Co-reporter:Wenfeng Lu, Yongqiang Liu, Haikuo Ma, Jiyue Zheng, Sheng Tian, Zhijian Sun, Lusong Luo, Jiajun Li, Hongjian Zhang, Zeng-Jie Yang, and Xiaohu Zhang
ACS Chemical Neuroscience September 20, 2017 Volume 8(Issue 9) pp:1980-1980
Publication Date(Web):June 15, 2017
DOI:10.1021/acschemneuro.7b00153
Medulloblastoma is one of the most prevalent brain tumors in children. Aberrant hedgehog (Hh) pathway signaling is thought to be involved in the initiation and development of medulloblastoma. Vismodegib, the first FDA-approved cancer therapy based on inhibition of aberrant hedgehog signaling, targets smoothened (Smo), a G-protein coupled receptor (GPCR) central to the Hh pathway. Although vismodegib exhibits promising therapeutic efficacy in tumor treatment, concerns have been raised from its nonlinear pharmacokinetic (PK) profiles at high doses partly due to low aqueous solubility. Many patients experience adverse events such as muscle spasms and weight loss. In addition, drug resistance often arises among tumor cells during treatment with vismodegib. There is clearly an urgent need to explore novel Smo antagonists with improved potency and efficacy. Through a scaffold hopping strategy, we have identified a series of novel tetrahydropyrido[4,3-d]pyrimidine derivatives, which exhibited effective inhibition of Hh signaling. Among them, compound 24 is three times more potent than vismodegib in the NIH3T3-GRE-Luc reporter gene assay. Compound 24 has a lower melting point and much greater solubility compared with vismodegib, resulting in linear PK profiles when dosed orally at 10, 30, and 100 mg/kg in rats. Furthermore, compound 24 showed excellent PK profiles with a 72% oral bioavailability in beagle dogs. Compound 24 demonstrated overall favorable in vitro safety profiles with respect to CYP isoform and hERG inhibition. Finally, compound 24 led to significant regression of subcutaneous tumor generated by primary Ptch1-deficient medulloblastoma cells in SCID mouse. In conclusion, tetrahydropyrido[4,3-d]pyrimidine derivatives represent a novel set of Smo inhibitors that could potentially be utilized to treat medulloblastoma and other Hh pathway related malignancies.Keywords: antagonist; cancer therapy; GPCR; hedgehog signaling pathway; medulloblastoma; scaffold hopping; Smoothened;
Co-reporter:Zhixiang Xu, Jiajun Li, Yiyuan Wu, Zhijian Sun, Lusong Luo, Zhilin Hu, Sudan He, Jiyue Zheng, Hongjian Zhang, Xiaohu Zhang
European Journal of Medicinal Chemistry 2016 Volume 108() pp:154-165
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.11.026
•A novel series of Wnt antagonists were developed by a scaffold hybridization strategy.•Many compounds exhibited Wnt inhibition at low nM range.•Compound 59 showed excellent chemical, plasma, and liver microsomal stability.•Compound 59 demonstrated good PK profiles with 30% oral bioavailability in rat.The Wnt signaling pathway is a critical developmental pathway which operates through control of cellular functions such as proliferation and differentiation. Aberrant Wnt signaling has been linked to the formation and metastasis of tumors. Porcupine, a member of the membrane-bound O-acyltransferase family of proteins, is an important component of the Wnt pathway. Porcupine catalyzes the palmitoylation of Wnt proteins, a process needed for their secretion and activity. Here we report a novel series of compounds obtained by a scaffold hybridization strategy from a known porcupine inhibitor class. The leading compound 59 demonstrated subnanomolar inhibition of Wnt signaling in a paracrine cellular assay. Compound 59 also showed excellent chemical, plasma and liver microsomal stabilities. Furthermore, compound 59 exhibited good pharmacokinetic profiles with 30% oral bioavailability in rat. Collectively, these results strongly support further optimization of this novel scaffold to develop better Wnt pathway inhibitors.
