Co-reporter:Zhen Han, Chau-wen Chou, Xiangkun Yang, Michael G. Bartlett, and Y. George Zheng
ACS Chemical Biology June 16, 2017 Volume 12(Issue 6) pp:1547-1547
Publication Date(Web):April 20, 2017
DOI:10.1021/acschembio.7b00114
p300 and GCN5 are two representative lysine acetyltransferases (KATs) in mammalian cells. It was recently reported that they possess multiple acyltransferase activities including acetylation, propionylation, and butyrylation of the ε-amino group of lysine residues of histones and non-histone protein substrates. Although thousands of acetylated substrates and acetylation sites have been identified by mass spectrometry-based proteomic screening, our knowledge about the causative connections between individual KAT members and their corresponding sub-acylomes remain very limited. Herein, we applied 3-azidopropionyl CoA (3AZ-CoA) as a bioorthogonal surrogate of acetyl-, propionyl- and butyryl-CoA for KAT substrate identification. We successfully attached the azide as a chemical warhead to cellular substrates of wild-type p300 and engineered GCN5. The substrates were subsequently labeled with biotin tag through the copper-catalyzed azide–alkyne cycloaddition (CuAAC). Following protein enrichment on streptavidin-coated resin, we conducted LC-MS/MS studies from which more than four hundred proteins were identified as GCN5 or p300 substrate candidates. These proteins are either p300- or GCN5-unique or shared by the two KATs and are extensively involved in various biological events including gene expression, cell cycle, and cellular metabolism. We also experimentally validated two novel substrates of GCN5, that is, IQGAP1 and SMC1. These results demonstrate extensive engagement of GCN5 and p300 in cellular pathways and provide new insights into understanding their functions in specific biological processes.
Co-reporter:Jing Zhang;Kun Qian;Chunli Yan;Maomao He;Brenson A. Jassim;Ivaylo Ivanov
MedChemComm (2010-Present) 2017 vol. 8(Issue 2) pp:440-444
Publication Date(Web):2017/02/22
DOI:10.1039/C6MD00573J
Protein arginine methyltransferase 1 (PRMT1) is a key player for the dynamic regulation of arginine methylation. Its dysregulation and aberrant expression are implicated in various pathological conditions, and a plethora of evidence suggests that PRMT1 inhibition is of significant therapeutic value. Herein, we reported the modification of a series of diamidine compounds with varied lengths in the middle alkyl linker for PRMT1 inhibition. Decamidine (2j), which possesses the longest linker in the series, displayed 2- and 4-fold increase in PRMT1 inhibition (IC50 = 13 μM), compared with furamidine and stilbamidine. The inhibitory activity toward PRMT1 was validated by secondary orthogonal assays. Docking studies showed that the increased activity is due to the extra interaction of the amidine group with the SAM binding pocket, which is absent when the linker is not long enough. These results provide structural insights into developing the amidine type of PRMT1 inhibitors.
Co-reporter:Jing Zhang and Yujun George Zheng
ACS Chemical Biology 2016 Volume 11(Issue 3) pp:583
Publication Date(Web):November 5, 2015
DOI:10.1021/acschembio.5b00812
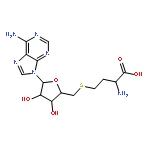
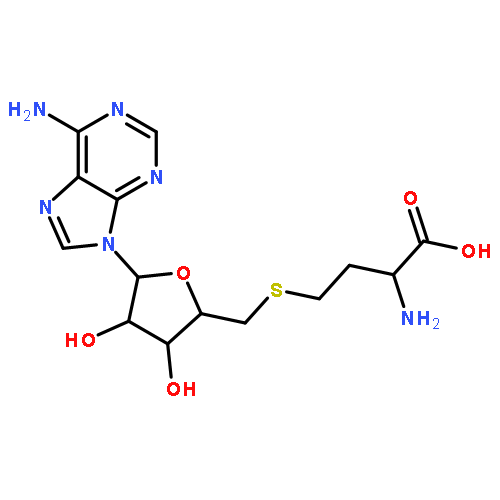
S-Adenosyl-L-methionine (SAM) is a sulfonium molecule with a structural hybrid of methionine and adenosine. As the second largest cofactor in the human body, its major function is to serve as methyl donor for SAM-dependent methyltransferases (MTases). The resultant transmethylation of biomolecules constitutes a significant biochemical mechanism in epigenetic regulation, cellular signaling, and metabolite degradation. Recently, numerous SAM analogs have been developed as synthetic cofactors to transfer the activated groups on MTase substrates for downstream ligation and identification. Meanwhile, new compounds built upon or derived from the SAM scaffold have been designed and tested as selective inhibitors for important MTase targets. Here, we summarized the recent development and application of SAM analogs as chemical biology tools for MTases.
Co-reporter:Yepeng Luan, Levi L. Blazer, Hao Hu, Taraneh Hajian, Jing Zhang, Hong Wu, Scott Houliston, Cheryl H. Arrowsmith, Masoud Vedadi and Yujun George Zheng
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 2) pp:631-638
Publication Date(Web):29 Oct 2015
DOI:10.1039/C5OB01794G
The histone methyltransferase MLL1 has been linked to translocation-associated gene fusion in childhood leukemias and is an attractive drug target. High-throughput biochemical analysis of MLL1 methyltransferase activity requires the production of at least a trimeric complex of MLL1, RbBP5 and WDR5 to elicit robust activity. Production of trimeric and higher order MLL1 complexes in the quantities and reproducibility required for high-throughput screening presents a significant impediment to MLL1 drug discovery efforts. We present here a small molecule fluorescent ligand (FL-NAH, 6) that is able to bind to the S-adenosylmethionine (SAM) binding site of MLL1 in a manner independent of the associated complex members. We have used FL-NAH to develop a fluorescence polarization-based SAM displacement assay in a 384-well format targeting the MLL1 SET domain in the absence of associated complex members. FL-NAH competes with SAM and is displaced from the MLL1 SET domain by other SAM-binding site ligands with Kdisp values similar to the higher-order complexes, but is unaffected by the H3 peptide substrate. This assay enables screening for SAM-competitive MLL1 inhibitors without requiring the use of trimeric or higher order MLL1 complexes, significantly reducing screening time and cost.
Co-reporter:Hao Hu; Eric A. Owens; Hairui Su; Leilei Yan; Andrew Levitz; Xinyang Zhao; Maged Henary
Journal of Medicinal Chemistry 2015 Volume 58(Issue 3) pp:1228-1243
Publication Date(Web):January 5, 2015
DOI:10.1021/jm501452j
Protein arginine methyltransferase 1 (PRMT1) is involved in many biological activities, such as gene transcription, signal transduction, and RNA processing. Overexpression of PRMT1 is related to cardiovascular diseases, kidney diseases, and cancers; therefore, selective PRMT1 inhibitors serve as chemical probes to investigate the biological function of PRMT1 and drug candidates for disease treatment. Our previous work found trimethine cyanine compounds that effectively inhibit PRMT1 activity. In our present study, we systematically investigated the structure–activity relationship of cyanine structures. A pentamethine compound, E-84 (compound 50), showed inhibition on PRMT1 at the micromolar level and 6- to 25-fold selectivity over CARM1, PRMT5, and PRMT8. The cellular activity suggests that compound 50 permeated the cellular membrane, inhibited cellular PRMT1 activity, and blocked leukemia cell proliferation. Additionally, our molecular docking study suggested compound 50 might act by occupying the cofactor binding site, which provided a roadmap to guide further optimization of this lead compound.
Co-reporter:Ran Zhou, Yiqian Xie, Hao Hu, Guang Hu, Viral Sanjay Patel, Jin Zhang, Kunqian Yu, Yiran Huang, Hualiang Jiang, Zhongjie Liang, Yujun George Zheng, and Cheng Luo
