Co-reporter:Sarah E. St. John, Matthew D. Therkelsen, Prasanth R. Nyalapatla, Heather L. Osswald, Arun K. Ghosh, Andrew D. Mesecar
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5072-5077
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.10.023
Co-reporter:Yahira M. Báez-Santos ; Scott J. Barraza ; Michael W. Wilson ; Michael P. Agius ; Anna M. Mielech ; Nicole M. Davis ; Susan C. Baker ; Scott D. Larsen
Journal of Medicinal Chemistry 2014 Volume 57(Issue 6) pp:2393-2412
Publication Date(Web):February 25, 2014
DOI:10.1021/jm401712t
Structure-guided design was used to generate a series of noncovalent inhibitors with nanomolar potency against the papain-like protease (PLpro) from the SARS coronavirus (CoV). A number of inhibitors exhibit antiviral activity against SARS-CoV infected Vero E6 cells and broadened specificity toward the homologous PLP2 enzyme from the human coronavirus NL63. Selectivity and cytotoxicity studies established a more than 100-fold preference for the coronaviral enzyme over homologous human deubiquitinating enzymes (DUBs), and no significant cytotoxicity in Vero E6 and HEK293 cell lines is observed. X-ray structural analyses of inhibitor-bound crystal structures revealed subtle differences between binding modes of the initial benzodioxolane lead (15g) and the most potent analogues 3k and 3j, featuring a monofluoro substitution at para and meta positions of the benzyl ring, respectively. Finally, the less lipophilic bis(amide) 3e and methoxypyridine 5c exhibit significantly improved metabolic stability and are viable candidates for advancing to in vivo studies.
Co-reporter:Katrina Moll;Qing Zhou
Acta Crystallographica Section F 2014 Volume 70( Issue 3) pp:283-287
Publication Date(Web):
DOI:10.1107/S2053230X14002519
A sparse-matrix screen for new crystallization conditions for the USP7 catalytic domain (USP7CD) led to the identification of a condition in which crystals grow reproducibly in 24–48 h. Variation of the halide metal, growth temperature and seed-stock concentration resulted in a shift in space group from P21 with two molecules in the asymmetric unit to C2 with one molecule in the asymmetric unit. Representative structures from each space group were determined to 2.2 Å resolution and these structures support previous findings that the catalytic triad and switching loop are likely to be in unproductive conformations in the absence of ubiquitin (Ub). Importantly, the new structures reveal previously unobserved electron density for blocking loop 1 (BL1) residues 410–419. The new structures indicate a distinct rearrangement of the USP7 BL1 compared with its position in the presence of bound Ub.
Co-reporter:Jon Jacobs ; Valerie Grum-Tokars ; Ya Zhou ; Mark Turlington ; S. Adrian Saldanha ; Peter Chase ; Aimee Eggler ▽; Eric S. Dawson ; Yahira M. Baez-Santos ▽; Sakshi Tomar ▽; Anna M. Mielech ; Susan C. Baker ; Craig W. Lindsley ; Peter Hodder ; Andrew Mesecar ▽;Shaun R. Stauffer
Journal of Medicinal Chemistry 2013 Volume 56(Issue 2) pp:534-546
Publication Date(Web):December 11, 2012
DOI:10.1021/jm301580n
A high-throughput screen of the NIH molecular libraries sample collection and subsequent optimization of a lead dipeptide-like series of severe acute respiratory syndrome (SARS) main protease (3CLpro) inhibitors led to the identification of probe compound ML188 (16-(R), (R)-N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)furan-2-carboxamide, Pubchem CID: 46897844). Unlike the majority of reported coronavirus 3CLpro inhibitors that act via covalent modification of the enzyme, 16-(R) is a noncovalent SARS-CoV 3CLpro inhibitor with moderate MW and good enzyme and antiviral inhibitory activity. A multicomponent Ugi reaction was utilized to rapidly explore structure–activity relationships within S1′, S1, and S2 enzyme binding pockets. The X-ray structure of SARS-CoV 3CLpro bound with 16-(R) was instrumental in guiding subsequent rounds of chemistry optimization. 16-(R) provides an excellent starting point for the further design and refinement of 3CLpro inhibitors that act by a noncovalent mechanism of action.
Co-reporter:Scott D. Pegan, Kamolchanok Rukseree, Glenn C. Capodagli, Erica A. Baker, Olga Krasnykh, Scott G. Franzblau, and Andrew D. Mesecar
Biochemistry 2013 Volume 52(Issue 5) pp:
Publication Date(Web):January 8, 2013
DOI:10.1021/bi300928u
Class II fructose 1,6-bisphosphate aldolases (FBAs, EC 4.1.2.13) comprise one of two families of aldolases. Instead of forming a Schiff base intermediate using an ε-amino group of a lysine side chain, class II FBAs utilize Zn(II) to stabilize a proposed hydroxyenolate intermediate (HEI) in the reversible cleavage of fructose 1,6-bisphosphate, forming glyceraldehyde 3-phosphate and dihydroxyacetone phosphate (DHAP). As class II FBAs have been shown to be essential in pathogenic bacteria, focus has been placed on these enzymes as potential antibacterial targets. Although structural studies of class II FBAs from Mycobacterium tuberculosis (MtFBA), other bacteria, and protozoa have been reported, the structure of the active site loop responsible for catalyzing the protonation–deprotonation steps of the reaction for class II FBAs has not yet been observed. We therefore utilized the potent class II FBA inhibitor phosphoglycolohydroxamate (PGH) as a mimic of the HEI- and DHAP-bound form of the enzyme and determined the X-ray structure of the MtFBA–PGH complex to 1.58 Å. Remarkably, we are able to observe well-defined electron density for the previously elusive active site loop of MtFBA trapped in a catalytically competent orientation. Utilization of this structural information and site-directed mutagenesis and kinetic studies conducted on a series of residues within the active site loop revealed that E169 facilitates a water-mediated deprotonation–protonation step of the MtFBA reaction mechanism. Also, solvent isotope effects on MtFBA and catalytically relevant mutants were used to probe the effect of loop flexibility on catalytic efficiency. Additionally, we also reveal the structure of MtFBA in its holoenzyme form.