Co-reporter:Zhaohui Yang, Linlang Li, Jiyue Zheng, Haikuo Ma, Sheng Tian, Jiajun Li, Hongjian Zhang, Xuechu Zhen, and Xiaohu Zhang
ACS Chemical Neuroscience 2016 Volume 7(Issue 11) pp:1575
Publication Date(Web):August 28, 2016
DOI:10.1021/acschemneuro.6b00218
Adenosine receptor A2A antagonists have emerged as potential treatment for Parkinson’s disease in the past decade. We have recently reported a series of adenosine receptor antagonists using heterocycles as bioisosteres for a potentially unstable acetamide. These compounds, while showing excellent potency and ligand efficiency, suffered from moderate cytochrome P450 inhibition and high clearance. Here we report a new series of adenosine receptor A2A antagonists based on a 4-amino-5-carbonitrile pyrimidine template. Compounds from this new template exhibit excellent potency and ligand efficiency with low cytochrome P450 inhibition. Although the clearance remains moderate to high, the leading compound, when dosed orally as low as 3 mg/kg, demonstrated excellent efficacy in the haloperidol induced catalepsy rat model for Parkinson’s disease.Keywords: Adenosine receptor; antagonist; GPCR; lead identification; Parkinson’s disease
Co-reporter:Zhixiang Xu, Xiangxiang Xu, Ruadhan O’Laoi, Haikuo Ma, Jiyue Zheng, Shuaishuai Chen, Lusong Luo, Zhilin Hu, Sudan He, Jiajun Li, Hongjian Zhang, Xiaohu Zhang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 22) pp:5861-5872
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmc.2016.09.041
The Wnt signaling pathway is an essential signal transduction pathway which leads to the regulation of cellular processes such as proliferation, differentiation and migration. Aberrant Wnt signaling is known to have an association with multiple cancers. Porcupine is an enzyme that catalyses the addition of palmitoleate to a serine residue in Wnt proteins, a process which is required for the secretion of Wnt proteins. Here we report the synthesis and structure–activity-relationship of the novel porcupine inhibitors based on a ‘reversed’ amide scaffold. The leading compound 53 was as potent as the clinical compound LGK974 in a cell based STF reporter gene assay. Compound 53 potently inhibited the secretion of Wnt3A, therefore was confirmed to be a porcupine inhibitor. Furthermore, compound 53 showed excellent chemical and plasma stabilities. However, the clearance of compound 53 in liver microsomal tests was moderate to high, and the solubility of compound 53 was suboptimal. Collective efforts toward further optimization of this novel tricyclic template to develop better porcupine inhibitors will be subsequently undertaken and reported in due course.Download high-res image (92KB)Download full-size image
Co-reporter:Haikuo Ma, Wenfeng Lu, Zhijian Sun, Lusong Luo, Delong Geng, Zhaohui Yang, Enqin Li, Jiyue Zheng, Meiyu Wang, Hongjian Zhang, Shilin Yang, Xiaohu Zhang
European Journal of Medicinal Chemistry 2015 Volume 89() pp:721-732
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.11.006
•A novel series of SMO antagonists were developed by a scaffold hopping strategy.•Many compounds exhibited improved biological activities compared to Vismodegib.•Compound 30 showed improved physical–chemical properties compared to Vismodegib.•Compound 30 demonstrated good PK profiles with a 77% oral bioavailability.The Smoothened (Smo) receptor is an important component of the hedgehog (Hh) signaling pathway, which plays a critical role during embryonic development. In adults, Hh signaling is curtailed and has limited functions such as stem cell maintenance and tissue repair. However, aberrant activity of the Hh signaling in adults has been linked to numerous human cancers. Inhibition of Smo leads to the blockade of Hh signaling, and therefore represents a promising approach toward novel anticancer therapy. Through scaffold morphing of a few known Smo antagonists, a series of novel tetrahydrothiazolopyridine derivatives were developed. Compounds from this new scaffold demonstrated excellent Hh signaling inhibition which was comparable to or better than that of Vismodegib. Further, compound 30 exhibited a lower melting point and a moderately improved solubility compared with those of Vismodegib; compounds 11 and 30 showed good pharmacokinetic profiles with 34% and 77% oral bioavailability in rat, respectively. Collectively, these results strongly support further optimization of this novel scaffold to develop better Smo antagonists.A novel series of smoothened antagonists were developed by a scaffold hopping strategy. Compounds from this new template demonstrated improved biological activities and physical–chemical properties compared with those of Vismodegib.