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 12) pp:2623-2632
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jcim.5b00454
Protein arginine methyltransferases (PRMTs) catalyze the posttranslational methylation of arginine, which is important in a range of biological processes, including epigenetic regulation, signal transduction, and cancer progression. Although previous studies of PRMT1 mutants suggest that the dimerization arm and the N-terminal region of PRMT1 are important for activity, the contributions of these regions to the structural architecture of the protein and its catalytic methylation activity remain elusive. Molecular dynamics (MD) simulations performed in this study showed that both the dimerization arm and the N-terminal region undergo conformational changes upon dimerization. Because a correlation was found between the two regions despite their physical distance, an allosteric pathway mechanism was proposed based on a network topological analysis. The mutation of residues along the allosteric pathways markedly reduced the methylation activity of PRMT1, which may be attributable to the destruction of dimer formation and accordingly reduced S-adenosyl-L-methionine (SAM) binding. This study provides the first demonstration of the use of a combination of MD simulations, network topological analysis, and biochemical assays for the exploration of allosteric regulation upon PRMT1 dimerization. These findings illuminate the results of mechanistic studies of PRMT1, which have revealed that dimer formation facilitates SAM binding and catalytic methylation, and provided direction for further allosteric studies of the PRMT family.
Co-reporter:Zhen Han;Dr. Yepeng Luan; Yujun George Zheng
ChemBioChem 2015 Volume 16( Issue 18) pp:2605-2609
Publication Date(Web):
DOI:10.1002/cbic.201500427
Abstract
Histone acetyltransferases (HATs) are key players in the epigenetic regulation of gene function. The recent discovery of diverse HAT substrates implies a broad spectrum of cellular functions of HATs. Many pathological processes are also intimately associated with the dysregulation of HAT levels and activities. However, detecting the enzymatic activity of HATs has been challenging, and this has significantly impeded drug discovery. To advance the field, we developed a convenient one-pot, mix-and-read strategy that is capable of directly detecting the acylated histone product through a fluorescent readout. The strategy integrates three technological platforms—bioorthogonal HAT substrate labeling, alkyne–azide click chemistry, and quenching FRET—into one system for effective probing of HAT enzyme activity.
Co-reporter:Leilei Yan ; Chunli Yan ; Kun Qian ; Hairui Su ; Stephanie A. Kofsky-Wofford ; Wei-Chao Lee ; Xinyang Zhao ; Meng-Chiao Ho ; Ivaylo Ivanov
Journal of Medicinal Chemistry 2014 Volume 57(Issue 6) pp:2611-2622
Publication Date(Web):February 24, 2014
DOI:10.1021/jm401884z
Protein arginine methylation is a posttranslational modification critical for a variety of biological processes. Misregulation of protein arginine methyltransferases (PRMTs) has been linked to many pathological conditions. Most current PRMT inhibitors display limited specificity and selectivity, indiscriminately targeting many methyltransferase enzymes that use S-adenosyl-l-methionine as a cofactor. Here we report diamidine compounds for specific inhibition of PRMT1, the primary type I enzyme. Docking, molecular dynamics, and MM/PBSA analysis together with biochemical assays were conducted to understand the binding modes of these inhibitors and the molecular basis of selective inhibition for PRMT1. Our data suggest that 2,5-bis(4-amidinophenyl)furan (1, furamidine, DB75), one leading inhibitor, targets the enzyme active site and is primarily competitive with the substrate and noncompetitive toward the cofactor. Furthermore, cellular studies revealed that 1 is cell membrane permeable and effectively inhibits intracellular PRMT1 activity and blocks cell proliferation in leukemia cell lines with different genetic lesions.
Co-reporter:Dr. Chao Yang;Liza Ngo;Dr. Y. George Zheng
ChemMedChem 2014 Volume 9( Issue 3) pp:537-541
Publication Date(Web):
DOI:10.1002/cmdc.201300478
Abstract
Tip60, the 60 kDa HIV-1 Tat-interactive protein, is a key member of the MYST family of histone acetyltransferases (HATs) and plays critical roles in apoptosis and DNA repair. Potent and selective inhibitors of Tip60 are valuable tools for studying the functions of this potential drug target. In this work, we designed, synthesized and evaluated a new set of substrate-based inhibitors containing multiple binding modalities. In addition to the coenzyme A (CoA) moiety and the histone H3 peptide backbone, mono- and tri-methyl marks were incorporated at Lys 4 and/or Lys 9 sites in the H3 peptide substrate. The biochemical assay results showed that the presence of methyl group(s) on the substrate resulted in more potent inhibitors of Tip60, relative to the parent H3-CoA bisubstrate inhibitor. Importantly, by comparing the inhibitory properties of the ligands against full-length Tip60 and the HAT domain, we determined that the K4me1 and K9me3 marks contributed to the potency augmentation by interacting with the catalytic region of the enzyme.
Co-reporter:Yepeng Luan, Levi L. Blazer, Hao Hu, Taraneh Hajian, Jing Zhang, Hong Wu, Scott Houliston, Cheryl H. Arrowsmith, Masoud Vedadi and Yujun George Zheng
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 2) pp:NaN638-638
Publication Date(Web):2015/10/29
DOI:10.1039/C5OB01794G
The histone methyltransferase MLL1 has been linked to translocation-associated gene fusion in childhood leukemias and is an attractive drug target. High-throughput biochemical analysis of MLL1 methyltransferase activity requires the production of at least a trimeric complex of MLL1, RbBP5 and WDR5 to elicit robust activity. Production of trimeric and higher order MLL1 complexes in the quantities and reproducibility required for high-throughput screening presents a significant impediment to MLL1 drug discovery efforts. We present here a small molecule fluorescent ligand (FL-NAH, 6) that is able to bind to the S-adenosylmethionine (SAM) binding site of MLL1 in a manner independent of the associated complex members. We have used FL-NAH to develop a fluorescence polarization-based SAM displacement assay in a 384-well format targeting the MLL1 SET domain in the absence of associated complex members. FL-NAH competes with SAM and is displaced from the MLL1 SET domain by other SAM-binding site ligands with Kdisp values similar to the higher-order complexes, but is unaffected by the H3 peptide substrate. This assay enables screening for SAM-competitive MLL1 inhibitors without requiring the use of trimeric or higher order MLL1 complexes, significantly reducing screening time and cost.
.jpg)
![1H-Benz[e]indolium,3-butyl-2-[3-(3-butyl-1,3-dihydro-1,1-dimethyl-2H-benz[e]indol-2-ylidene)-1-propenyl]-1,1-dimethyl-, iodide](http://img.cochemist.com/ccimg/886100/886046-46-4.png)
![1H-Benz[e]indolium,3-butyl-2-[3-(3-butyl-1,3-dihydro-1,1-dimethyl-2H-benz[e]indol-2-ylidene)-1-propenyl]-1,1-dimethyl-, iodide](http://img.cochemist.com/ccimg/886100/886046-46-4_b.png)














