Co-reporter:Roger Guilard, Gerhard Erker, Paul Raithby, Qiang Xu
Coordination Chemistry Reviews 2017 Volume 350(Volume 350) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.ccr.2017.10.007
Co-reporter:Simon A. Cotton, Paul R. Raithby
Coordination Chemistry Reviews 2017 Volume 340(Volume 340) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.ccr.2017.01.011
•Systematic study of four series of lanthanide coordination complexes.•Systematic analysis of the effect of the lanthanide contraction.•Emphasises the importance of the solvent in obtaining crystalline complexes.•Emphasises the complex equilibria in the solution chemistry of the lanthanide complexes.Download high-res image (127KB)Download full-size image
Co-reporter:Rayya A. Al-Balushi, Ashanul Haque, Maharaja Jayapal, Mohammed K. Al-Suti, John Husband, Muhammad S. Khan, Olivia F. Koentjoro, Kieran C. Molloy, Jonathan M. Skelton, and Paul R. Raithby
Inorganic Chemistry 2016 Volume 55(Issue 13) pp:6465
Publication Date(Web):June 10, 2016
DOI:10.1021/acs.inorgchem.6b00523
A series of trimethylsilyl-protected monoalkynes (Me3SiC≡C–R) and bis-alkynes (Me3 SiC≡C–R–C≡CSiMe3) incorporating carbazole spacer groups (R = carbazole-2-yl, carbazole-3-yl, carbazole-2,7-diyl, N-(2-ethylhexyl)carbazole-2,7-diyl, carbazole-3,6-diyl, N-(2-ethylhexyl)carbazole-3,6-diyl), together with the corresponding terminal monoalkynes (H–C≡C–R) and bis-alkynes (H–C≡C–R–C≡C–H), have been synthesized and characterized. The CuI-catalyzed dehydrohalogenation reaction between trans-[(Ph)(Et3P)2PtCl], trans-[(Et3P)2PtCl2], and trans-[(PnBu3)2PtCl2] and the terminal alkynes in iPr2NH/CH2Cl2 affords a series of Pt(II) mono- and diynes, while the dehydrohalogenation polycondensation reactions with trans-[(PnBu3)2PtCl2] under similar reaction conditions yields four Pt(II) poly-ynes of the form trans-[(PnBu3)2Pt–C≡C–R–C≡C−]n. The acetylide-functionalized carbazole ligands and the mono-, di-, and polynuclear Pt(II) σ-acetylide complexes have been characterized spectroscopically, with a subset analyzed using single-crystal X-ray diffraction. The Pt(II) mono-, di-, and poly-ynes incorporating the carbazole spacers are soluble in common organic solvents, and solution absorption spectra show a consistent red-shift between the 2- and 2,7- as well as 3- and 3,6-carbazole complexes. Computational modeling is used to explain the observed spectral shifts, which are related to the enhanced electronic delocalization in the latter systems. These results also indicate that the inclusion of carbazole-2,7-diyl units into rigid-rod organometallic polymers should enhance electronic transport along the chains.
Co-reporter:Rayya A. Al-Balushi, Ashanul Haque, Maharaja Jayapal, Mohammed K. Al-Suti, John Husband, Muhammad S. Khan, Jonathan M. Skelton, Kieran C. Molloy, and Paul R. Raithby
Inorganic Chemistry 2016 Volume 55(Issue 21) pp:10955
Publication Date(Web):October 18, 2016
DOI:10.1021/acs.inorgchem.6b01509
Trimethylsilyl-protected dialkynes incorporating azobenzene linker groups, Me3SiC≡CRC≡CSiMe3 (R = azobenzene-3,3′-diyl, azobenzene-4,4′-diyl, 2,5-dioctylazobenzene-4,4′-diyl), and the corresponding terminal dialkynes, HC≡CRC≡CH, have been synthesized and characterized. The CuI-catalyzed dehydrohalogenation reaction between trans-[Ph(Et3P)2PtCl] and the deprotected dialkynes in a 2:1 ratio in iPr2NH/CH2Cl2 gives the platinum(II) diynes trans-[Ph(Et3P)2PtC≡CRC≡CPt(PEt3)2Ph], while the dehydrohalogenation polycondensation reaction between trans-[(nBu3P)2PtCl2] and the dialkynes in a 1:1 molar ratio under similar reaction conditions affords the platinum(II) polyynes, [−Pt(PnBu3)2–C≡CRC≡C−]n. The materials have been characterized spectroscopically, with the diynes also studied using single-crystal X-ray diffraction. The platinum(II) diynes and polyynes are all soluble in common organic solvents. Optical-absorption measurements show that the compounds incorporating the para-alkynylazobenzene spacers have a higher degree of electronic delocalisation than their meta-alkynylazobenzene counterparts. Reversible photoisomerization in solution was observed spectroscopically for the alkynyl-functionalized azobenzene ligands and, to a lesser extent, for the platinum(II) complexes. Complementary quantum-chemical modeling was also used to analyze the optical properties and isomerization energetics.
Co-reporter:Muhammad S. Khan, Mohammed K. Al-Suti, Jayapal Maharaja, Ashanul Haque, Rayya Al-Balushi, Paul R. Raithby
Journal of Organometallic Chemistry 2016 Volume 812() pp:13-33
Publication Date(Web):15 June 2016
DOI:10.1016/j.jorganchem.2015.10.003
•Thiophene-based conjugated poly-ynes and poly(metalla-ynes) have been reviewed.•The materials possess low band gap with broad absorption range.•Poly(platina-ynes) have lower PCE values compared to their organic counterparts.•The performance depends on chemical structure, morphology and device architecture.Solar cells (SCs) are of considerable current research interest because of their potential as a clean alternative to fossil fuels. Researchers across the globe are developing novel polymeric materials with enhanced power conversion efficiency (PCE). Conjugated poly-ynes and poly(metalla-ynes) incorporating late transition metals and thiophene-based spacers have played a very important role in this strategic area of materials research. The performance of the SCs can be optimized by varying the conjugated spacers and/or the metal ions along the polymer backbone. Therefore, an analysis of structure-photovoltaic property relationships in poly-ynes and poly(metalla-ynes) is desirable as a guide for the development of new functional materials for use in SCs. Keeping the importance of this strategic topic in mind, herein we present a brief review on conjugated poly-ynes and poly(metalla-ynes) incorporating thiophene-based spacers that have potential SC applications. Attempts have been made to correlate the photovoltaic performance of the SCs to the chemical structure of thiophene-incorporated poly-ynes and poly(metalla-ynes). The performance of SCs is also strongly influenced by other factors such as morphology and device structure.Poly(metalla-ynes), particularly those of platinum(II) supported by thiophene-based linker groups, have played a fundamental part in the development of the science leading to the fabrication of solar cells. The chemistry of these poly(metalla-ynes) is reviewed here.
Co-reporter:Jane V. Knichal, William J. Gee, Andrew D. Burrows, Paul R. Raithby, and Chick C. Wilson
Crystal Growth & Design 2015 Volume 15(Issue 1) pp:465-474
Publication Date(Web):December 2, 2014
DOI:10.1021/cg501535b
The influence of weak hydrogen bonds on the crystal packing of a series of heavy and transition metal coordination polymers synthesized using the ligand 5-ethynyl-1,3-benzenedicarboxylic acid (H2ebdc) has been evaluated. Five coordination polymers were prepared and crystallographically characterized. These comprise two 1D chains, [Pb(ebdc)(DMSO)2] (1) and [Pb(ebdc)(DMF)] (2), two 2D nets, [Cu3(ebdc)(H2O)1.5(MeOH)0.5]·6H2O (3) and [Pb2(ebdc)2(DMF)4]·H2O (4), and a single 3D framework, [HNEt3][Zn3(μ3-OH)(μ2-H2O)(ebdc)3(MeOH)0.67(H2O)0.33]·MeOH·1.33H2O (5). The crystal structure of the free acid ligand form, H2ebdc·H2O, is also reported. Within the lead(II) coordination structures, ethynyl-derived C–H···O interactions are consistently found to provide the dominant influence over the crystal packing, as determined by solid-state structural analysis in combination with vibrational spectroscopy. The influence of weak hydrogen-bonding effects on the crystal packing of the transition metal coordination polymers that contain lattice water and methanol molecules was found to be far less prominent, which is interpreted in terms of the greater prevalence of strong hydrogen-bond donors and acceptors forming O–H···O interactions within these crystalline lattices.
Co-reporter: Paul R. Raithby
Angewandte Chemie International Edition 2015 Volume 54( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/anie.201411930
No abstract is available for this article.
Co-reporter:Abu Ali Ibn Sina;S. M. Ibrahim Al-Rafia
Journal of Inorganic and Organometallic Polymers and Materials 2015 Volume 25( Issue 3) pp:427-436
Publication Date(Web):2015 May
DOI:10.1007/s10904-014-0071-7
A new family of luminescent platinum(II) acetylide complexes and polymers were formed by the copper(I) catalyzed reaction of cis-[PtCl2(PR3)2] (R=C6H5–p-CH3) with appropriate acetylide ligands. The reaction of metal precursors with 2.5 equivalents of monoterminal acetylide ligands provided metal complexes trans-[Pt(p-tolyl3P)2(C≡C-R)2] (R=C6H4-p-NO2 (1) C6H4 -p-CH3 (2)), and equimolar amounts of diterminal ligand and metal chloride precursor, under reflux, afforded the metal poly-yne polymers [-Pt(p-tolyl3P)2C≡C–R–C≡C–]n, (R=biphenyl and 2,5-dioctyloxybenzene). Characterization of the newly developed polymer and metal complexes was accomplished by FT-IR, multinuclear NMR (1H, 31P, 13C) and mass spectrometry, as well as elemental analysis. The molecular structure of the metal complex trans-[Pt(p-tolyl3P)2(C≡CC6H4–p-NO2)2] (1) was confirmed by single crystal X-ray crystallography. The electronic absorption and photoluminescence spectra of the metal complexes and polymers have been used to probe their photophysical properties. The studies reveal that the presence of heavy metal atom and substituent groups on the phenyl ring of the ligands can enhance the efficiency of intersystem crossing from the S1 singlet excited state to the T1 triplet excited state and thus give intense phosphorescence.
Co-reporter:Lauren E. Hatcher, Paul R. Raithby
Coordination Chemistry Reviews 2014 Volumes 277–278() pp:69-79
Publication Date(Web):1 October 2014
DOI:10.1016/j.ccr.2014.02.021
•Tracing the development of photocrystallography.•The determination of the three-dimensional crystal structures of metastable and short-lived species.•The determination of the structures of metastable linkage isomers.•The determination of the structures of complexes with lifetimes in the microsecond range.•The development of Laue diffraction techniques to facilitate the determination of structures with sub-microsecond lifetimes.The methods that have been developed to determine the three-dimensional crystal and molecular structures while they are in metastable or short-lived photoactivated states are described. The structural science of photocrystallography has developed over the last two decades because of the use of synchrotron radiation, coupled with advances in cryogenics, computer hardware and software, and laser technology. Initial studies have been carried out on metastable linkage isomers and LIESST-generated metastable structures and, more recently, by using the synchronisation of laser pulses with X-ray pulses, it has been possible to determine the structures of complexes with microsecond lifetimes. In the future X-ray Laue techniques and one-shot XFEL studies applied to molecular systems promise to make the study of sub-microsecond species a reality.The area of photocrystallography and time resolved crystallography is reviewed, and the methods for determining the three-dimensional crystal structures of metastable and transient species using synchrotron X-ray radiation are described.
Co-reporter:Christopher H. Woodall, Simon K. Brayshaw, Stefanie Schiffers, David R. Allan, Simon Parsons, Rafael Valiente and Paul R. Raithby
CrystEngComm 2014 vol. 16(Issue 11) pp:2119-2128
Publication Date(Web):15 Nov 2013
DOI:10.1039/C3CE41933A
Single crystals of the dithienylethene compounds, 1,2-bis(2-methylbenzothiophen-3-yl)perfluorocyclopentene 1 and 1,2-bis(2,5-dimethylthiophen-3-yl)perfluorocyclopentene 2 undergo pressure-induced single-crystal to single-crystal phase transitions between 4.45–5.35 GPa and 4.15–5.70 GPa, respectively. For 1, there is a smooth reduction in unit-cell volume of ~20% from ambient pressure to 4.45 GPa, followed by a dramatic reduction in volume that coincides with a 7.7% increase in the b axis length. Above the pressure of 5.38 GPa a smooth volume reduction continues. In contrast, for 2, there is a continuous change in unit-cell volume with an observed space group change from C2/c to P21/c, between the pressures of 4.15 and 5.70 GPa. In the crystals of 1 between 4.45 and 5.38 GPa adjacent molecules slide over each other and the dominant stacking interaction changes from a thiophene⋯thiophene interaction at 4.45 GPa to a benzothiophene⋯benzothiophene interaction at 5.38 GPa and, within each molecule, the benzothiophene groups show a significant reorientation at the phase transition. In 2 there is a loss of molecular symmetry, concomitant with the change in space group, at the phase transition with the asymmetric unit changing from containing half a unique molecule to two independent molecules. The molecules show significant reorientations of their ring systems. The nature of the observed transition in 1 was investigated using solid-state computational methods to prove the superior thermodynamic stability of the high-pressure phase to the lower pressure phase at pressures above 5.38 GPa. Solid state UV-Vis spectroscopy of 1, over the pressure range from ambient to 15.4 GPa showed that the compound displayed piezochromism with a significant red shift in the π–π* absorption band and a colour change in the crystal from colourless to red with increasing pressure.