Co-reporter:Yan Dong, Kehuang Li, Zhixiang Xu, Haikuo Ma, Jiyue Zheng, Zhilin Hu, Sudan He, Yiyuan Wu, Zhijian Sun, Lusong Luo, Jiajun Li, Hongjian Zhang, Xiaohu Zhang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 21) pp:6855-6868
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmc.2015.09.048
The Wnt signaling pathway is a pivotal developmental pathway. It operates through control of cellular functions such as proliferation, differentiation, migration and polarity. Aberrant Wnt signaling has been implicated in the formation and metastasis of tumors. Porcupine is a component of the Wnt signaling pathway. It is a member of the membrane-bound O-acyltransferase family of proteins. Porcupine catalyzes the palmitoylation of Wnt proteins, a process which is essential to their secretion and activity. Here we report a novel series of compounds obtained by a scaffold hybridization strategy from two known porcupine inhibitor classes. The leading compound 62 demonstrated subnanomolar (IC50 0.11 nM) inhibition of Wnt signaling in a paracrine cellular reporter gene assay. Compound 62 also potently inhibited Wnt secretion into culture medium, an indication of direct inhibition of the porcupine protein. Furthermore, compound 62 showed excellent chemical, plasma and liver microsomal stabilities. Collectively, these results strongly support further optimization of this novel scaffold to develop better Wnt pathway inhibitors.
Co-reporter:Zhaohui Yang, Haikuo Ma, Zhijian Sun, Lusong Luo, Sheng Tian, Jiyue Zheng, Xiaohu Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3665-3670
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.06.049
Vismodegib is the first FDA approved cancer therapy based on inhibition of aberrant hedgehog signaling. Like most cancer therapies, vismodegib suffered from resistance, even during clinical development. Numerous reports demonstrated that simultaneous blockage of hedgehog and PI3K/AKT/mTOR pathways resulted in significantly superior outcomes compared with single agent alone in a number of animal disease models. The dual hedgehog and PI3K/AKT/mTOR inhibition represented a promising approach not only to overcoming the resistance but also to delaying its onset. Here we report a series of compounds based on a 6-(pyridin-3-yl)benzo[d]thiazole template which have demonstrated significant inhibition of both hedgehog and PI3K/AKT/mTOR signaling pathways. This new scaffold can serve as a lead for further optimization.Vismodegib is the first FDA approved cancer therapy based on inhibition of aberrant hedgehog signaling. Like most cancer therapies, vismodegib suffered from resistance, even during clinical development. Numerous reports demonstrated that simultaneous blockage of hedgehog and PI3K/AKT/mTOR pathways resulted in significantly superior outcomes compared with single agent alone in a number of animal disease models. The dual hedgehog and PI3K/AKT/mTOR inhibition represented a promising approach not only to overcoming the resistance but also to delaying its onset. Here we report a series of compounds based on a 6-(pyridin-3-yl)benzo[d]thiazole template which have demonstrated significant inhibition of both hedgehog and PI3K/AKT/mTOR signaling pathways. This new scaffold can serve as a lead for further optimization.
Co-reporter:Jiyue Zheng, Zhaohui Yang, Xuan Li, Linlang Li, Haikuo Ma, Meiyu Wang, Hongjian Zhang, Xuechu Zhen, and Xiaohu Zhang
ACS Chemical Neuroscience 2014 Volume 5(Issue 8) pp:674
Publication Date(Web):June 12, 2014
DOI:10.1021/cn5000716
Parkinson’s disease is a neurodegenerative disease characterized by the motor symptoms of bradykinesia, tremor, and rigidity. Current therapies are based mainly on dopaminergic replacement strategies by administration of either dopamine agonists or dopamine precursor levodopa (L-Dopa). These treatments provide symptomatic relief without slowing or stopping the disease progression, and long-term usage of these drugs is associated with diminished efficacy, motor fluctuation, and dyskinisia. Unfortunately, there had been few novel treatments developed in the past decades. Among nondopaminergic strategies for the treatment of Parkinson’s disease, antagonism of the adenosine A2A receptor has emerged to show great potential. Here we report the optimization of a new chemical scaffold, which achieved exceptional receptor binding affinity and ligand efficiency against adenosine A2A receptor. The leading compounds demonstrated excellent efficacy in the haloperidol induced catalepsy model for Parkinson’s disease.Keywords: Adenosine receptor; antagonist; GPCR; lead optimization; Parkinson’s disease
Co-reporter:Zhaohui Yang, Xuan Li, Haikuo Ma, Jiyue Zheng, Xuechu Zhen, Xiaohu Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 1) pp:152-155
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmcl.2013.11.051
We have previously reported a series of 2,4,6-trisubstituted pyrimidines as potent A2A receptor antagonists. The leading compounds often feature a potentially labile acetamide functional group which tends to hydrolyze under acidic conditions. Here we report the replacement of the acetamide functional group with bioisosteres. This effort led us to a new series of adenosine A2A receptor antagonists with improved potency and chemical stability.We have previously reported a series of 2,4,6-trisubstituted pyrimidines as potent A2A receptor antagonists. The leading compounds often feature a potentially labile acetamide functional group which tends to hydrolyze under acidic conditions. Here we report the replacement of the acetamide functional group with bioisosteres. This effort led us to a new series of adenosine A2A receptor antagonists with improved potency and chemical stability.
