Co-reporter:Michael De Rosa, David Arnold, Douglas Hartline, Linda Truong, Roman Verner, Tianwei Wang, and Christian Westin
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12288-12299
Publication Date(Web):November 17, 2015
DOI:10.1021/acs.joc.5b02192
Reaction of 3-aminopyrrole (as its salt) with trifluoromethyl-β-diketones gave γ-1H-pyrrolo[3,2-b]pyridines via reaction at the less reactive carbonyl group. The trifluoromethyl group increased the electrophilicity of the adjacent carbonyl group and decreased the basicity of the hydroxyl group of the CF3 amino alcohol formed. This amino alcohol was formed faster, but its subsequent dehydration to the β-enaminone was slow resulting in the preferential formation of the γ-regioisomer. Reaction of 4,4,4-trifluoro-1-phenyl-1,3-butadione with 3-aminopyrrole was carried out using a series of 6 amine buffers. Yields of the α-1H-pyrrolo[3,2-b]pyridine increased as the pKa of the amine buffer decreased. Surprisingly the yield went down at higher pKas. There was a change in mechanism as the reaction mixture became more basic. With strong amines trifluoromethyl-β-diketones were present mainly or completely as the enolate. Under reductive conditions (3-nitropyrrole/Sn/AcOH/trifluoromethyl-β-diketone) the α-1H-pyrrolo[3,2-b]pyridine was the major product as a result of Lewis acid catalysis by Sn2+. Similar α-regiochemistry was observed when the reaction of the 3-aminopyrrole salt with trifluoromethyl-β-diketones was carried out in the presence of base and tin(II) acetate.
Co-reporter:Michael De Rosa, David Arnold, Hemant Yennawar
Tetrahedron Letters 2014 Volume 55(Issue 40) pp:5491-5494
Publication Date(Web):1 October 2014
DOI:10.1016/j.tetlet.2014.08.004
Initial reaction of 3-aminopyrrole with the conjugate acid of 3,6-diphenyl-1,2,4,5-tetrazine gave an intermediate that rearranged to a 1H-1,2,4-(triazol-3-yl)pyrimidine via an unprecedented cascade. In this cascade, the s-tetrazine-ring opened, contracted to a 1,2,4-triazole-ring, and the pyrrole ring expanded to a pyrimidine. Similar results were obtained with three other electronically different s-tetrazines.
Co-reporter:Michael De Rosa and David Arnold
The Journal of Organic Chemistry 2013 Volume 78(Issue 3) pp:1107-1112
Publication Date(Web):December 28, 2012
DOI:10.1021/jo302457y
Protonation of 3-aminopyrrole at C-2 gave the σ-complex 1H-pyrrol-3(2H)-iminium cation, whereas protonation at the exoamino group gave its 1H-pyrrol-3-aminium tautomer. Both tautomers were isolated as their respective tetrakis(pentafluorophenyl)borate salt, an example of desmotropy. In solution, the NH3-tautomer was favored in hydrogen-bonding solvents and the CH2-tautomer in CH2Cl2. A combination of effects on the aromaticity of the aminopyrrole ring increased the relative stability of the σ-complexes (conjugate acids) such that they can be readily observed or isolated.
Co-reporter:Michael De Rosa, Nieves Canudas, David Arnold, and Hemant Yennawar
The Journal of Organic Chemistry 2013 Volume 78(Issue 14) pp:7264-7267
Publication Date(Web):June 27, 2013
DOI:10.1021/jo400776p
The chlorotropy observed by NMR in this study occurred by the rapid intermolecular transfer of a chloro group between 1-chlorobenzimidazole and benzimidazole in CCl4/CH3OH/K2CO3 solution.
Co-reporter:Michael De Rosa, David Arnold, and Douglas Hartline
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8614-8623
Publication Date(Web):July 30, 2013
DOI:10.1021/jo4012915
Reaction of 3-aminopyrrole with seven 1,3,5-triazines was studied in a one-step reaction (in situ formation of 3-aminopyrrole) and a two-step reaction (using the tetraphenylborate salt and an amine base). An inverse-electron demand Diels–Alder reaction (IEDDA) was observed with R1 = CF3, CO2Et, and H with the formation of 5H-pyrrolo[3,2-d]pyrimidine derivatives. SNAr was observed when 2,4,6-trifluoro- or 2,4,6-trichloro-1,3,5-triazine were used—1,3,5-triazines that had leaving groups. If excess 1,3,5-triazine was present the initial SNAr product reacted further, in the presence of acid and water, with another equivalent of 1,3,5-triazine to give compounds containing three linked heterocyclic rings. No reaction was observed with R1 = C6H5 and OCH3. Four mechanisms are proposed to explain the experimental results: uncatalyzed and acid catalyzed inverse electron demand Diels–Alder cascades leading to cycloaddition, and uncatalyzed and acid-catalyzed SNAr reactions leading, respectively, to single and double substitution products. Acid catalysis was a factor when there was reduced reactivity in either reactant.
Co-reporter:Michael De Rosa, David Arnold, Bernie O’Hare
Tetrahedron Letters 2009 50(1) pp: 12-14
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.10.100