Co-reporter:Hakikulla H. Shah, Rayya A. Al-Balushi, Mohammed K. Al-Suti, Muhammad S. Khan, Frank Marken, Anna L. Sudlow, Gabriele Kociok-Köhn, Christopher H. Woodall, Paul R. Raithby and Kieran C. Molloy
Dalton Transactions 2014 vol. 43(Issue 25) pp:9497-9507
Publication Date(Web):30 Apr 2014
DOI:10.1039/C3DT52914B
Three new neutral di-ferrocenyl-ethynylpyridinyl copper complexes, [L2(CuCl)2(PPh3)2] (2), [L2(CuBr)2(PPh3)2] (3), and [L2(CuI)2(PPh3)2] (4) were synthesized from the ferrocenyl-ethynylpyridine ligand (L) (1), the appropriate copper halide CuX (with X = Cl−, Br−, I−) and triphenylphosphine. These neutral complexes were fully characterized by spectroscopic methods and by single crystal X-ray crystallography. Cyclic voltammetry in dichloroethane revealed chemically reversible ferrocenyl oxidation signals followed by characteristic “stripping reduction peaks” showing evidence for oxidation-product electro-crystallization. Scanning electron microscopy confirmed spontaneous formation of crystalline oxidation products with three distinct morphologies for X = Cl−, Br−, I−. Energy dispersive X-ray elemental analysis data show Fe:P ratios of 1:2.0, 1:2.1 and 1:2.1 for electro-crystallization products of complexes 2, 3, and 4, respectively, indicating the presence of two [PF6]− anions in the vicinity of the dioxidized complexes, and suggesting product formulae [2]2+[PF6]−2, [3]2+[PF6]−2 and [4]2+[PF6]−2.
Co-reporter:Lauren E. Hatcher, Edward J. Bigos, Mathew J. Bryant, Emily M. MacCready, Thomas P. Robinson, Lucy K. Saunders, Lynne H. Thomas, Christine M. Beavers, Simon J. Teat, Jeppe Christensen and Paul R. Raithby
CrystEngComm 2014 vol. 16(Issue 35) pp:8263-8271
Publication Date(Web):20 Jun 2014
DOI:10.1039/C4CE00675E
The known complex [Ni(medpt)(η1-NO2)(η2-ONO)] 1 (medpt = 3,3′-diamino-N-methyldipropylamine) crystallises in the monoclinic space group P21/m with 1.5 molecules in the asymmetric unit with two different η1-NO2 ligand environments in the crystal structure. At 298 K the molecule (A) sitting in a general crystallographic site displays a mixture of isomers, 78% of the η1-NO2 isomer and 22% of an endo-nitrito–(η1-ONO) form. The molecule (B) sitting on a crystallographic mirror plane adopts the η1-NO2 isomeric form exclusively. However, a variable temperature crystallographic study showed that the two isomers were in equilibrium and upon cooling to 150 K the η1-ONO isomer converted completely to the η1-NO2 isomer, so that both independent molecules in the asymmetric unit were 100% in the η1-NO2 form. A kinetic analysis of the equilibrium afforded values of ΔH = −9.6 (±0.4) kJ mol−1, ΔS = −21.5 (±1.8) J K−1 mol−1 and EA = −1.6 (±0.05) kJ mol−1. Photoirradiation of single crystals of 1 with 400 nm light, at 100 K, resulted in partial isomerisation of the η1-NO2 isomer to the metastable η1-ONO isomer, with 89% for molecule (A), and 32% for molecule (B). The crystallographic space group also reduced in symmetry to P21 with Z′ = 3. The metastable state existed up to a temperature of 150 K above which temperature it reverted to the ground state. An analysis of the crystal packing in the ground and metastable states suggests that hydrogen bonding is responsible for the difference in the conversion between molecules (A) and (B).
Co-reporter:Dr. Christopher H. Woodall;Dr. Sara Fuertes;Dr. Christine M. Beavers;Lauren E. Hatcher;Andrew Parlett;Dr. Helena J. Shepherd;Dr. Jeppe Christensen;Dr. Simon J. Teat;Mourad Intissar;Dr. Alexre Rodrigue-Witchel;Dr. Yan Suffren; Christian Reber;Christopher H. Hendon;Dr. Davide Tiana; Aron Walsh; Paul R. Raithby
Chemistry - A European Journal 2014 Volume 20( Issue 51) pp:16933-16942
Publication Date(Web):
DOI:10.1002/chem.201404058
Abstract
A systematic investigation into the relationship between the solid-state luminescence and the intermolecular Au⋅⋅⋅Au interactions in a series of pyrazolate-based gold(I) trimers; tris(μ2-pyrazolato-N,N′)-tri-gold(I) (1), tris(μ2-3,4,5- trimethylpyrazolato-N,N′)-tri-gold(I) (2), tris(μ2-3-methyl-5-phenylpyrazolato-N,N′)-tri-gold(I) (3) and tris(μ2-3,5-diphenylpyrazolato-N,N′)-tri-gold(I) (4) has been carried out using variable temperature and high pressure X-ray crystallography, solid-state emission spectroscopy, Raman spectroscopy and computational techniques. Single-crystal X-ray studies show that there is a significant reduction in the intertrimer Au⋅⋅⋅Au distances both with decreasing temperature and increasing pressure. In the four complexes, the reduction in temperature from 293 to 100 K is accompanied by a reduction in the shortest intermolecular Au⋅⋅⋅Au contacts of between 0.04 and 0.08 Å. The solid-state luminescent emission spectra of 1 and 2 display a red shift with decreasing temperature or increasing pressure. Compound 3 does not emit under ambient conditions but displays increasingly red-shifted luminescence upon cooling or compression. Compound 4 remains emissionless, consistent with the absence of intermolecular Au⋅⋅⋅Au interactions. The largest pressure induced shift in emission is observed in 2 with a red shift of approximately 630 cm−1 per GPa between ambient and 3.80 GPa. The shifts in all the complexes can be correlated with changes in Au⋅⋅⋅Au distance observed by diffraction.
Co-reporter:Dr. Mark R. Warren;Dr. Timothy L. Easun;Dr. Simon K. Brayshaw; Robert J. Deeth; Michael W. George;Dr. Andrew L. Johnson;Dr. Stefanie Schiffers;Dr. Simon J. Teat;Dr. Anna J. Warren;Dr. John E. Warren; Chick C. Wilson;Dr. Christopher H. Woodall; Paul R. Raithby
Chemistry - A European Journal 2014 Volume 20( Issue 18) pp:5468-5477
Publication Date(Web):
DOI:10.1002/chem.201302053
Abstract
The solid-state, low-temperature linkage isomerism in a series of five square planar group 10 phosphino nitro complexes have been investigated by a combination of photocrystallographic experiments, Raman spectroscopy and computer modelling. The factors influencing the reversible solid-state interconversion between the nitro and nitrito structural isomers have also been investigated, providing insight into the dynamics of this process. The cis-[Ni(dcpe)(NO2)2] (1) and cis-[Ni(dppe)(NO2)2] (2) complexes show reversible 100 % interconversion between the η1-NO2 nitro isomer and the η1-ONO nitrito form when single-crystals are irradiated with 400 nm light at 100 K. Variable temperature photocrystallographic studies for these complexes established that the metastable nitrito isomer reverted to the ground-state nitro isomer at temperatures above 180 K. By comparison, the related trans complex [Ni(PCy3)2(NO2)2] (3) showed 82 % conversion under the same experimental conditions at 100 K. The level of conversion to the metastable nitrito isomers is further reduced when the nickel centre is replaced by palladium or platinum. Prolonged irradiation of the trans-[Pd(PCy3)2(NO2)2] (4) and trans-[Pt(PCy3)2(NO2)2] (5) with 400 nm light gives reversible conversions of 44 and 27 %, respectively, consistent with the slower kinetics associated with the heavier members of group 10. The mechanism of the interconversion has been investigated by theoretical calculations based on the model complex [Ni(dmpe)Cl(NO2)].
Co-reporter:Lauren E. Hatcher;Dr. Jeppe Christensen;Dr. Michelle L. Hamilton;Dr. Jose Trincao;Dr. David R. Allan;Dr. Mark R. Warren;Dr. Ian P. Clarke;Dr. Michael Towrie;Dr Sara Fuertes;Dr. Charles C. Wilson;Dr. Christopher H. Woodall;Dr. Paul R. Raithby
Chemistry - A European Journal 2014 Volume 20( Issue 11) pp:3128-3134
Publication Date(Web):
DOI:10.1002/chem.201304172
Abstract
At temperatures below 150 K, the photoactivated metastable endo-nitrito linkage isomer [Ni(Et4dien)(η2-O,ON)(η1-ONO)] (Et4dien=N,N,N′,N′-tetraethyldiethylenetriamine) can be generated with 100 % conversion from the ground state nitro-(η1-NO2) isomer on irradiation with 500 nm light, in the single crystal by steady-state photocrystallographic techniques. Kinetic studies show the system is no longer metastable above 150 K, decaying back to the ground state nitro-(η1-NO2) arrangement over several hours at 150 K. Variable-temperature kinetic measurements in the range of 150–160 K show that the rate of endo-nitrito decay is highly dependent on temperature, and an activation energy of Eact=+48.6(4) kJ mol−1 is calculated for the decay process. Pseudo-steady-state experiments, where the crystal is continually pumped by the light source for the duration of the X-ray experiment, show the production of a previously unobserved, exo-nitrito-(η1-ONO) linkage isomer only at temperatures close to the metastable limit (ca. 140–190 K). This exo isomer is considered to be a transient excited-state species, as it is only observed in data collected by pseudo-steady-state methods.
Co-reporter:Hakikulla H. Shah, Rayya A. Al-Balushi, Mohammed K. Al-Suti, Muhammad S. Khan, Christopher H. Woodall, Kieran C. Molloy, Paul R. Raithby, Thomas P. Robinson, Sara E. C. Dale, and Frank Marken
Inorganic Chemistry 2013 Volume 52(Issue 9) pp:4898-4908
Publication Date(Web):April 17, 2013
DOI:10.1021/ic3024887
A new series of bis(ferrocenylethynyl) complexes, 3–7, and a mono(ferrocenylethynyl) complex, 8, have been synthesized incorporating conjugated heterocyclic spacer groups, with the ethynyl group facilitating an effective long-range intramolecular interaction. The complexes were characterized by NMR, IR, and UV–vis spectroscopy as well as X-ray crystallography. The redox properties of these complexes were investigated using cyclic voltammetry and spectroelectrochemistry. Although there is a large separation of ∼14 Å between the two redox centers, ΔE1/2 values in this series of complexes ranged from 50 to 110 mV. The appearance of intervalance charge-transfer bands in the UV–vis–near-IR region for the monocationic complexes further confirmed effective intramolecular electronic communication. Computational studies are presented that show the degree of delocalization across the Fc–C≡C–C≡C–Fc (Fc = C5H5FeC5H4) highest occupied molecular orbital.
