Co-reporter:Emily J. Roncase, Clara Moon, Sandip Chatterjee, Gonzalo E. González-Páez, Charles S. Craik, Anthony J. O’Donoghue, and Dennis W. Wolan
ACS Chemical Biology June 16, 2017 Volume 12(Issue 6) pp:1556-1556
Publication Date(Web):April 17, 2017
DOI:10.1021/acschembio.7b00143
Cysteine proteases are among the most abundant hydrolytic enzymes produced by bacteria, and this diverse family of proteins have significant biological roles in bacterial viability and environmental interactions. Members of the clostripain-like (C11) family of cysteine proteases from commensal gut bacterial strains have recently been shown to mediate immune responses by inducing neutrophil phagocytosis and activating bacterial pathogenic toxins. Development of substrates, inhibitors, and probes that target C11 proteases from enteric bacteria will help to establish the role of these proteins at the interface of the host and microbiome in health and disease. We employed a mass spectrometry-based substrate profiling method to identify an optimal peptide substrate of PmC11, a C11 protease secreted by the commensal bacterium Parabacteroides merdae. Using this substrate sequence information, we synthesized a panel of fluorogenic substrates to calculate kcat and KM and to evaluate the importance of the P2 amino acid for substrate turnover. A potent and irreversible tetrapeptide inhibitor with a C-terminal acyloxymethyl ketone warhead, Ac-VLTK-AOMK, was then synthesized. We determined the crystal structure of PmC11 in complex with this inhibitor and uncovered key active-site interactions that govern PmC11 substrate recognition and specificity. This is the first C11 protease structure in complex with a substrate mimetic and is also the highest resolution crystal structure of a C11 protease to date at 1.12 Å resolution. Importantly, subjecting human epithelial cell lysates to PmC11 hydrolysis in combination with subtiligase-based N-terminal labeling and tandem mass spectrometry proteomics complemented the stringent substrate specificity observed in the in vitro substrate profiling experiment. The combination of chemical biological, biophysical, and biochemical techniques presented here to elucidate and characterize PmC11 substrate selectivity can be expanded to other proteases and the development of chemical tools to study these essential proteins in biologically relevant samples, such as the highly complex distal gut microbiome.
Co-reporter:Michael D. Mayers;Clara Moon;Gregory S. Stupp;Andrew I. Su
Journal of Proteome Research February 3, 2017 Volume 16(Issue 2) pp:1014-1026
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.jproteome.6b00938
Tandem mass spectrometry based shotgun proteomics of distal gut microbiomes is exceedingly difficult due to the inherent complexity and taxonomic diversity of the samples. We introduce two new methodologies to improve metaproteomic studies of microbiome samples. These methods include the stable isotope labeling in mammals to permit protein quantitation across two mouse cohorts as well as the application of activity-based probes to enrich and analyze both host and microbial proteins with specific functionalities. We used these technologies to study the microbiota from the adoptive T cell transfer mouse model of inflammatory bowel disease (IBD) and compare these samples to an isogenic control, thereby limiting genetic and environmental variables that influence microbiome composition. The data generated highlight quantitative alterations in both host and microbial proteins due to intestinal inflammation and corroborates the observed phylogenetic changes in bacteria that accompany IBD in humans and mouse models. The combination of isotope labeling with shotgun proteomics resulted in the total identification of 4434 protein clusters expressed in the microbial proteomic environment, 276 of which demonstrated differential abundance between control and IBD mice. Notably, application of a novel cysteine-reactive probe uncovered several microbial proteases and hydrolases overrepresented in the IBD mice. Implementation of these methods demonstrated that substantial insights into the identity and dysregulation of host and microbial proteins altered in IBD can be accomplished and can be used in the interrogation of other microbiome-related diseases.Keywords: activity-based probes; ComPIL; GO term enrichment; metaproteomics; microbiome; MudPIT; quantitative proteomics; SILAM;
Co-reporter:Zoë V.F. Wright, Nicholas C. Wu, Rameshwar U. Kadam, Ian A. Wilson, Dennis W. Wolan
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.06.074
Influenza is a highly contagious respiratory viral infection responsible for up to 50,000 deaths per annum in the US alone. The need for new therapeutics with novel modes of action is of paramount importance. We determined the X-ray structure of Arbidol with influenza hemagglutinin and found it was located in a distinct binding pocket. Herein, we report a structure-activity relationship study based on the co-complex combined with bio-layer interferometry to assess the binding of our compounds. Addition of a meta-hydroxy group to the thiophenol moiety of Arbidol to replace a structured water molecule in the binding pocket resulted in a dramatic increase in affinity against both H3 (1150-fold) and H1 (98-fold) hemagglutinin subtypes. Our analogues represent novel leads to yield more potent compounds against hemagglutinin that block viral entry.Download high-res image (70KB)Download full-size image
Co-reporter:Ana Y. Wang, Gonzalo E. González-Páez, and Dennis W. Wolan
Biochemistry 2015 Volume 54(Issue 28) pp:4365-4373
Publication Date(Web):July 1, 2015
DOI:10.1021/acs.biochem.5b00607
The secreted Streptococcus pyogenes cysteine protease SpeB is implicated in host immune system evasion and bacterial virulence. We present a small molecule inhibitor of SpeB 2477 identified from a high-throughput screen based on the hydrolysis of a fluorogenic peptide substrate Ac-AIK-AMC. 2477 inhibits other SpeB-related proteases but not human caspase-3, suggesting that the molecule targets proteases with the papain-like structural fold. A 1.59 Å X-ray crystal structure of 2477 bound to the SpeB active site reveals the mechanism of inhibition and the essential constituents of 2477 necessary for binding. An assessment against a panel of 2477 derivatives confirms our structural findings and shows that a carbamate and nitrile on 2477 are required for SpeB inhibition, as these moieties provide an extensive network of electrostatic and hydrogen-bonding interactions with SpeB active site residues. Surprisingly, despite 2477 having a reduced inhibitory potential against papain, the majority of 2477-related compounds inhibit papain to a much greater and broader extent than SpeB. These findings indicate that SpeB is more stringently selective than papain for this panel of small molecule inhibitors. On the basis of our structural and biochemical characterization, we propose modifications to 2477 for subsequent rounds of inhibitor design that will impart specificity to SpeB over other papain-like proteases, including alterations of the compound to exploit the differences in CA protease active site pocket sizes and electrostatics.