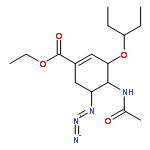
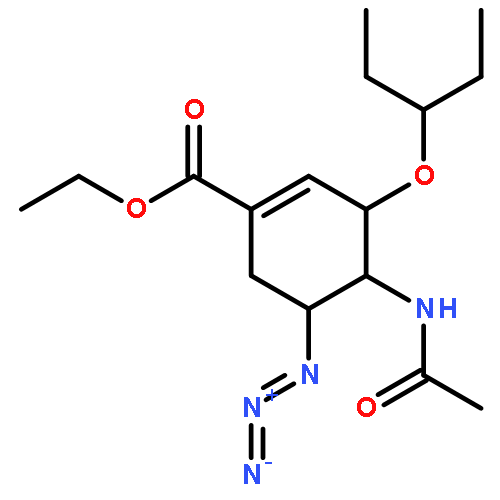


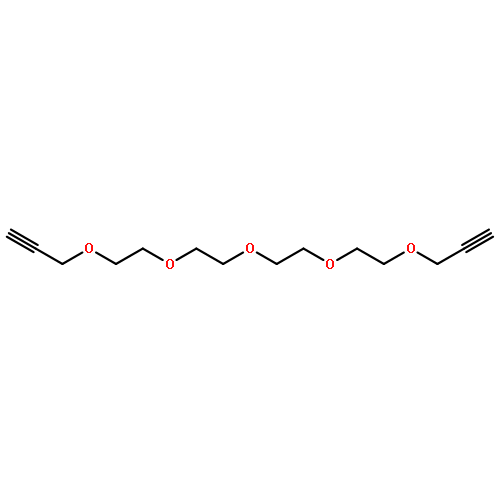
![4-Pentynamide, N-[3-[2-[2-(3-aminopropoxy)ethoxy]ethoxy]propyl]-](/data/chemimg/181200/1246993-52-1.png)
![4-Pentynamide, N-[3-[2-[2-(3-aminopropoxy)ethoxy]ethoxy]propyl]-](/data/chemimg/181200/1246993-52-1_b.png)
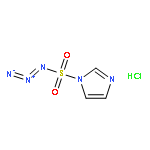
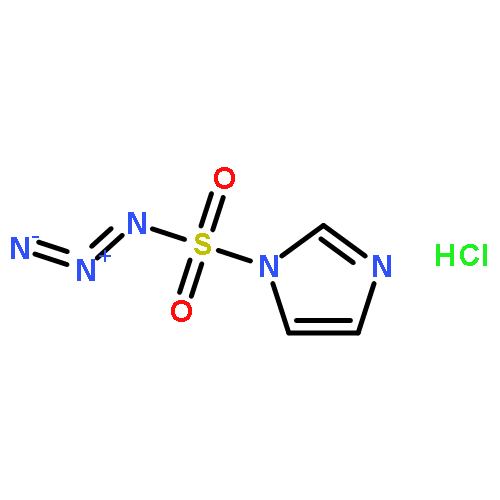
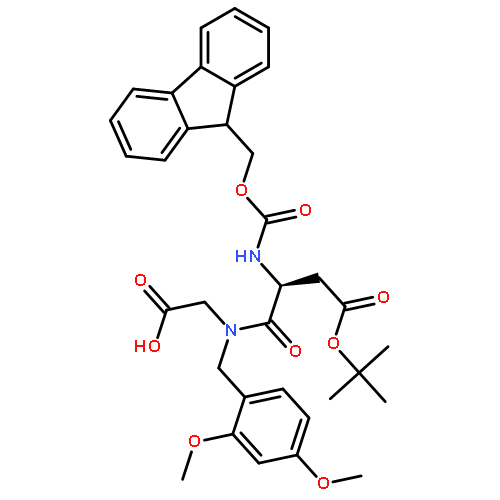