Co-reporter:Hakikulla H. Shah ; Rayya A. Al-Balushi ; Mohammed K. Al-Suti ; Muhammad S. Khan ; Christopher H. Woodall ; Anna L. Sudlow ; Paul R. Raithby ; Gabriele Kociok-Köhn ; Kieran C. Molloy ;Frank Marken
Inorganic Chemistry 2013 Volume 52(Issue 20) pp:12012-12022
Publication Date(Web):October 9, 2013
DOI:10.1021/ic401803p
Three new tetra-ferrocenylethynylpyridinyl copper complexes, L4(CuI)4 (3), L4(CuBr)2 (4), and L4(CuCl)2 (5) have been prepared from the reaction of ferrocenylethynylpyridine (L)(2) with copper halides CuX (with X = I–, Br–, Cl–).The ligand 2 and the complexes 3–5 have been fully characterized by spectroscopic methods. The structures of 2–4 have been confirmed by single-crystal X-ray crystallography. 2 forms a dimer in the crystalline-state through C–H··N hydrogen bonds. 4 and 5 are dimers and 3 a tetramer, in all cases linked through Cu–X··Cu bridging interactions. Cyclic voltammetry in dichloroethane showed chemically reversible multiferrocenyl oxidation signals with evidence for product electro-crystallization. The oxidation products were isolated by electrodeposition onto a Pt disc electrode and investigated by scanning electron microscopy which confirmed the spontaneous formation of crystalline oxidation products with distinctive morphologies. Energy dispersive X-ray elemental analysis shows the presence of hexafluorophosphate (counterion) with the P:Fe ratio of 1:1, 0.5:1, and 1:1 for the electrocrystallized products 3, 4, and 5, respectively, suggesting the formulas [3]4+(PF6–)4, [4]2+(PF6–)2, and [5]4+(PF6–)4 for the electro-crystallized products.
Co-reporter:Dr. Christopher H. Woodall;Dr. Christine M. Beavers;Dr. Jeppe Christensen;Lauren E. Hatcher;Mourad Intissar;Andrew Parlett;Dr. Simon J. Teat; Christian Reber; Paul R. Raithby
Angewandte Chemie International Edition 2013 Volume 52( Issue 37) pp:9691-9694
Publication Date(Web):
DOI:10.1002/anie.201302825
Co-reporter:Somia E. Bajwa, Thomas E. Storr, Lauren E. Hatcher, Thomas J. Williams, Christoph G. Baumann, Adrian C. Whitwood, David R. Allan, Simon J. Teat, Paul R. Raithby and Ian J. S. Fairlamb
Chemical Science 2012 vol. 3(Issue 5) pp:1656-1661
Publication Date(Web):23 Feb 2012
DOI:10.1039/C2SC01050J
Pd3(OAc)5NO2, an impurity in “Pd(OAc)2” {formally Pd3(OAc)6}, emerges as a serious issue in the synthesis of pure PdII complexes derived from Pd(OAc)2, for example in our C–H activation precatalyst, Pd(OAc)2(pip)2 (pip = piperidine). A previous proposal that nitrite anion can be formed by oxidation of CH3CN by metallic Pd and air, leading to cyclo(ortho)palladated complexes containing nitrite anion, e.g. Pd(NO2)(C^N)L (C^N = papaverine; L = CH3CN or DMSO) can be explained by Pd3(OAc)5NO2 acting as the nitrite source. Finally, photocrystallographic metastable linkage isomerisation and complete conversion to an oxygen-bound nitrito complex Pd(η1-ONO)(C^N)PPh3 has been observed.
Co-reporter:Simon K. Brayshaw, Timothy L. Easun, Michael W. George, Alexandra M. E. Griffin, Andrew L. Johnson, Paul R. Raithby, Teresa L. Savarese, Stefanie Schiffers, John E. Warren, Mark R. Warren and Simon J. Teat
Dalton Transactions 2012 vol. 41(Issue 1) pp:90-97
Publication Date(Web):20 Oct 2011
DOI:10.1039/C1DT11379H
Single crystal photocrystallographic experiments and solid state Raman spectroscopy have been used to determine the low temperature, metastable structures of the nickel(II) nitrito complexes [Ni(aep)2(η1-ONO)2] 1#O (aep = 1-(2-aminoethyl)piperidine), [Ni(aem)2(η1-ONO)2] 2#O (aem = 1-(2-aminoethyl)morpholine), and [Ni(aepy)2(η1-ONO)2] 3#O (aepy = 1-(2-aminoethyl)pyrrolidine and where the #O denotes the oxygen-bound nitrito metastable molecule). These linkage isomers of the equivalent nitro complexes [Ni(aep)2(η1-NO2)2] 1, [Ni(aem)2(η1-NO2)2] 2 and [Ni(aepy)2(η1-NO2)2] 3 are formed by LED irradiation at temperatures below 120 K. The behavior of the three complexes upon irradiation is generally similar, but some subtle differences have been observed. From the crystallographic studies all three complexes 1–3 exhibit the endo-nitrito linkage isomer upon irradiation, however, for 3# (a crystal structure that contains components of both 3 and 3#O) an exo-nitrito isomer is also observed. Under conditions of 90–100 K, with blue light, the conversion percentages to the nitrito isomers, 1#O, 2#O and 3#O were 16%, 22% and 30%, respectively. At temperatures below 110 K all three nitrito isomers were stable for over four hours but while 2#O and 3#O could be detected at temperatures down to 30 K, at temperatures below 60 K the metastable structure 1#O appeared to be quenched and only the nitro isomer 1 was identified in the crystal. The solid state Raman spectra for 1#, 2# and 3# confirmed the photocrystallographic results with the nitrito isomers being identified from the O–N–O deformation vibrations.
Co-reporter:Mark R. Warren, Simon K. Brayshaw, Lauren E. Hatcher, Andrew L. Johnson, Stefanie Schiffers, Anna J. Warren, Simon J. Teat, John E. Warren, Christopher H. Woodall and Paul R. Raithby
Dalton Transactions 2012 vol. 41(Issue 42) pp:13173-13179
Publication Date(Web):10 May 2012
DOI:10.1039/C2DT30314K
Low temperature, single crystal photocrystallographic studies have been carried out on four square planar Group 10 complexes [Ni(PEt3)2(NO2)2] 1, [Pd(PPh3)2(NO2)2] 2, [Pd(AsPh3)2(NO2)2] 3 and [Pt(PPh3)2(NO2)2] 4, in which the two nitro groups adopt the trans configuration. Irradiation with UV light, at 100 K, of single crystals of complexes 1–3 photoisomerise from the η1-NO2 nitro form to the η1-ONO nitrito form occurred. Complex 1 underwent 25% conversion to the nitrito form before crystal decomposition occurred. 2 and 3 underwent 46% and 39% conversion, respectively, to the nitrito form when a photostationary state was reached. While under the same experimental conditions 4 showed no isomerisation. The photocrystallographic results can be correlated with the results of DFT calculations and with the observed trends in the solution UV/visible absorption spectroscopy obtained for these complexes. The results suggest that while steric factors in the isomerization processes are important there may also be a kinetic effect relating to the lability of the metal involved.
Co-reporter:Sara Fuertes, Simon K. Brayshaw, Paul R. Raithby, Stephanie Schiffers, and Mark R. Warren
Organometallics 2012 Volume 31(Issue 1) pp:105-119
Publication Date(Web):December 23, 2011
DOI:10.1021/om200589q
The reaction of a new ligand, ethyl 2,6-diphenylisonicotinate (EtO2C–C∧N∧C–H2), with K2PtCl4 in acetic acid affords the monocyclometalated complex [{(EtO2C–C∧N∧C–H)Pt(μ-Cl)}2] (1), which transforms to the bis-cyclometalated derivative [Pt(EtO2C–C∧N∧C)(DMSO)] (2) when heated in hot DMSO. Complex 2 is the precursor for preparing a new series of neutral mononuclear bis-cyclometalated complexes [Pt(EtO2C–C∧N∧C)(L)] (L = tht (3), PPh3 (4), CN-tBu (5), py (6), py-tBu (7), py-NH2 (8), py-CN (9), py-CONH2 (10)). These new complexes have been characterized spectroscopically, and structures of 2–10 have been determined crystallographically. Within each crystal structure the individual molecules pack in a head-to-tail arrangement. Noncovalent interactions, including π···π, C–H···O, C–H···N, N–H···Pt, N–H···π, C–H···π, and N–H···O, contribute significantly to the supramolecular structures displayed by these complexes in the solid state. All complexes display UV–vis absorptions in dichloromethane solution. Excitation and emission studies as well as lifetime measurements are described and can be correlated to the solid-state structures of the complexes. DFT and TDDFT computational studies have been performed on 5 and 8 which support the conclusions drawn from the photophysical studies.
Co-reporter:Sara Fuertes ; Christopher H. Woodall ; Paul R. Raithby ;Violeta Sicilia
Organometallics 2012 Volume 31(Issue 11) pp:4228-4240
Publication Date(Web):May 30, 2012
DOI:10.1021/om300170j
The mononuclear complex [(EtO2C-C∧N∧C)Pt(dmpyz)] (1) (dmpyz = 2,5-dimethylpyrazine) has been synthesized by reaction of [(EtO2C-C∧N∧C)Pt(dmso)] (A) with dmpyz in a 1:1 molar ratio in dichloromethane. Complex 1 is the precursor for preparing the homodinuclear complex [{(EtO2C-C∧N∧C)Pt}2(μ-dmpyz)] (2) and the heterotrinuclear clusters [{(EtO2C-C∧N∧C)Pt(dmpyz)}2M]X (M = Cu, X = PF6 (3); M = Ag, X = BF4 (4)). Compounds 1, 2, and 4 were studied by X-ray diffraction methods. In the crystal packing of 1 and 2, the molecules display short intermolecular π···π contacts, which control the solid-state emissive behavior. X-ray study on 4 shows two [Pt2Ag] sandwich-type clusters in the asymmetric unit, both with the two square-planar “(R-CNC)Pt(dmpyz)” moieties stabilized by two Pt → Ag donor–acceptor bonds as well as by η1- and η2-Ag–C interactions. Intramolecular π–π contacts were found between the pyridine rings of the CNC ligands within the same Pt2Ag cluster. 1H and 195Pt NMR studies confirm that the Pt2M cluster is also retained in solution at room temperature. 195Pt NMR spectra of 3 and 4 show signals shifted significantly downfield when comparing with that for the monomer (1), which is attributed to the presence of Pt–M dative bonds. At lower temperatures (T = 193 K), the copper derivative definitely falls apart, whereas the silver one still holds up unbroken. In the solid state at 77 K, compounds 1–4 give red emissions arising from 3ππ excited states due to the intra- or intermolecular π–π contacts observed in the crystal structures. As expected, in glassy solutions (77 K), compound 3 displays analogous emissions to those from the starting material (1). Complexes 1 and 2 show structured emission bands that are particularly sensitive to the λex (HE and LE). In contrast, 4 displays an unstructured emission at 680 nm with a shoulder at 556 nm; both are not dependent on the λex. DFT and TDDFT computational studies have been performed on 1 and 2, which support the conclusions drawn from the photophysical studies.
Co-reporter:Muhammad S. Khan, Mohammed K. Al-Suti, Hakkikulla H. Shah, Said Al-Humaimi, Fathiya R. Al-Battashi, Jens K. Bjernemose, Louise Male, Paul R. Raithby, Ning Zhang, Anna Köhler and John E. Warren
Dalton Transactions 2011 vol. 40(Issue 39) pp:10174-10183
Publication Date(Web):06 Sep 2011
DOI:10.1039/C1DT11010A
A series of trimethylsilyl-protected di-alkynes incorporating 3,4-ethylenedioxythiophene (EDOT) linker groups Me3Si–CC–R–CC–SiMe3 (R = ethylenedioxythiophene-3,4-diyl 1a, 2,2′-bis-3,4-ethylenedioxythiophene-5,5′-diyl 2a, 2,2′,5′,2′′-ter-3,4-ethylenedioxythiophene-5,5′′-diyl 3a) and the corresponding terminal di-alkynes, H–CC–R–CC–H 1b–2b has been synthesized and characterized and the single crystal X-ray structure of 1a has been determined. CuI-catalyzed dehydrohalogenation reaction between trans-[(Ph)(Et3P)2PtCl] and the terminal di-alkynes 1b–2b in iPr2NH/CH2Cl2 (2:1 mole ratio) gives the Pt(II) di-ynes trans-[(Et3P)2(Ph)Pt–CC–R–CC–Pt(Ph)(Et3P)2] 1M–2M while the dehydrohalogenation polycondensation reaction between trans-[(nBu3P)2PtCl2] and 1b–2b (1:1 mole ratio) under similar reaction conditions affords the Pt(II) poly-ynes trans-[Pt(PnBu3)2–CC–R–CC-]n1P–2P. The di-ynes and poly-ynes have been characterized spectroscopically and, for 1M and 2M, by single-crystal X-ray which confirms the “rigid rod” di-yne backbone. The materials possess excellent thermal stability, are soluble in common organic solvents and readily cast into thin films. Optical absorption spectroscopic measurements reveal that the EDOT spacers create stronger donor-acceptor interactions between the platinum(II) centres and conjugated ligands along the rigid backbone of the organometallic polymers compared to the related non-fused and fused oligothiophene spacers.