Co-reporter:Kanny K. Wan;Dr. Kotaro Iwasaki;Jeffrey C. Umotoy;Dr. Dennis W. Wolan;Dr. Ryan A. Shenvi
Angewandte Chemie International Edition 2015 Volume 54( Issue 8) pp:2410-2415
Publication Date(Web):
DOI:10.1002/anie.201411493
Abstract
A nitrosopurine ene reaction easily assembles the asmarine pharmacophore and transmits remote stereochemistry to the diazepine-purine hetereocycle. This reaction generates potent cytotoxins which exceed the potency of asmarine A (1.2 μM IC50) and supersede the metabolites as useful leads for biological discovery.
Co-reporter:Kanny K. Wan;Dr. Kotaro Iwasaki;Jeffrey C. Umotoy;Dr. Dennis W. Wolan;Dr. Ryan A. Shenvi
Angewandte Chemie 2015 Volume 127( Issue 8) pp:2440-2445
Publication Date(Web):
DOI:10.1002/ange.201411493
Abstract
A nitrosopurine ene reaction easily assembles the asmarine pharmacophore and transmits remote stereochemistry to the diazepine-purine hetereocycle. This reaction generates potent cytotoxins which exceed the potency of asmarine A (1.2 μM IC50) and supersede the metabolites as useful leads for biological discovery.
Co-reporter:Chris J. Vickers, Gonzalo E. González-Páez, Kevin M. Litwin, Jeffrey C. Umotoy, Evangelos A. Coutsias, and Dennis W. Wolan
ACS Chemical Biology 2014 Volume 9(Issue 10) pp:2194
Publication Date(Web):July 31, 2014
DOI:10.1021/cb5004256
Caspases are fundamental to many essential biological processes, including apoptosis, differentiation, and inflammation. Unregulated caspase activity is also implicated in the development and progression of several diseases, such as cancer, neurodegenerative disorders, and sepsis. Unfortunately, it is difficult to determine exactly which caspase(s) of the 11 isoforms that humans express is responsible for specific biological functions. This lack of resolution is primarily due to highly homologous active sites and overlapping substrates. Currently available peptide-based inhibitors and probes are based on specificity garnered from peptide substrate libraries. For example, the canonical tetrapeptide LETD was discovered as the canonical sequence that is optimally recognized by caspase-8; however, LETD-based inhibitors and substrates promiscuously bind to other isoforms with equal affinity, including caspases-3, -6, and -9. In order to mitigate this problem, we report the identification of a new series of compounds that are >100-fold selective for inhibiting the initiator caspases-8 and -9 over the executioner caspases-3, -6, and -7.
Co-reporter:Chris J. Vickers, Gonzalo E. González-Páez, and Dennis W. Wolan
ACS Chemical Biology 2014 Volume 9(Issue 10) pp:2199
Publication Date(Web):August 18, 2014
DOI:10.1021/cb500586p
Caspases are a family of cysteine proteases that are well-known for their roles in apoptosis and inflammation. Recent studies provide evidence that caspases are also integral to many additional cellular processes, such as differentiation and proliferation. Likewise, aberrant caspase activity has been implicated in the progression of several diseases, including neurodegenerative disorders, cancer, cardiovascular disease, and sepsis. These observations establish the importance of caspases to a diverse array of physiological functions and future endeavors will undoubtedly continue to elucidate additional processes that require caspase activity. Unfortunately, the existence of 11 functional human caspases, with overlapping substrate specificities, confounds the ability to confidently assign one or more isoforms to biological phenomena. Herein, we characterize a first-in-class FRET substrate that is selectively recognized by active caspase-3 over other initiator and executioner caspases. We further apply this substrate to specifically image caspase-3 activity in live cells undergoing apoptosis.