![D-Allothreonine,N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/130700/130674-54-3.png)
![D-Allothreonine,N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/130700/130674-54-3_b.png)





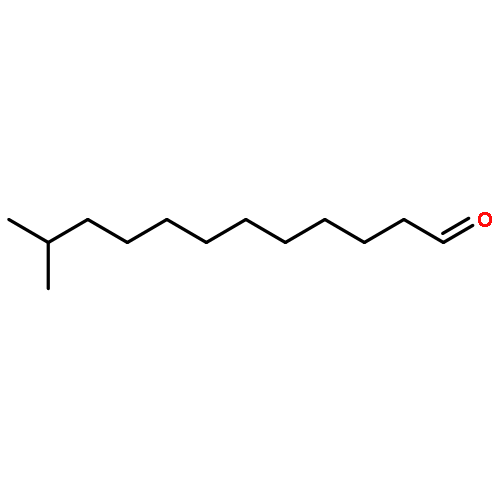
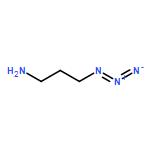
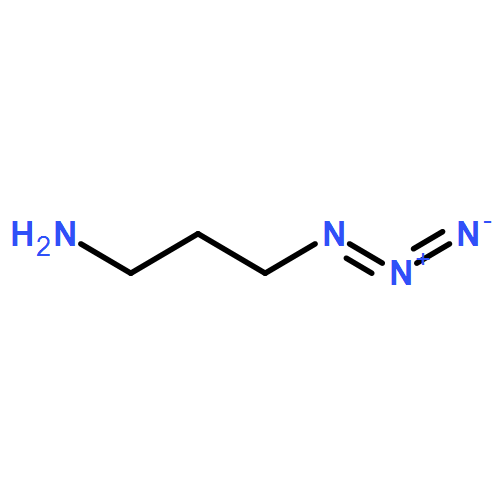


![Butanamide,N-cyclohexyl-4-[(1,2-dihydro-2-oxo-6-quinolinyl)oxy]-N-methyl-](http://img.cochemist.com/ccimg/68600/68550-75-4.png)
![Butanamide,N-cyclohexyl-4-[(1,2-dihydro-2-oxo-6-quinolinyl)oxy]-N-methyl-](http://img.cochemist.com/ccimg/68600/68550-75-4_b.png)