Co-reporter:Yahira M. Báez-Santos, Sarah E. St. John, Andrew D. Mesecar
Antiviral Research (March 2015) Volume 115() pp:21-38
Publication Date(Web):March 2015
DOI:10.1016/j.antiviral.2014.12.015
Co-reporter:Thomas J. Wubben, Andrew D. Mesecar
Journal of Molecular Biology (26 November 2010) Volume 404(Issue 2) pp:202-219
Publication Date(Web):26 November 2010
DOI:10.1016/j.jmb.2010.09.002
Phosphopantetheine adenylyltransferase (PPAT) catalyzes the penultimate step in the coenzyme A (CoA) biosynthetic pathway, reversibly transferring an adenylyl group from ATP to 4′-phosphopantetheine (PhP) to form dephosphocoenzyme A. This reaction sits at the branch point between the de novo pathway and the salvage pathway, and has been shown to be a rate-limiting step in the biosynthesis of CoA. Importantly, bacterial and mammalian PPATs share little sequence homology, making the enzyme a potential target for antibiotic development. A series of steady-state kinetic, product inhibition, and direct binding studies with Mycobacterium tuberculosis PPAT (MtPPAT) was conducted and suggests that the enzyme utilizes a nonrapid-equilibrium random bi–bi mechanism. The kinetic response of MtPPAT to the binding of ATP was observed to be sigmoidal under fixed PhP concentrations, but substrate inhibition was observed at high PhP concentrations under subsaturating ATP concentrations, suggesting a preferred pathway to ternary complex formation. Negative cooperativity in the kinetic response of MtPPAT to PhP binding was observed under certain conditions and confirmed thermodynamically by isothermal titration calorimetry, suggesting the formation of an asymmetric quaternary structure during sequential ligation of substrates. Asymmetry in binding was also observed in isothermal titration calorimetry experiments with dephosphocoenzyme A and CoA. X-ray structures of MtPPAT in complex with PhP and the nonhydrolyzable ATP analogue adenosine-5′-[(α,β)-methyleno]triphosphate were solved to 1.57 Å and 2.68 Å, respectively. These crystal structures reveal small conformational changes in enzyme structure upon ligand binding, which may play a role in the nonrapid-equilibrium mechanism. We suggest that the proposed kinetic mechanism and asymmetric character in MtPPAT ligand binding may provide a means of reaction and pathway regulation in addition to that of the previously determined CoA feedback.Graphical AbstractDownload high-res image (106KB)Download full-size imageResearch Highlights► MtPPAT utilizes a nonrapid-equilibrium random bi–bi kinetic mechanism. ► Conformational changes may play a role in the rate-determining step. ► Calorimetry studies suggest that substrate binding is asymmetric in nature. ► Kinetic, thermodynamic, and structural studies suggest cooperative substrate binding. ► Cooperativity may help regulate MtPPAT reaction in cells.
Co-reporter:Nicole M. Hjortland, Andrew D. Mesecar
Archives of Biochemistry and Biophysics (15 December 2016) Volume 612() pp:35-45
Publication Date(Web):15 December 2016
DOI:10.1016/j.abb.2016.10.008
•USP17 is highly upregulated in cancer and is a viable anti-cancer drug target.•USP17 was purified from E. coli inclusion bodies and is highly active.•USP17 appears to function as a monomer in solution.•USP17 can recognize and cleave Lys11, Lys33, Lys48 and Lys63 ubiquitin linkages.•Purified USP17 can be used for high-throughput screening to identify inhibitors.USP17 is a deubiquitinating enzyme that is upregulated in numerous cancers and therefore a drug target. We developed a robust expression, purification, and assay system for USP17 enabling its enzymatic and structural characterization. USP17 was expressed in E. coli as inclusion bodies and then solubilized, refolded, and purified using affinity and size-exclusion chromatography. Milligram quantities of pure USP17 can be produced that is catalytically more efficient (kcat/Km = 1500 (x103) M−1sec−1) than other human USPs studied to date. Analytical size-exclusion chromatography, analytical ultracentrifugation, and dynamic light scattering studies suggest that the quaternary structure of USP17 is a monomer. Steady-state kinetic studies show that USP17 efficiently hydrolyzes both ubiquitin-AMC (kcat = 1.5 sec−1 and Km = 1.0 μM) and ubiquitin-rhodamine110 (kcat = 1.8 sec−1 and Km = 2.0 μM) substrates. Ubiquitin chain cleavage assays reveal that USP17 efficiently cleaves di-ubiquitin chains with Lys11, Lys33, Lys48 and Lys63 linkages and tetra-ubiquitin chains with Lys11, Lys48 and Lys63 linkages but is inefficient in cleaving di-ubiquitin chains with Lys6, Lys27, or Lys29 linkages or linear ubiquitin chains. The substrate specificity of USP17 is most similar to that of USP1, where both USPs display higher specificity than other characterized members of the USP family.Download high-res image (179KB)Download full-size image
.jpg)
![6,8-diamino-7-chloro-1-methyl-2-oxo-1,2-dihydropyrrolo[4,3,2-de]quinoline-4-carboxamide](http://img.cochemist.com/ccimg/1096400/1096365-01-3.png)
![6,8-diamino-7-chloro-1-methyl-2-oxo-1,2-dihydropyrrolo[4,3,2-de]quinoline-4-carboxamide](http://img.cochemist.com/ccimg/1096400/1096365-01-3_b.png)




