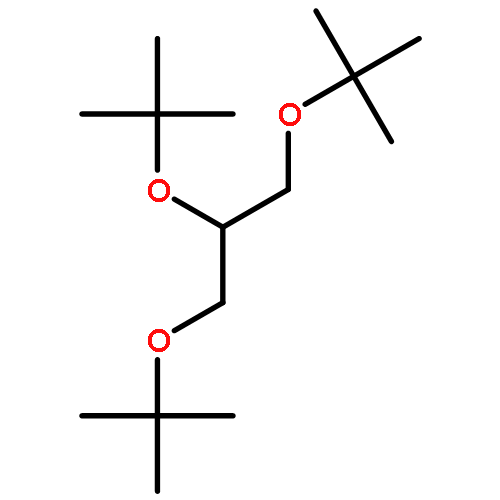
![1,3-BIS[(2-METHYLPROPAN-2-YL)OXY]PROPAN-2-OL](http://img.cochemist.com/ccimg/79900/79808-30-3.png)
![1,3-BIS[(2-METHYLPROPAN-2-YL)OXY]PROPAN-2-OL](http://img.cochemist.com/ccimg/79900/79808-30-3_b.png)

![2-[(2-METHYLPROPAN-2-YL)OXY]PROPANE-1,3-DIOL](http://img.cochemist.com/ccimg/73800/73757-68-3.png)
![2-[(2-METHYLPROPAN-2-YL)OXY]PROPANE-1,3-DIOL](http://img.cochemist.com/ccimg/73800/73757-68-3_b.png)