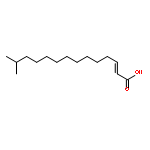








![(2WEI ,5WEI ,7WEI ,13WEI )-4-ACETOXY-13-{[(2R,3S)-3-(BENZOYLAMINO)-2-HYDROXY-<WBR />3-PHENYLPROPANOYL]OXY}-1,7-DIHYDROXY-9,10-DIOXO-5,20-EPOXYTAX-11-<WBR />EN-2-YL BENZOATE](http://img.cochemist.com/ccimg/26300/26296-50-4.png)
![(2WEI ,5WEI ,7WEI ,13WEI )-4-ACETOXY-13-{[(2R,3S)-3-(BENZOYLAMINO)-2-HYDROXY-<WBR />3-PHENYLPROPANOYL]OXY}-1,7-DIHYDROXY-9,10-DIOXO-5,20-EPOXYTAX-11-<WBR />EN-2-YL BENZOATE](http://img.cochemist.com/ccimg/26300/26296-50-4_b.png)
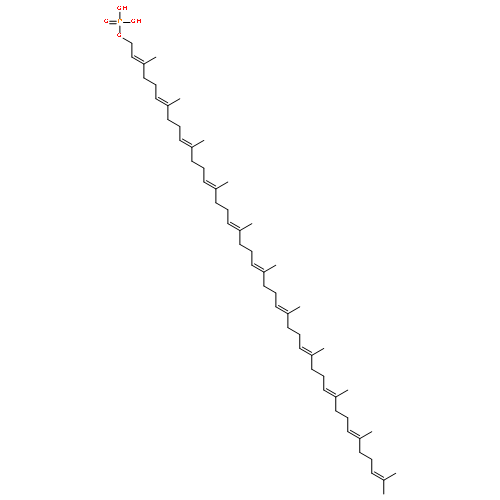

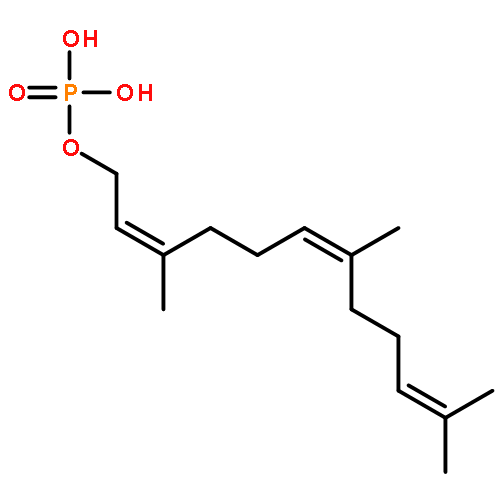


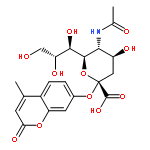
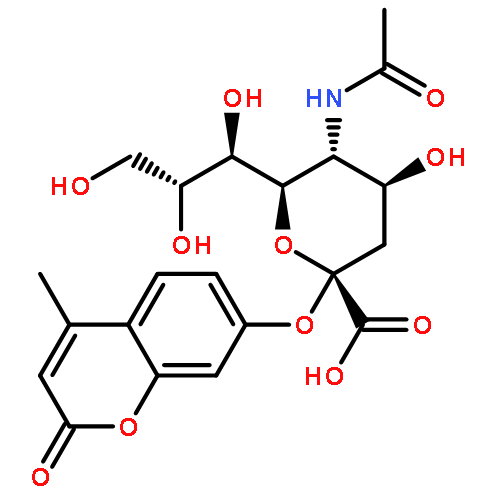
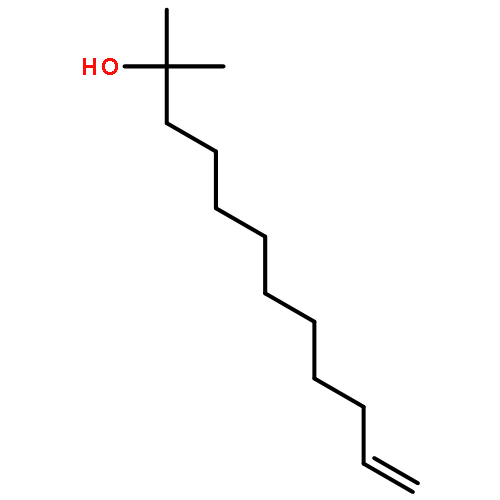
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)
![9-Octadecenoic acid(9Z)-, 1,1'-[1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/2500/2462-63-7.png)
![9-Octadecenoic acid(9Z)-, 1,1'-[1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/2500/2462-63-7_b.png)