Co-reporter:Wenfeng Lu, Delong Geng, Zhijian Sun, Zhaohui Yang, Haikuo Ma, Jiyue Zheng, Xiaohu Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 10) pp:2300-2304
Publication Date(Web):15 May 2014
DOI:10.1016/j.bmcl.2014.03.079
The hedgehog (Hh) signaling pathway is a key regulator during embryonic development, while in adults, it has limited functions such as stem cell maintenance and tissue repair. The aberrant activity of the Hh signaling in adults has been linked to numerous human cancers. Inhibition of Hh signaling therefore represents a promising approach toward novel anticancer therapies. The Smoothened (Smo) receptor mediates Hh signaling. Here we report a new series of Smo antagonists which were obtained by a scaffold hopping strategy. Compounds from this new scaffold demonstrated decent inhibition of Hh pathway signaling. The new scaffold can serve as a starting point for further optimization.The hedgehog (Hh) signaling pathway is a key regulator during embryonic development, while in adults, it has limited functions such as stem cell maintenance and tissue repair. The aberrant activity of the Hh signaling in adults has been linked to numerous human cancers. Inhibition of Hh signaling therefore represents a promising approach toward novel anticancer therapies. The Smoothened (Smo) receptor mediates Hh signaling. Here we report a new series of Smo antagonists which were obtained by a scaffold hopping strategy. Compounds from this new scaffold demonstrated decent inhibition of Hh pathway signaling. The new scaffold can serve as a starting point for further optimization.
Co-reporter:Zhixiang Xu, Jing Wu, Jiyue Zheng, Haikuo Ma, Hongjian Zhang, Xuechu Zhen, Long Tai Zheng, Xiaohu Zhang
International Immunopharmacology (April 2015) Volume 25(Issue 2) pp:528-537
Publication Date(Web):1 April 2015
DOI:10.1016/j.intimp.2015.02.033
•A number of non-steroidal anti-inflammatory drug (NSAID) conjugates were synthesized.•Among the tested analogues, diclofenac-cysteamine conjugate exhibited potent inhibitory activities on NO production.•Diclofenac-cysteamine conjugate suppressed the pro-inflammatory gene expression by inhibiting MAPKs/AP-1 pathway.•Diclofenac-cysteamine conjugate displayed neuroprotective activity in vitro.Neuroinflammation is involved in the process of several central nervous system (CNS) diseases such as Parkinson's disease, Alzheimer's disease, ischemia and multiple sclerosis. As the macrophages in the central nervous system, microglial cell function in the innate immunity of the brain and are largely responsible for the inflammation-mediated neurotoxicity. Prevention of microglia activation might alleviate neuronal damage and degeneration under the inflammatory conditions, and therefore, represents a possible therapeutic approach to the aforementioned CNS diseases. Here we report the synthesis of a number of non-steroidal anti-inflammatory drug (NSAID) conjugates, and the evaluation of their anti-inflammatory effects in lipopolysaccharide (LPS)-stimulated BV-2 microglial cells and primary mouse microglial cells. Among the tested analogues, compounds 8 and 11 demonstrated potent inhibition of nitric oxide production with no or weak cell toxicity. Compound 8 also significantly suppressed the expression of tumor necrosis factor (TNF)-α, interleukin (IL)-6, cyclooxygenase (COX)-2 as well as inducible nitric oxide synthase (iNOS) in LPS-stimulated BV-2 microglial cells. Further mechanistic studies indicated that compound 8 significantly suppressed phosphorylation of mitogen-activated protein kinases (MAPKs) and subsequent activation of activator of transcription 1 (AP-1). Furthermore, in a co-culture system, compound 8 inhibited the cytotoxicity generated by LPS-activated microglia toward HT-22 neuroblastoma cells. Collectively, these experimental results demonstrated that compound 8 possessed potent anti-neuroinflammatory activity via inhibition of microglia activation, and might serve as a potential lead for the therapeutic treatment of neuroinflammatory diseases.