Co-reporter:Simon K. Brayshaw, Lionel P. Clarke, Pertti Homanen, Olivia F. Koentjoro, John E. Warren, and Paul R. Raithby
Organometallics 2011 Volume 30(Issue 15) pp:3955-3965
Publication Date(Web):July 8, 2011
DOI:10.1021/om200056c
The reaction between 2- and 3-thienyl-substituted 1,3-butadiynes and the electron-deficient osmium cluster Os3H2(CO)10 yields trinuclear coordination products, associated with transformations of the diacetylene ligands. Depending on the heteroaryl end groups, osmium clusters with both a closed and open Os-triangle core were formed. The reaction between Os3H2(CO)10 and 1,4-bis(2-thienyl)butadiyne yielded [Os3(μ-H)(CO)10{(μ-η-(C4H3S)(C8H4S)}] (1) and [Os3(μ-H)(CO)10{(μ3-η2-η1-η1-(SC7H4)C(SC4H3)}] (2), whereas in the analogous case of 1,4-bis(3-thienyl)butadiyne the main coordination product was found to be [Os3(μ-H)(CO)10{(μ-η-(C4H3S)(C8H4S)}] (3). Compounds 1–3 were stable in air, but lost carbon monoxide upon prolonged heating. Thermal decarbonylation of 1 under N2 yielded a mixture of [Os3(μ-H)(CO)9{(μ3-η3-(C4H3S)(C8H4S)}] (4) and [Os3(μ-H)2(CO)9{(μ3-η1-η1-(C4H3S)(C8H3S)}] (5). Thermal decarbonylation of 2 yielded [Os3(μ-H)(CO)9{(μ3-η3-(C4H3S)(C8H4S)}] (6), while thermal decarbonylation of 3 yielded [Os3(μ-H)(CO)9{(μ3-η3-(C4H3S)(C8H4S)}] (7). A reaction involving 3 with CF3COOH affords as the main cluster product the known cluster [Os3(μ-H)(CO)10(O2CF3)] (8) and, unusually, permits the isolation and characterization of the novel organic molecule [(C4H3S)(C8H4S)(OCF3)] (9) cleaved from the parent cluster. The structures of the new compounds were established by single-crystal X-ray studies and spectroscopic methods and supported by density functional theory.
Co-reporter:Dr. Simon K. Brayshaw;Stephanie Schiffers;Anna J. Stevenson;Dr. Simon J. Teat;Mark R. Warren;Robert D. Bennett;Dr. Igor V. Sazanovich;Dr. Alastair R. Buckley;Dr. Julia A. Weinstein;Dr. Paul R. Raithby
Chemistry - A European Journal 2011 Volume 17( Issue 16) pp:4385-4395
Publication Date(Web):
DOI:10.1002/chem.201003487
Abstract
We introduce a new highly efficient photochromic organometallic dithienylethene (DTE) complex, the first instance of a DTE core symmetrically modified by two PtII chromophores [Pt(PEt3)2(CC)(DTE)(CC)Pt(PEt3)2Ph] (1), which undergoes ring-closure when activated by visible light in solvents of different polarity, in thin films and even in the solid state. Complex 1 has been synthesised and fully photophysically characterised by (resonance) Raman and transient absorption spectroscopy complemented by calculations. The ring-closing photoconversion in a single crystal of 1 has been followed by X-ray crystallography. This process occurs with the extremely high yield of 80 %—considerably outperforming the other DTE derivatives. Remarkably, the photocyclisation of 1 occurs even under visible light (>400 nm), which is not absorbed by the non-metallated DTE core HCC(DTE)CCH (2) itself. This unusual behaviour and the high photocyclisation yields in solution are attributed to the presence of a heavy atom in 1 that enables a triplet-sensitised photocyclisation pathway, elucidated by transient absorption spectroscopy and DFT calculations. The results of resonance Raman investigation confirm the involvement of the alkynyl unit in the frontier orbitals of both closed and open forms of 1 in the photocyclisation process. The changes in the Raman spectra upon cyclisation have permitted the identification of Raman marker bands, which include the acetylide stretching vibration. Importantly, these bands occur in the spectral region unobstructed by other vibrations and can be used for non-destructive monitoring of photocyclisation/photoreversion processes and for optical readout in this type of efficiently photochromic thermally stable systems. This study indicates a strategy for generating efficient solid-state photoswitches in which modification of the PtII units has the potential to tune absorption properties and hence operational wavelength across the visible range.
Co-reporter:Dr. Simon K. Brayshaw;Stephanie Schiffers;Anna J. Stevenson;Dr. Simon J. Teat;Mark R. Warren;Robert D. Bennett;Dr. Igor V. Sazanovich;Dr. Alastair R. Buckley;Dr. Julia A. Weinstein;Dr. Paul R. Raithby
Chemistry - A European Journal 2011 Volume 17( Issue 16) pp:
Publication Date(Web):
DOI:10.1002/chem.201190076
Co-reporter:Lauren E. Hatcher;Mark R. Warren;Dr. David R. Allan;Dr. Simon K. Brayshaw;Dr. Andrew L. Johnson;Dr. Sara Fuertes;Dr. Stefanie Schiffers;Anna J. Stevenson;Dr. Simon J. Teat;Christopher H. Woodall; Paul R. Raithby
Angewandte Chemie International Edition 2011 Volume 50( Issue 36) pp:8371-8374
Publication Date(Web):
DOI:10.1002/anie.201102022
Co-reporter:Lauren E. Hatcher;Mark R. Warren;Dr. David R. Allan;Dr. Simon K. Brayshaw;Dr. Andrew L. Johnson;Dr. Sara Fuertes;Dr. Stefanie Schiffers;Anna J. Stevenson;Dr. Simon J. Teat;Christopher H. Woodall; Paul R. Raithby
Angewandte Chemie 2011 Volume 123( Issue 36) pp:8521-8524
Publication Date(Web):
DOI:10.1002/ange.201102022
Co-reporter:Saifun Nahar;John E. Davies;Gregory P. Shields
Journal of Cluster Science 2010 Volume 21( Issue 3) pp:379-396
Publication Date(Web):2010 September
DOI:10.1007/s10876-010-0331-9
Reaction of the [Rh(η5-C5Me5)(NCMe)3]2+ (1) dication with the hexaosmium [Os6(CO)17]2− (2) dianion leads to the initial formation of [Os6(CO)17Rh(η5-C5Me5)] (3). This cluster readily adds CO to form [Os6(CO)18Rh(η5-C5Me5)] (4) which has been characterised crystallographically. 3 also adds dihydrogen to give [Os6H2(CO)17Rh(η5-C5Me5)] (5) and undergoes a substitution reaction with PPh3 to form [Os6(CO)16(PPh3)Rh(η5-C5Me5)] (6). With the [Ru6(CO)18]2− (7) dianion, [Rh(η5-C5Me5)(NCMe)3]2+ (1) reacts to form three mixed-metal clusters [Ru5(CO)15Rh(η5-C5Me5)] (8), [Ru6(CO)18Rh(η5-C5Me5)] (9) and [Ru6(CO)18Rh2(η5-C5Me5)2] (10). The clusters have been characterised spectroscopically and the structures of 8 and 10 have been confirmed crystallographically. The cluster 8 undergoes a substitution reaction with P(OMe)3 to form the disubstituted product [Ru5(CO)13(P(OMe)3)2Rh((η5-C5Me5)] (11) which has also been characterised crystallographically.
Co-reporter:Lekshmi Sudha Devi, Mohammed K. Al-Suti, Ning Zhang, Simon J. Teat, Louise Male, Hazel A. Sparkes, Paul R. Raithby, Muhammad S. Khan and Anna Köhler
Macromolecules 2009 Volume 42(Issue 4) pp:1131-1141
Publication Date(Web):January 22, 2009
DOI:10.1021/ma802399a
The synthesis and characterization of the thieno[3,2-b]thiophene and dithieno[3,2-b:2′,3′-d]thiophene containing platinum(II) poly-ynes and their molecular precursors is described and the electronic structure is established by absorption, luminescence and photoinduced absorption measurements. A comparison of the electronic structure of the fused and the nonfused oligothiophenes, thieno[3,2-b]thiophene, dithieno[3,2-b:2′,3′-d]thiophene, 2,2′-bithiophene, and 2,2′:5′,2′′-terthiophene incorporated in platinum(II) poly-ynes is reported. We find the singlet S1 and triplet T1 and Tn excited states to be at higher energy in thin films made from the fused systems than from the nonfused systems. For ligands with the same number of rings, we attribute this to the decreased number of double bonds in the fused system and to the presence of an additional sulfur atom in spacers with the same number of double bonds.
Co-reporter:MarkR. Warren;SimonK. Brayshaw Dr.;AndrewL. Johnson Dr.;Stefanie Schiffers;PaulR. Raithby ;TimothyL. Easun Dr.;MichaelW. George ;JohnE. Warren Dr.;SimonJ. Teat Dr.
Angewandte Chemie 2009 Volume 121( Issue 31) pp:5821-5824
Publication Date(Web):
DOI:10.1002/ange.200901706
Co-reporter:MarkR. Warren;SimonK. Brayshaw Dr.;AndrewL. Johnson Dr.;Stefanie Schiffers;PaulR. Raithby ;TimothyL. Easun Dr.;MichaelW. George ;JohnE. Warren Dr.;SimonJ. Teat Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 31) pp:5711-5714
Publication Date(Web):
DOI:10.1002/anie.200901706
Co-reporter:Peiyi Li, Birte Ahrens, Andrew D. Bond, John E. Davies, Olivia F. Koentjoro, Paul R. Raithby and Simon J. Teat
Dalton Transactions 2008 (Issue 12) pp:1635-1646
Publication Date(Web):24 Jan 2008
DOI:10.1039/B716664H
A series of novel digold complexes incorporating ethynyl pyridine derivatives as a spacer unit, [(R3P)Au(CC)X(CC)Au(PR3)] (R = Ph, X = 2,5-pyridine (1); R = Cy (cyclohexane), X = 2,5-pyridine (2); R = Ph, X = 2,6-pyridine (3); R = Ph, X = 2,5′-bipyridine (4); R = Ph, X = 2,6′-bipyridine (5)), has been synthesised. All the complexes have been characterised spectroscopically and the structures determined by single-crystal X-ray crystallography. The central (CC)(X)(CC) unit is essentially linear for complexes 1, 2 and 4 and kinked for complexes 3 and 5, but only in 1, with the shortest spacer group and the less bulky phosphine ligand, is there evidence of d10⋯d10Au⋯Au interactions (Au–Au 3.351(2) Å). The solution UV/visible absorption and emission spectra for all the complexes are similar to those of the free ligands suggesting that the spectra are dominated by π–π* ligand-centred transitions and this is confirmed by DFT calculations.
Co-reporter:Simon A. Cotton, Victoria M.A. Fisher, Paul R. Raithby, Stefanie Schiffers, Simon J. Teat
Inorganic Chemistry Communications 2008 Volume 11(Issue 7) pp:822-824
Publication Date(Web):July 2008
DOI:10.1016/j.inoche.2008.04.007
Reaction of hydrated scandium nitrate with 1,10-phenanthroline (phen) in methanol leads to formation of the unusual dimeric complex [(phen)(NO3)2Sc(μ-OMe)2Sc(NO3)2(phen)], in which the scandium centres are eight co-ordinate. The complex features two bridging methoxy ligands, as well as bidentate nitrates and chelating 1,10-phenanthroline ligands.Reaction of hydrated scandium nitrate with 1,10-phenanthroline led to the formation and structural characterisation of the first di-methoxy bridged dimeric complex [(phen)(NO3)2Sc(μ-OMe)2Sc(NO3)2(phen)], in which the scandium centres are eight co-ordinate.