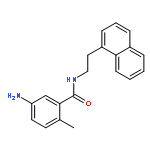
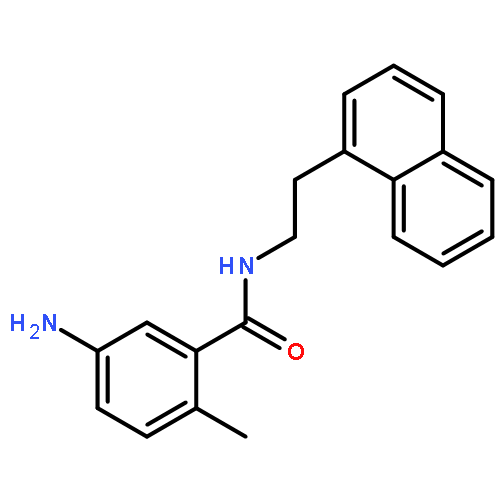
![1-[(e)-2-(4-chlorophenyl)vinyl]-3,5-dimethoxybenzene](http://img.cochemist.com/ccimg/1032600/1032508-03-4.png)
![1-[(e)-2-(4-chlorophenyl)vinyl]-3,5-dimethoxybenzene](http://img.cochemist.com/ccimg/1032600/1032508-03-4_b.png)

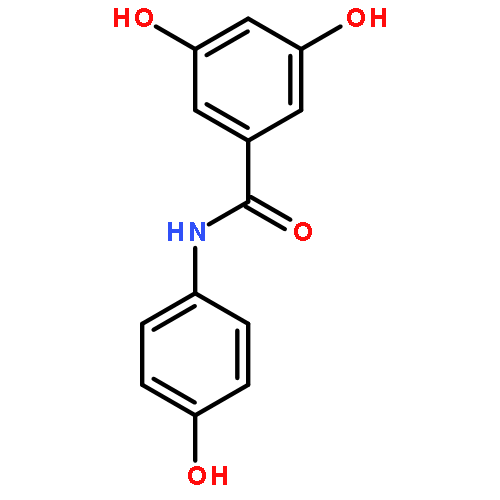
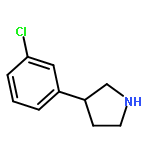
![Phenol, 4-[(1E)-2-(3,4-dimethoxyphenyl)ethenyl]-, acetate](http://img.cochemist.com/ccimg/880400/880354-47-2.png)
![Phenol, 4-[(1E)-2-(3,4-dimethoxyphenyl)ethenyl]-, acetate](http://img.cochemist.com/ccimg/880400/880354-47-2_b.png)
![1,3-Benzenediol, 5-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/823900/823804-68-8.png)
![1,3-Benzenediol, 5-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/823900/823804-68-8_b.png)
![Acetamide, N-[[3-(aminomethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/741300/741248-96-4.png)
![Acetamide, N-[[3-(aminomethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/741300/741248-96-4_b.png)