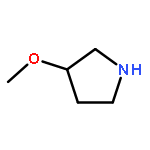

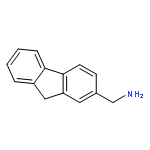
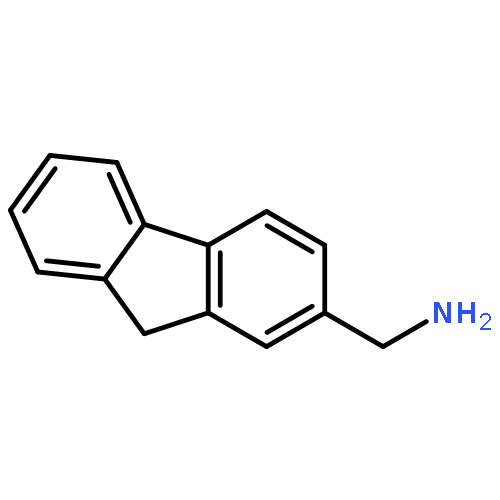


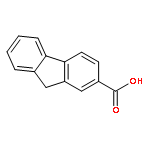
![Dibenzo[b,d]furan-3-carbonitrile](http://img.cochemist.com/ccimg/29100/29021-90-7.png)
![Dibenzo[b,d]furan-3-carbonitrile](http://img.cochemist.com/ccimg/29100/29021-90-7_b.png)








![IMIDAZO[1,2-A]PYRAZIN-2-AMINE](/data/chemimg/453000/1289267-53-3.png)
![IMIDAZO[1,2-A]PYRAZIN-2-AMINE](/data/chemimg/453000/1289267-53-3_b.png)
![tert-Butyl 2-(hydroxymethyl)-5,6-dihydroimidazo[1,2-a]pyrazine-7(8H)-carboxylate](http://img.cochemist.com/ccimg/1251000/1250996-70-3.png)
![tert-Butyl 2-(hydroxymethyl)-5,6-dihydroimidazo[1,2-a]pyrazine-7(8H)-carboxylate](http://img.cochemist.com/ccimg/1251000/1250996-70-3_b.png)
![6-CHLORO-1-CYCLOBUTYL-3-METHYL-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/1032100/1032047-47-4.png)
![6-CHLORO-1-CYCLOBUTYL-3-METHYL-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/1032100/1032047-47-4_b.png)
![Acetamide,N-[6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-benzothiazolyl]-](http://img.cochemist.com/ccimg/885100/885069-14-7.png)
![Acetamide,N-[6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-benzothiazolyl]-](http://img.cochemist.com/ccimg/885100/885069-14-7_b.png)














![Thiazolo[5,4-c]pyridin-2-amine, N-ethyl-4,5,6,7-tetrahydro-](http://img.cochemist.com/ccimg/124500/124458-24-8.png)
![Thiazolo[5,4-c]pyridin-2-amine, N-ethyl-4,5,6,7-tetrahydro-](http://img.cochemist.com/ccimg/124500/124458-24-8_b.png)





![4,5,6,7-Tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/97900/97817-23-7.png)
![4,5,6,7-Tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/97900/97817-23-7_b.png)
![Ethyl 5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine-2-carboxylate](http://img.cochemist.com/ccimg/91500/91476-82-3.png)
![Ethyl 5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine-2-carboxylate](http://img.cochemist.com/ccimg/91500/91476-82-3_b.png)


![ETHYL [2-(3-METHOXYPHENYL)ETHYL]CARBAMATE](http://img.cochemist.com/ccimg/33600/33543-63-4.png)
![ETHYL [2-(3-METHOXYPHENYL)ETHYL]CARBAMATE](http://img.cochemist.com/ccimg/33600/33543-63-4_b.png)


![N-(3-CHLORO-4-FLUOROPHENYL)-N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-1<WBR />,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/3300/3289-77-8.png)
![N-(3-CHLORO-4-FLUOROPHENYL)-N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-1<WBR />,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/3300/3289-77-8_b.png)