Co-reporter:Birte Ahrens, Lionel P. Clarke, Neil Feeder, Muhammad S. Khan, Peiyi Li, James N. Martin, Paul R. Raithby
Inorganica Chimica Acta 2008 Volume 361(Issue 11) pp:3117-3124
Publication Date(Web):27 July 2008
DOI:10.1016/j.ica.2007.12.020
The reaction of 2 equiv. of [Os3(CO)10(MeCN)2] with R–CC–L–CC–R (R = H, L = (C4H2S); R = SiMe3, L = (C4H2S–C4H2S), (C4H2S–C4H2S–C4H2S), (C4H2S)–(C14H8)–(C4H2S)) affords the series of linked clusters [{Os3(CO)10}(HCC(C4H2S)CCH){Os3(CO)10}] (1), [{Os3(CO)10}(Me3SiCC(C4H2S–C4H2S)CCSiMe3){Os3(CO)10}] (2), [{Os3(CO)10}(Me3SiCC(C4H2S–C4H2S–C4H2S)CCSiMe3){Os3(CO)10}] (4) and [{Os3(CO)10}(Me3SiCC(C4H2S)–(C14H8)–(C4H2S)CCSiMe3){Os3(CO)10}] (6) as the major products. The complexes have been characterised by a range of spectroscopic methods and, in the case of 1 and 2 by single crystal X-ray crystallography. The alkyne groups cap the osmium triangles in the expected μ3-η2-||-bonding mode and each triangle is coordinated by nine terminal and one μ2-carbonyl group. Solution UV–Vis spectra of the complexes were similar to those observed for the free ligands consistent with there being little delocalisation between the cluster units and the thiophene groups.The series of ethynyl thiophene ligands, R–CC–L–CC–R (R = H, L = (C4H2S); R = SiMe3, L = (C4H2S–C4H2S), (C4H2S–C4H2S–C4H2S), (C4H2S)–(C14H8)–(C4H2S)), have been used to link two “Os3(CO)10” cluster carbonyl units together successfully.
Co-reporter:Simon K. Brayshaw Dr.;Jennifer C. Green ;Ruth Edge Dr.;Eric J. L. McInnes ;Paul R. Raithby ;John E. Warren Dr.;Andrew S. Weller Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 41) pp:
Publication Date(Web):6 SEP 2007
DOI:10.1002/anie.200703069
Heptarhodium wagon wheel [Rh7(PiPr3)6H18][BArF4]2 (shown), which resembles a planar Rh(111) surface with 18 hydride ligands, was obtained together with [Rh8(PiPr3)6H16][BArF4]2 from the reaction of [Rh(PiPr3)2(nbd)][BArF4] and [Rh(nbd)2][BArF4] with hydrogen (ArF=C6H3(CF3)2, nbd=norbornadiene). Some of the H ligands are located in threefold hollows between Rh centers and thus mimic the orientation of atomic hydrogen adsorbed on Rh(111).
Co-reporter:Simon K. Brayshaw Dr.;Jennifer C. Green ;Ruth Edge Dr.;Eric J. L. McInnes ;Paul R. Raithby ;John E. Warren Dr.;Andrew S. Weller Dr.
Angewandte Chemie 2007 Volume 119(Issue 41) pp:
Publication Date(Web):6 SEP 2007
DOI:10.1002/ange.200703069
Ein Heptarhodium-Speichenrad, [Rh7(PiPr3)6H18][BArF4]2 (siehe Bild), das einer planaren Rh(111)-Oberfläche mit 18 Hydridliganden ähnelt, entstand neben [Rh8(PiPr3)6H16][BArF4]2 bei der Reaktion von [Rh(PiPr3)2(nbd)][BArF4] und [Rh(nbd)2][BArF4] (ArF=C6H3(CF3)2, nbd=Norbornadien) mit Wasserstoff. Einige der H-Liganden befinden sich in dreizähligen Lücken zwischen Rh-Zentren und imitieren so die Orientierung von atomarem Wasserstoff auf Rh(111).
Co-reporter:Katharine F. Bowes, Jacqueline M. Cole, Shamus L. G. Husheer, Paul R. Raithby, Teresa L. Savarese, Hazel A. Sparkes, Simon J. Teat and John E. Warren
Chemical Communications 2006 (Issue 23) pp:2448-2450
Publication Date(Web):04 May 2006
DOI:10.1039/B604039J
The structure of a new metastable geometric isomer of [Ru(NH3)4(H2O)(SO2)][MeC6H4SO3]2 in which the SO2 group is coordinated through a single oxygen in an η1-OSO bonding mode has been determined at 13 K; the new isomer was obtained as a 36% component of the structure within a single crystal upon irradiation using a tungsten lamp.
Co-reporter:Angelo J. Amoroso, Brian F.G. Johnson, Jack Lewis, Chi-Keung Li, M. Carmen Ramirez de Arellano, Paul R. Raithby, Gregory P. Shields, Wing-Tak Wong
Inorganica Chimica Acta 2006 Volume 359(Issue 11) pp:3589-3595
Publication Date(Web):1 August 2006
DOI:10.1016/j.ica.2005.12.010
Reactions between the activated cluster [Os3(CO)10(NCMe)2] and malonic acid, succinic acid and dicarboxylic acetylene, respectively, lead to the formation of the linked cluster complexes [{Os3H(CO)10}2(CO2CH2CO2)] (1), [{Os3H(CO)10}2(CO2C2H4CO2)] (2), and [{Os3H(CO)10}2(C4O4)] (3) in good yield. Cluster 3 was subsequently treated with [Co2(CO)8] and this results in the addition of a “Co2(CO)6” group giving [{Os3H(CO)10}2(C2O4){Co2(CO)6}] (4). The X-ray crystal structures are reported for 2–4. In each structure the two triangular triosmium units are linked by the carboxylate groups and within each complex the carboxylate groups are chelating and bridge two osmium atoms.The reaction of 2 equiv. of [Os3(CO)10(NCMe)2] with a series of dicarboxylic acids leads to the formation of a series of hexanuclear linked clusters. The reaction of one of these clusters, [{Os3H(CO)10}2(C4O4)] with [Co2(CO)8] affords the mixed metal cluster [{Os3H(CO)10}2(C4O4){Co2(CO)6}].
Co-reporter:Peiyi Li, Birte Ahrens, Neil Feeder, Paul R. Raithby, Simon J. Teat and Muhammad S. Khan
Dalton Transactions 2005 (Issue 5) pp:874-883
Publication Date(Web):31 Jan 2005
DOI:10.1039/B415965A
A series of protected and terminal dialkynes with extended π-conjugation through the fused oligothienyl linker unit in the backbone, 2,5-bis(trimethylsilylethynyl)thieno[3,2-b]thiophene 1a, 5,5′-bis(trimethylsilylethynyl)dithieno[3,2-b:2′,3′-d]thiophene 1b, 2,5-bis(ethynyl)thieno[3,2-b]thiophene 2a, 5,5′-bis(ethynyl)dithieno[3,2-b:2′,3′-d]thiophene 2b, has been synthesized and characterised. The digold alkynyl complexes [(Ph3P)Au(CC)(C6H2S2)(CC)Au(PPh3)]
3a and [(Ph3P)Au(CC)(C8H2S3)(CC)Au(PPh3)]
3b have then been prepared by the reaction of two equivalents of Ph3PAuCl and a methanolic KOH solution of 1a and 1b, respectively. The complexes have been characterised spectroscopically. The crystal structures show that the gold centres adopt a linear two-coordinate geometry appropriate for Au(I) complexes. Within the crystals adjacent molecules are linked by Au⋯S intermolecular interactions in the range 3.48–3.89 Å, but there are no short Au⋯Au contacts. The absence of Au⋯Au interactions in solution is confirmed by UV/visible absorption and emission spectroscopy, the spectra being dominated by ligand-centred π–π* interactions.
Co-reporter:Ramkrishna Saha, Md. Abdul Qaium, Dipen Debnath, Muhammad Younus, Nazia Chawdhury, Nasim Sultana, Gabriele Kociok-Köhn, Li-ling Ooi, Paul R. Raithby and Masashi Kijima
Dalton Transactions 2005 (Issue 16) pp:2760-2765
Publication Date(Web):12 Jul 2005
DOI:10.1039/B505484B
A series of cis-platinum ethynyl complexes with the general formula cis–[Pt(dppe)(CCR)2]
(dppe = 1,2-bis(diphenylphosphino)ethane; R = C6H4-p-NO21, C6H4-p-CH32, C6H4-p-CCH 3 and C6H4-p-C6H4-p-CCH 4) have been prepared by the coupling reaction of cis-[Pt(dppe)Cl2] with two equivalents of the appropriate alkyne. The new complexes have been fully characterized by spectroscopic techniques, and the cis square planar arrangement at the platinum centre has been confirmed by single-crystal X-ray diffraction studies of complexes 1, 2 and 4. The absorption spectra of the complexes 1–4 are dominated by a π
→
π* band that contains some platinum (n + 1) p orbital character. The position of the band is dependent on the electron donating or withdrawing properties of the ethynyl substituents, R. Complex 1 displays a triplet emission in the green, at room temperature, while complexes 2–4, display singlet emissions in the blue. Again, the difference can be attributed to the nature of the R substituents.
Co-reporter:Katharine F. Bowes, Ian P. Clark, Jacqueline M. Cole, Matthew Gourlay, Alexandra M. E. Griffin, Mary F. Mahon, Liling Ooi, Anthony W. Parker, Paul R. Raithby, Hazel A. Sparkes and Mike Towrie
CrystEngComm 2005 vol. 7(Issue 43) pp:269-275
Publication Date(Web):06 Apr 2005
DOI:10.1039/B502275D
The crystal structure of a new monoclinic polymorph of 2,2′ : 6′,2″ terpyridine (terpy) that contains two independent but structurally similar molecules, and crystallises in a space group P21/c, is reported. Variable-temperature single-crystal X-ray diffraction studies at 100, 150, 200 and 250 K show that the b-axis length increases significantly with increasing temperature. The molecules pack in a sandwich herringbone arrangement and the intermolecular non-bonded contacts are described in detail. All structural results are contrasted with those obtained for the previously reported polymorph of terpyridine, which crystallises in the orthorhombic space group P212121. Photophysical properties of the new polymorph of terpyridine are also reported. The solid-state room temperature UV/vis absorption and emission spectra were measured. The sample was found to phosphoresce on the millisecond timescale, this being measured as a function of temperature (100–280 K). Its long-lived phosphorescent nature also allowed a pseudo-steady-state to be built up by continuous laser pumping (at 10 Hz repetition rate), which was investigated as a function of temperature (100–280 K).
Co-reporter:Frank H. Allen, Mary F. Mahon, Paul R. Raithby, Gregory P. Shields and Hazel A. Sparkes
New Journal of Chemistry 2005 vol. 29(Issue 1) pp:182-187
Publication Date(Web):08 Dec 2004
DOI:10.1039/B412989J
The mechanism of the solid state [2 + 2] cycloaddition of alkenes has been investigated using the structure correlation method based on geometrical data calculated from single crystal X-ray diffraction studies retrieved from the Cambridge Structural Database (CSD). Searches were carried out for non-bonded alkene⋯alkene reactant interactions, within a limiting C⋯C separation of the sum of van der Waals radii plus 20%, and for bonded cyclobutane product fragments. The results were visualised and interpreted using principal component analysis and symmetry deformation coordinates. The reaction pathway for [2 + 2] cycloaddition was established and it was shown that (a) the alkene moieties are not required to be parallel for the reaction to occur and (b) a large twist angle of the reacting alkene fragments is permissible to form a puckered cyclobutane reaction product, as long as similar intra-annular valence angles are maintained around the four-membered ring.
Co-reporter:Lionel P. Clarke, Jacqueline M. Cole, John E. Davies, Alexandra French, Olivia F. Koentjoro, Paul R. Raithby and Gregory P. Shields
New Journal of Chemistry 2005 vol. 29(Issue 1) pp:145-153
Publication Date(Web):01 Jan 2004
DOI:10.1039/B412578A
Reaction of [Os3(μ-H)2(CO)10] with 1,4-dipyridylbuta-1,3-diyne yields two clusters, [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}]
1 and [Os3(μ-H)(CO)10-{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}]
2, in which the diyne has rearranged to form a substituted indolizine ring system. Complex 1 converts slowly to 2 at room temperature, and may be decarbonylated to yield [Os3(μ-H)(CO)9{μ-η1:η2:η1-(C8H5N)–C–(C5H4N)}]
3. An analogous reaction involving [Os3(CO)10(MeCN)2] generates three products, [Os3(μ-H)(CO)10{μ-η1:η1-(NC5H3)–C2C2–(C5H5N)}]
4 and [{Os3(μ-H)(CO)10}2{μ-η1:η1-(NC5H3)–C2–}2]
5, both coordinated via orthometallated pyridyl rings, and a minor product [{Os3(CO)10}2{μ3-η1:η1:η1-C2-(NC5H4)}2]
6, coordinated via
μ-carbene and σ-N interactions, the linking ligand retaining its central CC bond. Complex 4 reacts with [Os3(μ-H)2(CO)10] to form the linked cluster [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}]
7, also forming an indolizine ring system. The structures of 1–3, 6, 6·2[CH2Cl2] and 7 have been established by X-ray crystallography.