![Benzene, 1-[(1E)-2-(4-fluorophenyl)ethenyl]-3,5-dimethoxy-](http://img.cochemist.com/ccimg/688400/688348-27-8.png)
![Benzene, 1-[(1E)-2-(4-fluorophenyl)ethenyl]-3,5-dimethoxy-](http://img.cochemist.com/ccimg/688400/688348-27-8_b.png)
![BENZENAMINE, 4-[(1E)-2-(3-METHOXYPHENYL)ETHENYL]-](http://img.cochemist.com/ccimg/676500/676473-10-2.png)
![BENZENAMINE, 4-[(1E)-2-(3-METHOXYPHENYL)ETHENYL]-](http://img.cochemist.com/ccimg/676500/676473-10-2_b.png)


![Benzenamine, 4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/586500/586410-15-3.png)
![Benzenamine, 4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/586500/586410-15-3_b.png)


![Silane,[(5-ethenyl-1,3-phenylene)bis(oxy)]bis[(1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/193700/193695-64-6.png)
![Silane,[(5-ethenyl-1,3-phenylene)bis(oxy)]bis[(1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/193700/193695-64-6_b.png)
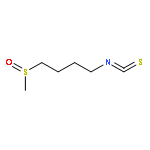
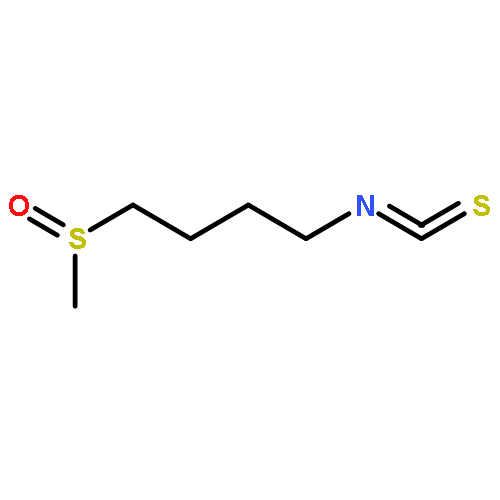
![Carbamodithioic acid, [4-(methylsulfinyl)butyl]-, methyl ester](http://img.cochemist.com/ccimg/187700/187612-30-2.png)
![Carbamodithioic acid, [4-(methylsulfinyl)butyl]-, methyl ester](http://img.cochemist.com/ccimg/187700/187612-30-2_b.png)
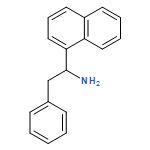