![(2Z)-1-BUTYL-2-[(E)-3-(1-BUTYL-3,3-DIMETHYLINDOL-1-IUM-2-YL)PROP-2-ENYLIDENE]-3,3-DIMETHYLINDOLE;IODIDE](http://img.cochemist.com/ccimg/132800/132752-00-2.png)
![(2Z)-1-BUTYL-2-[(E)-3-(1-BUTYL-3,3-DIMETHYLINDOL-1-IUM-2-YL)PROP-2-ENYLIDENE]-3,3-DIMETHYLINDOLE;IODIDE](http://img.cochemist.com/ccimg/132800/132752-00-2_b.png)


![Butanoic acid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-4-oxo-, 1,1-dimethylethyl ester, (2S)-](/data/chemimg/3758600/81323-59-3.png)
![Butanoic acid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-4-oxo-, 1,1-dimethylethyl ester, (2S)-](/data/chemimg/3758600/81323-59-3_b.png)
![1H-Benz[e]indolium, 3-ethyl-1,1,2-trimethyl-, iodide](http://img.cochemist.com/ccimg/80600/80566-25-2.png)
![1H-Benz[e]indolium, 3-ethyl-1,1,2-trimethyl-, iodide](http://img.cochemist.com/ccimg/80600/80566-25-2_b.png)