Co-reporter:Muhammad S. Khan, Muna R. A. Al-Mandhary, Mohammed K. Al-Suti, Fathiya R. Al-Battashi, Sultan Al-Saadi, Birte Ahrens, Jens K. Bjernemose, Mary F. Mahon, Paul R. Raithby, Muhammad Younus, Nazia Chawdhury, Anna Köhler, Elizabeth A. Marseglia, Emilio Tedesco, Neil Feeder and Simon J. Teat
Dalton Transactions 2004 (Issue 15) pp:2377-2385
Publication Date(Web):22 Jun 2004
DOI:10.1039/B405070C
A series of protected and terminal dialkynes with extended π-conjugation through a condensed aromatic linker unit in the backbone, 1,4-bis(trimethylsilylethynyl)naphthalene, 2a, 1,4-bis(ethynyl)naphthalene, 2b, 9,10-bis(trimethylsilylethynyl)anthracene 3a, 9,10-bis(ethynyl)anthracene 3b, have been synthesized and characterized spectroscopically. The solid-state structures of 2a and 3a have been confirmed by single crystal X-ray diffraction studies. Reaction of two equivalents of the complex trans-[Ph(Et3P)2PtCl] with an equivalent of the terminal dialkynes 1,4-bis(ethynyl)benzene 1b and 2b–3b, in iPr2NH–CH2Cl2, in the presence of CuI, at room temperature, afforded the platinum(II) di-ynes trans-[Ph(Et3P)2Pt–CC–R–CC–Pt(PEt3)2Ph]
(R = benzene-1,4-diyl 1c; naphthalene-1,4-diyl 2c and anthracene-9,10-diyl 3c) while reactions between equimolar quantities of trans-[(nBu3P)2PtCl2] and 2b–3b under similar conditions readily afforded the platinum(II) poly-ynes trans-[–(nBu3P)2Pt–CC–R–CC–]n
(R = naphthalene-1,4-diyl 2d and anthracene-9,10-diyl 3d). The Pt(II) diynes and poly-ynes have been characterized by analytical and spectroscopic methods, and the single crystal X-ray structures of 1c and 2c have been determined. These structures confirm the trans-square planar geometry at the platinum centres and the linear nature of the molecules. The di-ynes and poly-ynes are soluble in organic solvents and readily cast into thin films. Optical spectroscopic measurements reveal that the electron-rich naphthalene and anthracene spacers create strong donor–acceptor interactions between the Pt(II) centres and conjugated ligands along the rigid backbone of the organometallic polymers. Thermogravimetry shows that the di-ynes possess a somewhat higher thermal stability than the corresponding poly-ynes. Both the Pt(II) di-ynes and the poly-ynes exhibit increasing thermal stability along the series of spacers from phenylene through naphthalene to anthracene.
Co-reporter:Nazia Chawdhury, Nicholas J. Long, Mary F. Mahon, Li-ling Ooi, Paul R. Raithby, Stephanie Rooke, Andrew J.P. White, David J. Williams, Muhammad Younus
Journal of Organometallic Chemistry 2004 Volume 689(Issue 4) pp:840-847
Publication Date(Web):16 February 2004
DOI:10.1016/j.jorganchem.2003.11.035
A series of aromatic ethynyl-bridged ferrocenes with the general formula Fc–CC–R–CC–Fc (Fc=ferrocenyl, R=C6H2(-p-CH3)2 (1), C6H4-p-C6H4 (2), C5H3N (3), 9,10–C14H8 (4), C4H2S (5), (C4H2S)2 (6) and (C4H2S)3 (7)) has been synthesised by the reaction of ethynyl ferrocene with the appropriate dibromo-arenes. The new complexes have been characterised by spectroscopic techniques. The structures of 3 and 7 were determined via X-ray crystallography, and both show the trans–trans configuration of the two ethynyl ferrocene groups with respect to the central R group. The electronic properties of the compounds have been studied via optical spectroscopy and cyclic voltammetry.A series of aromatic ethynyl-bridged ferrocenes with the general formula Fc–CC–R–CC–Fc (Fc=ferrocenyl, R=C6H2(-p-CH3)2, C6H4-p-C6H4, C5H3N, 9,10–C14H8, C4H2S, (C4H2S)2 and (C4H2S)3) has been synthesised by the reaction of ethynyl ferrocene with appropriate dibromo-arenes. The structures of 3 and 7 have been determined via X-ray crystallography, and the electronic properties of the compounds have been studied via optical spectroscopy and cyclic voltammetry.
Co-reporter:Muhammad S. Khan, Muna R. A. Al-Mandhary, Mohammed K. Al-Suti, Birte Ahrens, Mary F. Mahon, Louise Male, Paul R. Raithby, Clare E. Boothby and Anna Köhler
Dalton Transactions 2003 (Issue 1) pp:74-84
Publication Date(Web):25 Nov 2002
DOI:10.1039/B208963G
A series of trimethylsilyl-protected and terminal mono- and bis-alkynes based on 9,9-dioctylfluorene, 2-(trimethylsilylethynyl)-9,9-dioctylfluorene 1a, 2-ethynyl-9,9-dioctylfluorene 1b, 2,7-bis(trimethylsilylethynyl)-9,9-dioctylfluorene 2a, 2,7-bis(ethynyl)-9,9-dioctylfluorene 2b, have been synthesised. Reaction of trans-[(PnBu3)2PtCl2] with 2 equivalents of the terminal ethyne 1b yields the mononuclear platinum(II) diyne 3, reaction of trans-[(Ph)(Et3P)2PtCl] with 0.5 equivalents of the diterminal ethyne 2b gives the dinuclear platinum(II) diyne 4 while 1 ∶ 1 reaction between trans-[(PnBu3)2PtCl2] and 2b gives the platinum(II) poly-yne 5. Treatment of 2,5-dioctyloxy-1,4-diiodobenzene with 1b in 1 ∶ 2 stoichiometry produces the organic di-yne 6 while 1 ∶ 1 reaction between 2,5-dioctyloxy-1,4-diiodobenzene and 2b, 2,7-bis(ethynyl)fluorene or 2,7-bis(ethynyl)fluoren-9-one produces the organic co-poly-ynes 7–9. All the new materials have been characterised by analytical and spectroscopic methods and the single crystal X-ray structures of 2a and 3 have been determined. The diynes and poly-ynes are soluble in organic solvents and are readily cast into thin films. Optical spectroscopic measurements reveal that the attachment of octyl side-chains on the fluorenyl spacer reduces inter-chain interaction in the poly-ynes while a fluorenonyl spacer creates a donor–acceptor interaction along the rigid backbone of the organometallic poly-ynes and organic co-poly-ynes.
Co-reporter:Muhammad S. Khan, Mohammed K. Al-Suti, Muna R. A. Al-Mandhary, Birte Ahrens, Jens K. Bjernemose, Mary F. Mahon, Louise Male, Paul R. Raithby, Richard H. Friend, Anna Köhler and Joanne S. Wilson
Dalton Transactions 2003 (Issue 1) pp:65-73
Publication Date(Web):25 Nov 2002
DOI:10.1039/B208494E
A new series of rigid rod protected and terminal dialkynes with extended π-conjugation through aromatic and hetero-aromatic linker units in the backbone, 2,5-bis(trimethylsilylethynyl)-1-(2-ethylhexyloxy)-4-methoxybenzene 1a, 2,5-bis(ethynyl)-1-(2-ethylhexyloxy)-4-methoxybenzene 1b, 5,8-bis(trimethylsilylethynyl)quinoline 2a, 5,8-bis(ethynyl)quinoline 2b, 2,3-diphenyl-5,8-bis(trimethylsilylethynyl)quinoxaline 3a, 2,3-diphenyl-5,8-bis(ethynyl)quinoxaline 3b, 4,7-bis(trimethysilylethynyl)-2,1,3-benzothiadiazole 4a and 4,7-bis(ethynyl)-2,1,3-benzothiadiazole 4b, has been synthesised. Treatment of the complex trans-[Pt(Ph)(Cl)(Et3P)2] with half an equivalent of the diterminal alkynes 1b–4b in iPr2NH–CH2Cl2, in the presence of CuI, at room temperature, afforded the platinum(II) di-yne complexes trans-[(Et3P)2(Ph)Pt–CC–R–CC–Pt(Ph)(Et3P)2] [R = 1-(2-ethylhexyloxy)-4-methoxybenzene-2,5-diyl 1c, quninoline-5,8-diyl 2c, 2,3-diphenylquinoxaline-5,8-diyl 3c, 2,1,3-benzothiadiazole-4,7-diyl 4c] in good yields. The new acetylide-functionalised ligands and the platinum(II)
σ-acetylide complexes have been characterised by analytical and spectroscopic methods and X-ray single crystal structure determinations for 2c–4c. The absorption spectra of the complexes 2c–4c show substantial donor–acceptor interaction between the platinum(II) centres and the conjugated ligands. The photoluminescence spectra of 1c–3c show characteristic singlet (S1) and triplet (T1) emissions. Both the singlet and triplet emissions as well as the absorption decrease in energy with increasing electronegativity of the spacer groups along the series 1c–4c.
Co-reporter:David R. Armstrong;Robert P. Davies;Robert Haigh;Mark A. Hendy;Paul R. Raithby;Ronald Snaith;Andrew E. H. Wheatley
European Journal of Inorganic Chemistry 2003 Volume 2003(Issue 18) pp:
Publication Date(Web):12 SEP 2003
DOI:10.1002/ejic.200300244
(Trimethylsilyl)diazomethane (1-H) reacts with nBuLi in THF at elevated temperature to afford (previously reported) 1-Li·3/2THF. However, reaction in hexane/TMEDA at low temperature affords instead the N-lithiate Me3SiCNNLi·TMEDA (9), which is a novel “open” pseudo-cubic tetramer in the solid state. Variable-temperature NMR spectroscopy suggests that N-metallated 9, apparently the kinetic product of the reaction, irreversibly rearranges at high temperature in solution to give the thermodynamically preferred C-lithiated isomer. These observations, supported by DFT calculations, influence our understanding of the reactivity of lithiated diazomethanes towards aryl isothiocyanates, suggesting as they do that previously observed product selectivity in these reactions is critically dependent on temperature control exercised during the process. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:Matthew G. Davidson;Paul R. Raithby;Andrew L. Johnson;Philip D. Bolton
European Journal of Inorganic Chemistry 2003 Volume 2003(Issue 18) pp:
Publication Date(Web):12 SEP 2003
DOI:10.1002/ejic.200300372
Complexation of the salt lithium triflamide [LiNTf2; NTf2 = N(SO2CF3)2], with a range of nitrogen donor ligands results in the formation of a series of coordination complexes [(Tf2N)Li·N(H)PPh2Me] (1), [(Tf2N)Li·N(H)PPh3] (2), [(Tf2N)Li·TMEDA] (3), [(Tf2N)Li·PMDTEA] (4) and [(Bz-TAC)2Li][NTf2] (5). The molecular structures of 1, 3, 4 and 5 have been determined by single-crystal X-ray diffraction. The resulting solvated structures are discussed in terms of solvent-separated ion pairs, contact ion pairs and higher aggregates, with the degree of aggregation and ion contact being dependent on the relative coordinating abilities and steric demands of the donor ligand and the anion, and provide an insight into the possible structures of lithium aggregates present in complex systems such as polymer electrolytes and synthetic reaction media. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:Simon A. Cotton, Oliver E. Noy, Florian Liesener, Paul R. Raithby
Inorganica Chimica Acta 2003 Volume 344() pp:37-42
Publication Date(Web):20 February 2003
DOI:10.1016/S0020-1693(02)01266-5
The complexes [M(terpy)(NO3)3·(H2O)] (M=Eu (1), Tb(2); terpy=2,2′:6′,2″-terpyridyl) are isomorphous and crystallise in the triclinic space group with Z=2, while the related complex [Eu(bipy)2(NO3)3] (3) (bipy=2,2′-bipyridyl) crystallises in the orthorhombic space group Pcan (non-standard setting of Pbcn) with Z=4 such that the two halves of the molecule are related by a crystallographic twofold axis. In each of the three complexes the metal atom is 10 co-ordinate and all three contain three bidentate nitrate groups. In complexes 1 and 2 the coordination sphere is completed by the three nitrogen donor atoms of the terpyridine ligand and by the oxygen donor atom of the coordinated water solvent molecule. In complex 3 the coordination sphere is completed by two nitrogen donor atoms from each of the two bipyridine ligands. These structures are compared with other lanthanide complexes of related ligands and the factors affecting the co-ordination geometry evaluated.In the complexes [M(terpy)(NO3)3·(H2O)] (M=Eu, Tb) and in [Eu(bipy)2(NO3)3] the lanthanide ions are 10 co-ordinate, and each complex contains three bidentate nitrate groups. In [M(terpy)(NO3)3·(H2O)] (M=Eu, Tb) the terpy occupies three of the sites in the equatorial plane as does the coordinated water molecule. Two of the nitrate groups are axial, and one is pseudo equatorial.