![Benzeneacetonitrile, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/167300/167263-57-2.png)
![Benzeneacetonitrile, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/167300/167263-57-2_b.png)
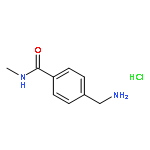
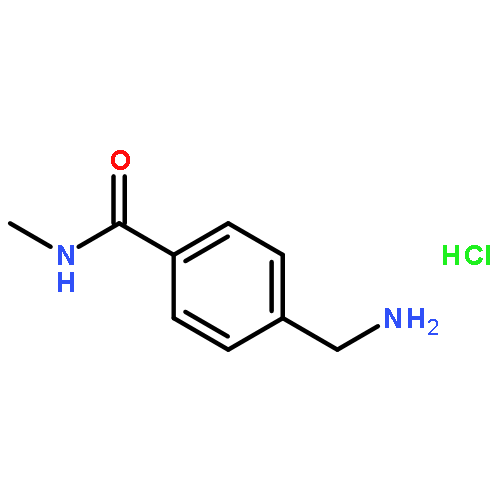
![Benzenamine, 4-[(1E)-2-(3,4-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/161900/161810-35-1.png)
![Benzenamine, 4-[(1E)-2-(3,4-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/161900/161810-35-1_b.png)


![Benzoic acid, 3,5-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/158200/158139-68-5.png)
![Benzoic acid, 3,5-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/158200/158139-68-5_b.png)
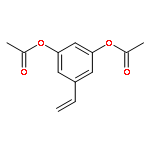



![Benzenamine, 4-[(1E)-2-(3,4,5-trimethoxyphenyl)ethenyl]-](/data/chemimg/1680900/134029-73-5.png)
![Benzenamine, 4-[(1E)-2-(3,4,5-trimethoxyphenyl)ethenyl]-](/data/chemimg/1680900/134029-73-5_b.png)
![Benzene, 1,2,3-trimethoxy-5-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1681200/134029-66-6.png)
![Benzene, 1,2,3-trimethoxy-5-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1681200/134029-66-6_b.png)


![Phenol,4-[(1E)-2-(3,4-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/128300/128294-47-3.png)
![Phenol,4-[(1E)-2-(3,4-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/128300/128294-47-3_b.png)
![Benzenamine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/111400/111359-74-1.png)
![Benzenamine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/111400/111359-74-1_b.png)
![Benzenamine, 4-[2-(3,5-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/111000/110997-91-6.png)
![Benzenamine, 4-[2-(3,5-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/111000/110997-91-6_b.png)
![Benzenamine, 4-[2-(3,4,5-trimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/111000/110997-90-5.png)
![Benzenamine, 4-[2-(3,4,5-trimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/111000/110997-90-5_b.png)
![Benzenamine, 4-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/111000/110997-83-6.png)
![Benzenamine, 4-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/111000/110997-83-6_b.png)