![4-[5-(4-CARBAMIMIDOYLPHENYL)THIOPHEN-2-YL]BENZENECARBOXIMIDAMIDE](http://img.cochemist.com/ccimg/73900/73819-28-0.png)
![4-[5-(4-CARBAMIMIDOYLPHENYL)THIOPHEN-2-YL]BENZENECARBOXIMIDAMIDE](http://img.cochemist.com/ccimg/73900/73819-28-0_b.png)




![3-ETHYL-2-METHYLBENZO[G][1,3]BENZOTHIAZOL-3-IUM;IODIDE](http://img.cochemist.com/ccimg/54600/54581-48-5.png)
![3-ETHYL-2-METHYLBENZO[G][1,3]BENZOTHIAZOL-3-IUM;IODIDE](http://img.cochemist.com/ccimg/54600/54581-48-5_b.png)
![Benzothiazolium,3-butyl-2-[3-(3-butyl-2(3H)-benzothiazolylidene)-1-propen-1-yl]-, iodide (1:1)](/data/chemimg/1135900/53213-85-7.png)
![Benzothiazolium,3-butyl-2-[3-(3-butyl-2(3H)-benzothiazolylidene)-1-propen-1-yl]-, iodide (1:1)](/data/chemimg/1135900/53213-85-7_b.png)


![Naphtho[2,1-d]thiazole,2-methyl-](/data/chemimg/2200/20686-62-8.png)
![Naphtho[2,1-d]thiazole,2-methyl-](/data/chemimg/2200/20686-62-8_b.png)






![Benzothiazolium,3-ethyl-2-[3-(3-ethyl-2(3H)-benzothiazolylidene)-1-propen-1-yl]-, iodide (1:1)](/data/chemimg/1013200/905-97-5.png)
![Benzothiazolium,3-ethyl-2-[3-(3-ethyl-2(3H)-benzothiazolylidene)-1-propen-1-yl]-, iodide (1:1)](/data/chemimg/1013200/905-97-5_b.png)
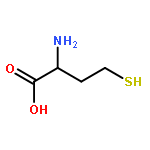
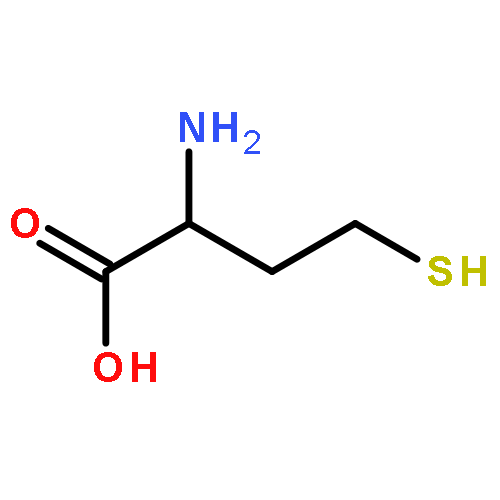


![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)