Co-reporter:Lionel P Clarke, John E Davies, Dmitrii V Krupenya, Paul R Raithby, Gregory P Shields, Galina L Starova, Sergey P Tunik
Journal of Organometallic Chemistry 2003 Volume 683(Issue 2) pp:313-323
Publication Date(Web):15 October 2003
DOI:10.1016/S0022-328X(03)00537-0
The reaction between [Os3H2(CO)10] and 1,4-diphenylbuta-1,3-diyne yields two isomers of the triosmium cluster [Os3(μ-H)(CO)10{μ3-η1:η3:η1-Ph(C)C9H6)}] 1, 2, the structures of which involve an ‘open’ metal triangle and exhibit an unusual pseudo-allylic interaction involving a fused six- and five-membered ring system obtained from a ring-closure reaction of the diyne. Thermal decarbonylation of 1 and 2 produces [Os3(μ-H)(CO)9{μ3-η1:η3:η1-Ph(C)C9H6)}] (3) which has a ‘closed’ Os3 triangular core and the same formal μ3-η1:η3:η1-allylic coordination mode. The reaction of 1 with trimethylamine-N-oxide in the presence of acetonitrile affords [Os3(μ-H)(CO)8(MeCN){μ3-η1:η3:η1-(Ph(C)C9H6)}] (4), an acetonitrile-substituted derivative of 3. The structures of clusters 1–4 have been established by X-ray crystallography, and all the new clusters have been characterised spectroscopically.Reaction of the unsaturated cluster [Os3H2(CO)10] with 1,4-diphenylbuta-1,3-diyne gives two isomeric products with ‘open’ triangular metal frameworks that show ring closure of the diyne on the cluster triangle. Decarbonylation of these clusters gives a single trinuclear product with a ‘closed’ triangular osmium core.
Co-reporter:Peiyi Li, Birte Ahrens, Ka-Ho Choi, Muhammad S. Khan, Paul R. Raithby, Paul J. Wilson and Wai-Yeung Wong
CrystEngComm 2002 vol. 4(Issue 69) pp:405-412
Publication Date(Web):19 Jul 2002
DOI:10.1039/B202283D
The acetylide-functionalised thiophene gold(I) complexes [(PPh3)Au(CC(C4H2S)(C4H3S))]
1, [(PPh3)Au(CC(C4H2S)CC)Au(PPh3)]
2, [(PR3)Au(CC(C4H2S)2CC)Au(PR3)]
(R = Ph 3, Cy 4) and [(PPh3)Au(CC(C4H2S)3CC)Au(PPh3)]
5 have been prepared by the reaction of the trimethylsilylethynyl oligothiophene
with KOH/MeOH, followed by the addition of a stoichiometric amount of the gold(I) phosphine chloride and NaOMe in MeOH. The products have been characterised spectroscopically and their single crystal X-ray structures determined. In the molecular structures of all the complexes the Au(I) centres adopt the expected linear, two-coordinate geometry, except that in 2 the material forms a polymer through additional Au⋯Au interactions [Au⋯Au 3.2915(10) and 3.2347(9)
Å] between the molecular units. With the longer acetylene-functionalised spacer groups no Au⋯Au interactions are present, but hydrogen bonding and π⋯π interactions are of significance. The solution absorption and emission spectra of these complexes have been recorded and the maxima are increasingly red-shifted as the length and planarity of the functionalised spacer groups increases which is
consistent with the absorptions and emissions being dominated by ligand-centred π–π* transitions.
Co-reporter:Elena A. Vinogradova, Olga Yu. Vassilyeva, Volodymyr N. Kokozay, Brian W. Skelton, Jens K. Bjernemose and Paul R. Raithby
Dalton Transactions 2002 (Issue 22) pp:4248-4252
Publication Date(Web):14 Oct 2002
DOI:10.1039/B205917G
Three kinds of CdII/CuII compounds of different nuclearity: tetranuclear [Cd2Cu2I4L4(dmso)2] 1, pentanuclear [Cd2Cu3Br6L4(dmso)2] 2 and hexanuclear [Cd4Cu2Cl6L6(H2O)2] 3, have been synthesised by the reaction of zerovalent copper and cadmium halides with non-aqueous (dmso, CH3CN) solutions of 2-dimethylaminoethanol (HL) in air. The choice of counteranion in the initial cadmium salt provides a useful method of altering the nuclearity and structure of the heterometallic Cd/Cu complexes. Crystallographic investigations reveal that 1 has a centrosymmetric tetranuclear structure with a zig-zag Cd–Cu–Cu–Cd skeleton. The molecular structure of 2 consists of a pentanuclear centrosymmetric core in which four metal atoms are linked together by bridging oxygen atoms of L groups and bromide anions to form a parallelogram Cu–Cd–Cu–Cd centred on the fifth Cu. The hexanuclear complex 3 is made up of two symmetry-related units with triangular CuCd2 cores linked by amino alkoxo bridges, involving cadmium centres of both units.
Co-reporter:Birte Ahrens, Simon A. Cotton, Neil Feeder, Oliver E. Noy, Paul R. Raithby and Simon J. Teat
Dalton Transactions 2002 (Issue 9) pp:2027-2030
Publication Date(Web):08 Apr 2002
DOI:10.1039/B200480C
Structural studies show that the solvolysis of a nitrate group in the heavy lanthanide complexes [Ln(terpy)(NO3)3] {Ln = Yb, Lu; terpy = 2,2′:6′,2″-terpyridine} is stereoselective. In the complexes [Ln(terpy)(NO3)3(EtOH)], the ‘equatorial nitrate group’, which lies in the same plane as the terpy ligand, coordinates in a monodentate fashion and the vacant coordination site is filled by an ethanol molecule. Similarly, in the unusual complexes [Yb(terpy)(NO3)2(H2O)2][NO3] and [Lu(terpy)(NO3)2(H2O)(EtOH)][NO3], two water molecules or a water and an ethanol molecule are bound to the metal in preference to the nitrate and lie in the same plane as the terpy ligand.
Co-reporter:Radchada Buntem, John F. Gallagher, Jack Lewis, Paul R. Raithby, Moira-Ann Rennie and Gregory P. Shields
Dalton Transactions 2000 (Issue 23) pp:4297-4303
Publication Date(Web):15 Nov 2000
DOI:10.1039/B006746F
Deprotonation of [Os3H2(CO)10(PPh3)], with DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), and subsequent treatment with the ionic coupling reagent [Ru(η5-C5H5)(MeCN)3][PF6] afforded the tetrahedral cluster [Os3H(CO)10(PPh3){Ru(η5-C5H5)}]. Reduction of the trinuclear osmium cluster [Os3(CO)11(PPh3)] with K/Ph2CO and subsequent coupling with [Ru(η5-C5H5)(MeCN)3][PF6] yielded the pentanuclear clusters [Os3(CO)11(PPh3){Ru(η5-C5H5)}2], [Os3H2(CO)11(PPh3){Ru(η5-C5H5)}2] and the butterfly cluster [Os3H(CO)11(PPh3){Ru(η5-C5H5)}]. In an analogous reaction using [Os3(CO)11{P(OMe)3}] only one complex [Os3(CO)11{P(OMe)3}{Ru(η5-C5H5)}2] was isolated. This undergoes an orthometallation when heated under reflux in toluene to yield the novel spiked tetrahedral cluster [Os3Ru2H(CO)11{P(OMe)3}(η5-C5H5)(μ3-η5-C5H4)]. All the new complexes have been characterised spectroscopically and the molecular and crystal structures of three have been determined by single-crystal X-ray diffraction. The structure of [Os3Ru2H(CO)11{P(OMe)3}(η5-C5H5)(μ3-η5-C5H4)] shows an uncommon μ3-η5-bonding mode for the deprotonated cyclopentadiene ring.
Co-reporter:Christopher J. Adams, Lionel P. Clarke, Ana M. Martín-Castro, Paul R. Raithby and Gregory P. Shields
Dalton Transactions 2000 (Issue 22) pp:4015-4017
Publication Date(Web):23 Oct 2000
DOI:10.1039/B006696F
Reaction of the labile triosmium cluster [Os3(CO)10(NCMe)2] with the thienyl substituted diyne [(C4H3S)C2C2(C4H3S)] affords [Os3(CO)10{μ3-η2-(C4H3S)C2C2(C4H3S)}] 1 and the novel co-linear cluster [Os3(CO)11{μ3-η4-(C4H3S)C2C2(C4H3S)}] 2 in reasonable yield; the X-ray structure of 2 shows that the diyne is coordinated to an ‘open’ triosmium unit as a 1,2,3-triene-1,4-diyl unit with essentially equal CC bond distances.
Co-reporter:Lionel P. Clarke, John E. Davies, Paul R. Raithby and Gregory P. Shields
Dalton Transactions 2000 (Issue 24) pp:4527-4533
Publication Date(Web):28 Nov 2000
DOI:10.1039/B006810L
The reaction of [Ru4H4(CO)12] with the 1,3-diynes RC2C2R (R = Me, SiMe3 or Ph) under reflux conditions, in heptane, yielded the tetraruthenium clusters [Ru4(CO)12{μ4-η1∶η2∶η2∶η1-(RCH2C3R)}] 1 and [Ru4(CO)12{μ4-η2∶η1∶η1∶η2-[RC(H)C]2}] 2 (R = Me a, SiMe3b or Ph c) in good yield. In 1 a 1,1-dihydrogenation occurs to generate an allene-1,3-diyl ligand, which coordinates to the butterfly Ru4 face in a novel η1∶η2∶η2∶η1 mode, whereas in 2 a 1,4-dihydrogenation occurs to yield a 1,3-diene-2,3-diyl ligand bound to the Ru4 butterfly via four C atoms in a η2∶η1∶η1∶η2 mode. In addition, the trinuclear cluster [Ru3(μ-H)(CO)9{μ3-η2∶η2∶η1-[RCH2C2CHPh)] 3c (R = Ph) is produced as a result of 1,1,4 trihydrogenation of the diyne and loss of one ruthenium vertex. The clusters have fully been characterised spectroscopically, and the crystal structures of 1a,1c 2a,2c and 3c determined.
Co-reporter:Peiyi Li, Birte Ahrens, Andrew D. Bond, John E. Davies, Olivia F. Koentjoro, Paul R. Raithby and Simon J. Teat
Dalton Transactions 2008(Issue 12) pp:NaN1646-1646
Publication Date(Web):2008/01/24
DOI:10.1039/B716664H
A series of novel digold complexes incorporating ethynyl pyridine derivatives as a spacer unit, [(R3P)Au(CC)X(CC)Au(PR3)] (R = Ph, X = 2,5-pyridine (1); R = Cy (cyclohexane), X = 2,5-pyridine (2); R = Ph, X = 2,6-pyridine (3); R = Ph, X = 2,5′-bipyridine (4); R = Ph, X = 2,6′-bipyridine (5)), has been synthesised. All the complexes have been characterised spectroscopically and the structures determined by single-crystal X-ray crystallography. The central (CC)(X)(CC) unit is essentially linear for complexes 1, 2 and 4 and kinked for complexes 3 and 5, but only in 1, with the shortest spacer group and the less bulky phosphine ligand, is there evidence of d10⋯d10Au⋯Au interactions (Au–Au 3.351(2) Å). The solution UV/visible absorption and emission spectra for all the complexes are similar to those of the free ligands suggesting that the spectra are dominated by π–π* ligand-centred transitions and this is confirmed by DFT calculations.