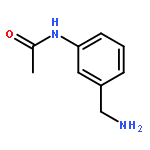
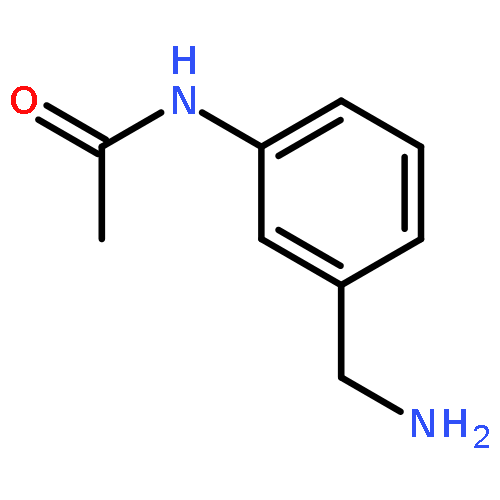
![Benzenamine, 3-[2-(4-aminophenyl)ethenyl]-, (E)- (9CI)](/data/chemimg/2073300/95522-37-5.png)
![Benzenamine, 3-[2-(4-aminophenyl)ethenyl]-, (E)- (9CI)](/data/chemimg/2073300/95522-37-5_b.png)

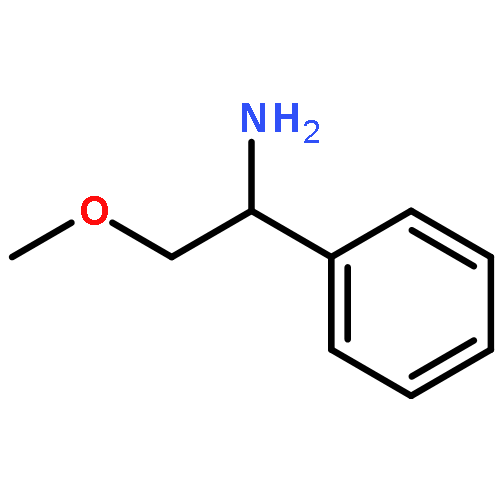
![BENZENAMINE, 3-[2-(4-AMINOPHENYL)ETHYL]-](http://img.cochemist.com/ccimg/85800/85765-58-8.png)
![BENZENAMINE, 3-[2-(4-AMINOPHENYL)ETHYL]-](http://img.cochemist.com/ccimg/85800/85765-58-8_b.png)
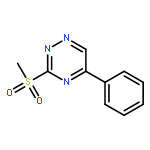
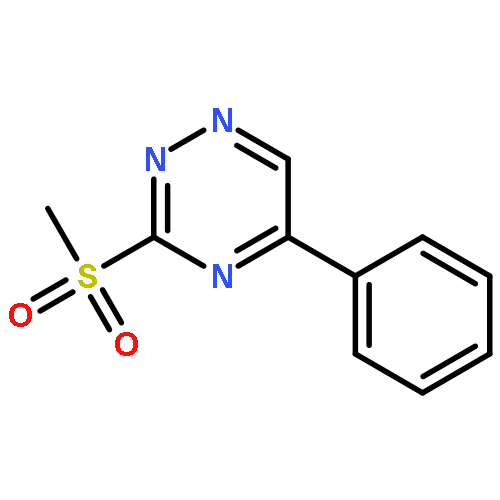
![Benzoic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/83500/83405-98-5.png)
![Benzoic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/83500/83405-98-5_b.png)
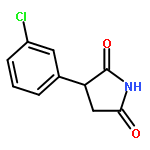
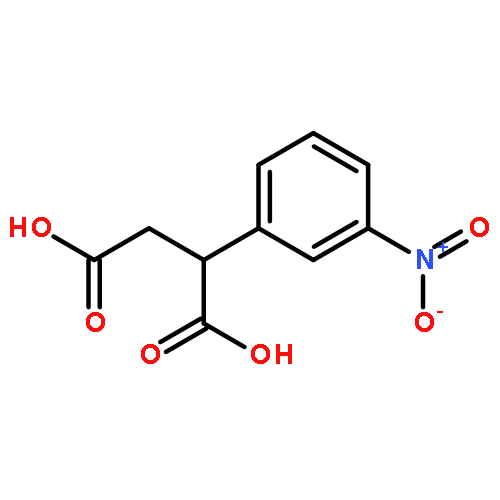
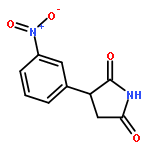
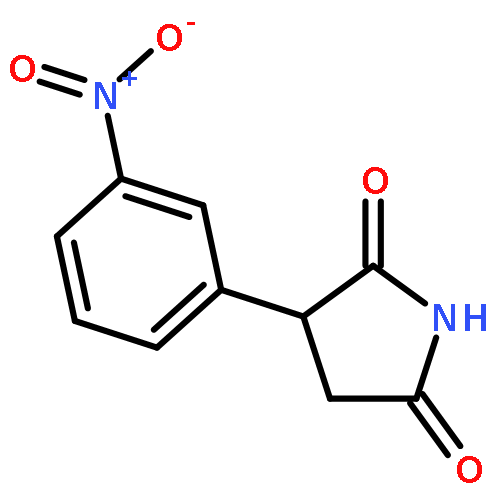


![Phenol,4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-, 1-acetate](http://img.cochemist.com/ccimg/63400/63366-83-6.png)
![Phenol,4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-, 1-acetate](http://img.cochemist.com/ccimg/63400/63366-83-6_b.png)
![Propanedioic acid, [[3-(phenylmethoxy)phenyl]methylene]-, diethyl ester](http://img.cochemist.com/ccimg/63000/62904-57-8.png)
![Propanedioic acid, [[3-(phenylmethoxy)phenyl]methylene]-, diethyl ester](http://img.cochemist.com/ccimg/63000/62904-57-8_b.png)
![2,5-Pyrrolidinedione, 3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/60100/60050-38-6.png)
![2,5-Pyrrolidinedione, 3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/60100/60050-38-6_b.png)
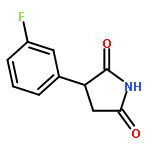
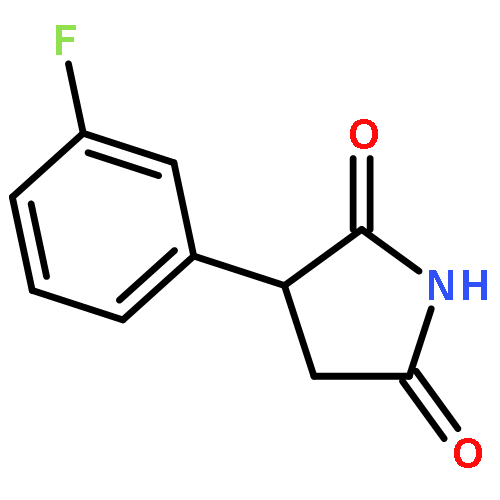


![1-[5-(3-NITROPHENYL)FURAN-2-YL]ETHANONE](http://img.cochemist.com/ccimg/56700/56656-35-0.png)
![1-[5-(3-NITROPHENYL)FURAN-2-YL]ETHANONE](http://img.cochemist.com/ccimg/56700/56656-35-0_b.png)
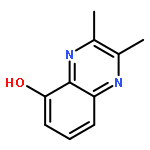
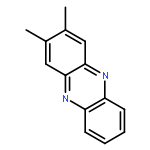
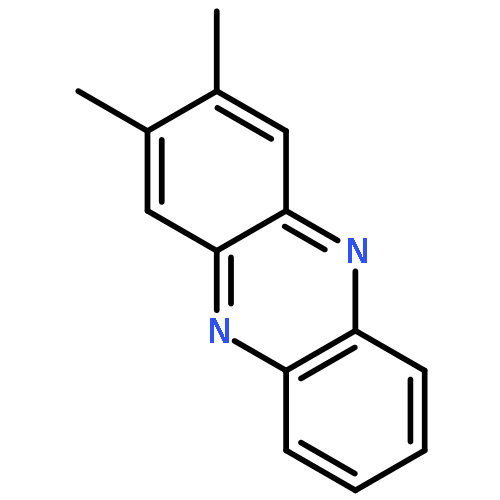
![Benzo[g]quinoxaline, 2,3-dimethyl-](http://img.cochemist.com/ccimg/52800/52736-74-0.png)
![Benzo[g]quinoxaline, 2,3-dimethyl-](http://img.cochemist.com/ccimg/52800/52736-74-0_b.png)
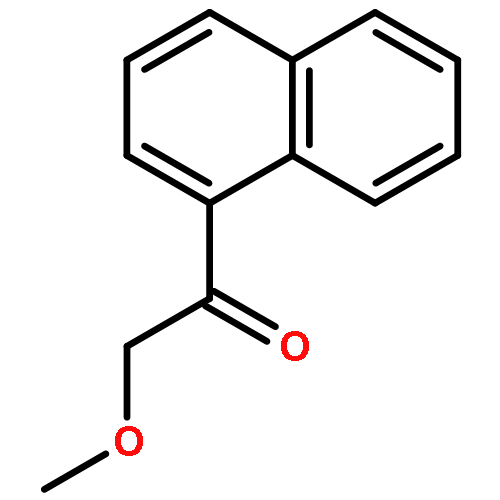
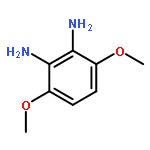
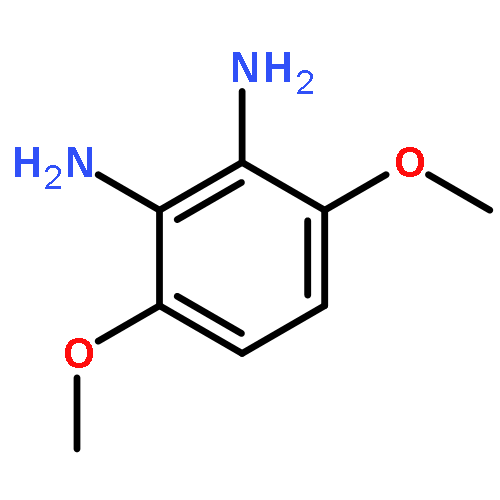
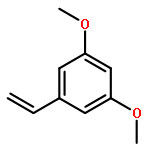