Co-reporter:Muhammad S. Khan, Mohammed K. Al-Suti, Hakkikulla H. Shah, Said Al-Humaimi, Fathiya R. Al-Battashi, Jens K. Bjernemose, Louise Male, Paul R. Raithby, Ning Zhang, Anna Köhler and John E. Warren
Dalton Transactions 2011 - vol. 40(Issue 39) pp:NaN10183-10183
Publication Date(Web):2011/09/06
DOI:10.1039/C1DT11010A
A series of trimethylsilyl-protected di-alkynes incorporating 3,4-ethylenedioxythiophene (EDOT) linker groups Me3Si–CC–R–CC–SiMe3 (R = ethylenedioxythiophene-3,4-diyl 1a, 2,2′-bis-3,4-ethylenedioxythiophene-5,5′-diyl 2a, 2,2′,5′,2′′-ter-3,4-ethylenedioxythiophene-5,5′′-diyl 3a) and the corresponding terminal di-alkynes, H–CC–R–CC–H 1b–2b has been synthesized and characterized and the single crystal X-ray structure of 1a has been determined. CuI-catalyzed dehydrohalogenation reaction between trans-[(Ph)(Et3P)2PtCl] and the terminal di-alkynes 1b–2b in iPr2NH/CH2Cl2 (2:1 mole ratio) gives the Pt(II) di-ynes trans-[(Et3P)2(Ph)Pt–CC–R–CC–Pt(Ph)(Et3P)2] 1M–2M while the dehydrohalogenation polycondensation reaction between trans-[(nBu3P)2PtCl2] and 1b–2b (1:1 mole ratio) under similar reaction conditions affords the Pt(II) poly-ynes trans-[Pt(PnBu3)2–CC–R–CC-]n1P–2P. The di-ynes and poly-ynes have been characterized spectroscopically and, for 1M and 2M, by single-crystal X-ray which confirms the “rigid rod” di-yne backbone. The materials possess excellent thermal stability, are soluble in common organic solvents and readily cast into thin films. Optical absorption spectroscopic measurements reveal that the EDOT spacers create stronger donor-acceptor interactions between the platinum(II) centres and conjugated ligands along the rigid backbone of the organometallic polymers compared to the related non-fused and fused oligothiophene spacers.
Co-reporter:Simon K. Brayshaw, Timothy L. Easun, Michael W. George, Alexandra M. E. Griffin, Andrew L. Johnson, Paul R. Raithby, Teresa L. Savarese, Stefanie Schiffers, John E. Warren, Mark R. Warren and Simon J. Teat
Dalton Transactions 2012 - vol. 41(Issue 1) pp:NaN97-97
Publication Date(Web):2011/10/20
DOI:10.1039/C1DT11379H
Single crystal photocrystallographic experiments and solid state Raman spectroscopy have been used to determine the low temperature, metastable structures of the nickel(II) nitrito complexes [Ni(aep)2(η1-ONO)2] 1#O (aep = 1-(2-aminoethyl)piperidine), [Ni(aem)2(η1-ONO)2] 2#O (aem = 1-(2-aminoethyl)morpholine), and [Ni(aepy)2(η1-ONO)2] 3#O (aepy = 1-(2-aminoethyl)pyrrolidine and where the #O denotes the oxygen-bound nitrito metastable molecule). These linkage isomers of the equivalent nitro complexes [Ni(aep)2(η1-NO2)2] 1, [Ni(aem)2(η1-NO2)2] 2 and [Ni(aepy)2(η1-NO2)2] 3 are formed by LED irradiation at temperatures below 120 K. The behavior of the three complexes upon irradiation is generally similar, but some subtle differences have been observed. From the crystallographic studies all three complexes 1–3 exhibit the endo-nitrito linkage isomer upon irradiation, however, for 3# (a crystal structure that contains components of both 3 and 3#O) an exo-nitrito isomer is also observed. Under conditions of 90–100 K, with blue light, the conversion percentages to the nitrito isomers, 1#O, 2#O and 3#O were 16%, 22% and 30%, respectively. At temperatures below 110 K all three nitrito isomers were stable for over four hours but while 2#O and 3#O could be detected at temperatures down to 30 K, at temperatures below 60 K the metastable structure 1#O appeared to be quenched and only the nitro isomer 1 was identified in the crystal. The solid state Raman spectra for 1#, 2# and 3# confirmed the photocrystallographic results with the nitrito isomers being identified from the O–N–O deformation vibrations.
Co-reporter:Hakikulla H. Shah, Rayya A. Al-Balushi, Mohammed K. Al-Suti, Muhammad S. Khan, Frank Marken, Anna L. Sudlow, Gabriele Kociok-Köhn, Christopher H. Woodall, Paul R. Raithby and Kieran C. Molloy
Dalton Transactions 2014 - vol. 43(Issue 25) pp:NaN9507-9507
Publication Date(Web):2014/04/30
DOI:10.1039/C3DT52914B
Three new neutral di-ferrocenyl-ethynylpyridinyl copper complexes, [L2(CuCl)2(PPh3)2] (2), [L2(CuBr)2(PPh3)2] (3), and [L2(CuI)2(PPh3)2] (4) were synthesized from the ferrocenyl-ethynylpyridine ligand (L) (1), the appropriate copper halide CuX (with X = Cl−, Br−, I−) and triphenylphosphine. These neutral complexes were fully characterized by spectroscopic methods and by single crystal X-ray crystallography. Cyclic voltammetry in dichloroethane revealed chemically reversible ferrocenyl oxidation signals followed by characteristic “stripping reduction peaks” showing evidence for oxidation-product electro-crystallization. Scanning electron microscopy confirmed spontaneous formation of crystalline oxidation products with three distinct morphologies for X = Cl−, Br−, I−. Energy dispersive X-ray elemental analysis data show Fe:P ratios of 1:2.0, 1:2.1 and 1:2.1 for electro-crystallization products of complexes 2, 3, and 4, respectively, indicating the presence of two [PF6]− anions in the vicinity of the dioxidized complexes, and suggesting product formulae [2]2+[PF6]−2, [3]2+[PF6]−2 and [4]2+[PF6]−2.
Co-reporter:Somia E. Bajwa, Thomas E. Storr, Lauren E. Hatcher, Thomas J. Williams, Christoph G. Baumann, Adrian C. Whitwood, David R. Allan, Simon J. Teat, Paul R. Raithby and Ian J. S. Fairlamb
Chemical Science (2010-Present) 2012 - vol. 3(Issue 5) pp:NaN1661-1661
Publication Date(Web):2012/02/23
DOI:10.1039/C2SC01050J
Pd3(OAc)5NO2, an impurity in “Pd(OAc)2” {formally Pd3(OAc)6}, emerges as a serious issue in the synthesis of pure PdII complexes derived from Pd(OAc)2, for example in our C–H activation precatalyst, Pd(OAc)2(pip)2 (pip = piperidine). A previous proposal that nitrite anion can be formed by oxidation of CH3CN by metallic Pd and air, leading to cyclo(ortho)palladated complexes containing nitrite anion, e.g. Pd(NO2)(C^N)L (C^N = papaverine; L = CH3CN or DMSO) can be explained by Pd3(OAc)5NO2 acting as the nitrite source. Finally, photocrystallographic metastable linkage isomerisation and complete conversion to an oxygen-bound nitrito complex Pd(η1-ONO)(C^N)PPh3 has been observed.
Co-reporter:Mark R. Warren, Simon K. Brayshaw, Lauren E. Hatcher, Andrew L. Johnson, Stefanie Schiffers, Anna J. Warren, Simon J. Teat, John E. Warren, Christopher H. Woodall and Paul R. Raithby
Dalton Transactions 2012 - vol. 41(Issue 42) pp:NaN13179-13179
Publication Date(Web):2012/05/10
DOI:10.1039/C2DT30314K
Low temperature, single crystal photocrystallographic studies have been carried out on four square planar Group 10 complexes [Ni(PEt3)2(NO2)2] 1, [Pd(PPh3)2(NO2)2] 2, [Pd(AsPh3)2(NO2)2] 3 and [Pt(PPh3)2(NO2)2] 4, in which the two nitro groups adopt the trans configuration. Irradiation with UV light, at 100 K, of single crystals of complexes 1–3 photoisomerise from the η1-NO2 nitro form to the η1-ONO nitrito form occurred. Complex 1 underwent 25% conversion to the nitrito form before crystal decomposition occurred. 2 and 3 underwent 46% and 39% conversion, respectively, to the nitrito form when a photostationary state was reached. While under the same experimental conditions 4 showed no isomerisation. The photocrystallographic results can be correlated with the results of DFT calculations and with the observed trends in the solution UV/visible absorption spectroscopy obtained for these complexes. The results suggest that while steric factors in the isomerization processes are important there may also be a kinetic effect relating to the lability of the metal involved.

![1H-1,2,3-Triazole, 4-phenyl-1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]-](/data/chemimg/3722700/1239481-06-1.png)
![1H-1,2,3-Triazole, 4-phenyl-1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]-](/data/chemimg/3722700/1239481-06-1_b.png)






![1,2-Cyclohexanediamine, N,N'-bis[(2-methoxyphenyl)methyl]-, (1R,2R)-](http://img.cochemist.com/ccimg/854600/854513-44-3.png)
![1,2-Cyclohexanediamine, N,N'-bis[(2-methoxyphenyl)methyl]-, (1R,2R)-](http://img.cochemist.com/ccimg/854600/854513-44-3_b.png)





![2-[2-(trimethylsilyl)ethynyl]-Furan](http://img.cochemist.com/ccimg/40300/40231-01-4.png)
![2-[2-(trimethylsilyl)ethynyl]-Furan](http://img.cochemist.com/ccimg/40300/40231-01-4_b.png)


![Benzene, [2-nitro-1-(nitromethyl)ethyl]-](http://img.cochemist.com/ccimg/117600/117538-84-8.png)
![Benzene, [2-nitro-1-(nitromethyl)ethyl]-](http://img.cochemist.com/ccimg/117600/117538-84-8_b.png)
![1,3,2-Dioxaborolane, 2-[4-(azidomethyl)phenyl]-4,4,5,5-tetramethyl-](/data/chemimg/422500/1239481-05-0.png)
![1,3,2-Dioxaborolane, 2-[4-(azidomethyl)phenyl]-4,4,5,5-tetramethyl-](/data/chemimg/422500/1239481-05-0_b.png)
![Ruthenium,tricarbonyl[(1,2,3,4-h)-1,3-cyclohexadiene]-](http://img.cochemist.com/ccimg/12200/12108-25-7.png)
![Ruthenium,tricarbonyl[(1,2,3,4-h)-1,3-cyclohexadiene]-](http://img.cochemist.com/ccimg/12200/12108-25-7_b.png)
![2-[[(2-HYDROXY-3,5-DIMETHYLPHENYL)METHYL-(2-HYDROXYETHYL)AMINO]METHYL]-4,6-DIMETHYLPHENOL](/data/chemimg/392300/118328-21-5.png)
![2-[[(2-HYDROXY-3,5-DIMETHYLPHENYL)METHYL-(2-HYDROXYETHYL)AMINO]METHYL]-4,6-DIMETHYLPHENOL](/data/chemimg/392300/118328-21-5_b.png)
![Phenol, 2,2',2''-[nitrilotris(methylene)]tris[4,6-dimethyl-](http://img.cochemist.com/ccimg/58000/57914-22-4.png)
![Phenol, 2,2',2''-[nitrilotris(methylene)]tris[4,6-dimethyl-](http://img.cochemist.com/ccimg/58000/57914-22-4_b.png)
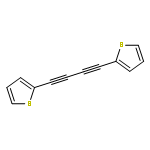
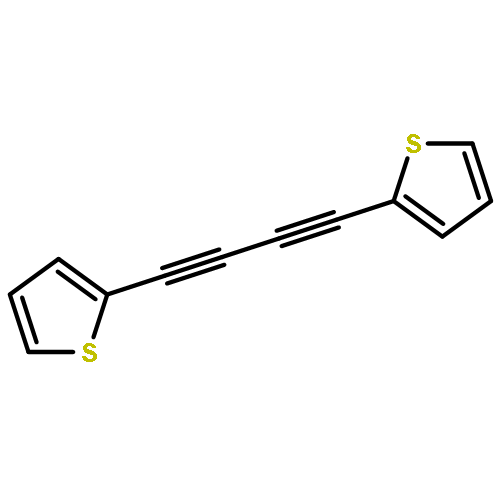








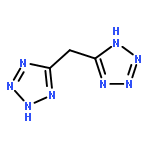




![Phenol,2,2',2'',2'''-[1,2-ethanediylbis[nitrilobis(methylene)]]tetrakis[4,6-dimethyl-(9CI)](http://img.cochemist.com/ccimg/142700/142647-87-8.png)
![Phenol,2,2',2'',2'''-[1,2-ethanediylbis[nitrilobis(methylene)]]tetrakis[4,6-dimethyl-(9CI)](http://img.cochemist.com/ccimg/142700/142647-87-8_b.png)








![2,2'-Bithiophene, 5,5'-bis[2-(trimethylsilyl)ethynyl]-](/data/chemimg/131900/139896-64-3.png)
![2,2'-Bithiophene, 5,5'-bis[2-(trimethylsilyl)ethynyl]-](/data/chemimg/131900/139896-64-3_b.png)