![Benzenamine,4-[(1E)-2-(2,5-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/23500/23435-31-6.png)
![Benzenamine,4-[(1E)-2-(2,5-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/23500/23435-31-6_b.png)
![3-[3-(Trifluoromethyl)phenyl]pyrrolidine HCl](http://img.cochemist.com/ccimg/21800/21767-35-1.png)
![3-[3-(Trifluoromethyl)phenyl]pyrrolidine HCl](http://img.cochemist.com/ccimg/21800/21767-35-1_b.png)










![Propanedioic acid, [(3-chlorophenyl)methylene]-, diethyl ester](http://img.cochemist.com/ccimg/6800/6768-21-4.png)
![Propanedioic acid, [(3-chlorophenyl)methylene]-, diethyl ester](http://img.cochemist.com/ccimg/6800/6768-21-4_b.png)
![1,3-dimethyl-5-{[2-methyl-1-(prop-2-en-1-yl)-1H-indol-3-yl]methylidene}-2-thioxodihydropyrimidine-4,6(1H,5H)-dione](http://img.cochemist.com/ccimg/6400/6331-45-9.png)
![1,3-dimethyl-5-{[2-methyl-1-(prop-2-en-1-yl)-1H-indol-3-yl]methylidene}-2-thioxodihydropyrimidine-4,6(1H,5H)-dione](http://img.cochemist.com/ccimg/6400/6331-45-9_b.png)



![Ethanone, 1-(3'-nitro[1,1'-biphenyl]-4-yl)-](http://img.cochemist.com/ccimg/5100/5002-11-9.png)
![Ethanone, 1-(3'-nitro[1,1'-biphenyl]-4-yl)-](http://img.cochemist.com/ccimg/5100/5002-11-9_b.png)
![Benzenamine,4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4400/4309-66-4.png)
![Benzenamine,4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4400/4309-66-4_b.png)
![3-AMINO-3-[5-(3-CHLORO-4-METHOXY-PHENYL)-2-FURYL]PROPANAMIDE](http://img.cochemist.com/ccimg/3500/3464-18-4.png)
![3-AMINO-3-[5-(3-CHLORO-4-METHOXY-PHENYL)-2-FURYL]PROPANAMIDE](http://img.cochemist.com/ccimg/3500/3464-18-4_b.png)



















![Benzo[b]phenazine](http://img.cochemist.com/ccimg/300/257-97-6.png)
![Benzo[b]phenazine](http://img.cochemist.com/ccimg/300/257-97-6_b.png)