Co-reporter:Chris J. Vickers ; Gonzalo E. González-Páez
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12869-12876
Publication Date(Web):August 5, 2013
DOI:10.1021/ja406399r
Caspases are a family of cysteine-aspartyl proteases that are well recognized for their essential roles in apoptosis and inflammation. Recently, caspases have also been linked to the promotion of other biologically important phenomena, such as cellular differentiation and proliferation. Dysregulation of the multifaceted and indispensable activities of caspases has been globally linked to several diseases, including cancer and neurodegenerative disorders; however, the specific caspase members responsible for these diseases have yet to be assigned. Activity-based probes (ABPs) and peptide-based inhibitors are instrumental in the detection and control of protease activity and serve as alternative methods to genetic approaches. Such molecules aid in the interrogation of specific proteases within cellular and animal models as well as help elucidate aberrant proteolytic function correlated to disease phenotypes. No ABPs or inhibitors have been discovered that specifically target one of the eleven human caspases in a cellular context. Therefore, ascribing distinct contributions to an individual caspase activity within naturally occurring biological systems is not possible. Herein, we describe a peptide series optimized for the selective detection and inhibition of active caspase-3 in cells. These compounds exhibit low nanomolar potency against caspase-3 with >120-fold selectivity over caspase-7 which shares 77% active site identity. Our ability to individually target wild-type active caspase-3 for detection and cell permeable inhibition is a valuable proof-of-concept methodology that can be readily employed to probe the significance of caspase-3 in apoptosis, neurological disorders, cardiovascular diseases, and sepsis.
Co-reporter:Chris J. Vickers, Gonzalo E. González-Páez, and Dennis W. Wolan
ACS Chemical Biology 2013 Volume 8(Issue 7) pp:1558
Publication Date(Web):April 24, 2013
DOI:10.1021/cb400209w
Caspases are required for essential biological functions, most notably apoptosis and pyroptosis, but also cytokine production, cell proliferation, and differentiation. One of the most well studied members of this cysteine protease family includes executioner caspase-3, which plays a central role in cell apoptosis and differentiation. Unfortunately, there exists a dearth of chemical tools to selectively monitor caspase-3 activity under complex cellular and in vivo conditions due to its close homology with executioner caspase-7. Commercially available activity-based probes and substrates rely on the canonical DEVD tetrapeptide sequence, which both caspases-3 and -7 recognize with similar affinity, and thus the individual contributions of caspase-3 and/or -7 toward important cellular processes are irresolvable. Here, we analyzed a variety of permutations of the DEVD peptide sequence in order to discover peptides with biased activity and recognition of caspase-3 versus caspases-6, -7, -8, and -9. Through this study, we identify fluorescent and biotinylated probes capable of selective detection of caspase-3 using key unnatural amino acids. Likewise, we determined the X-ray crystal structures of caspases-3, -7, and -8 in complex with our lead peptide inhibitor to elucidate the binding mechanism and active site interactions that promote the selective recognition of caspase-3 over other highly homologous caspase family members.
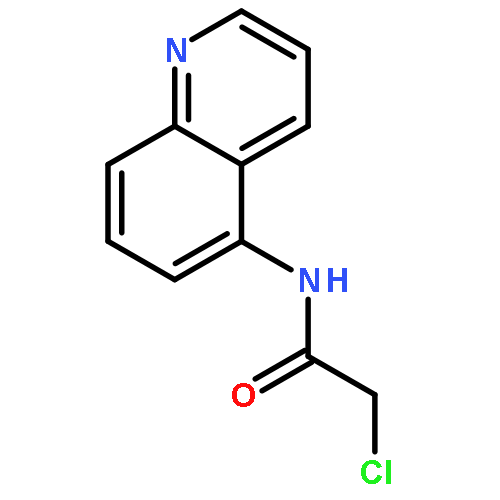
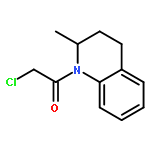
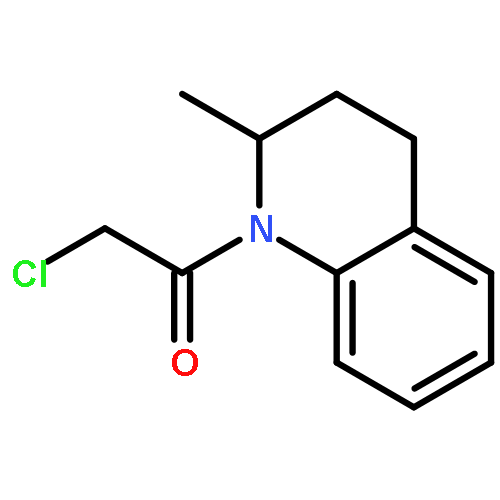
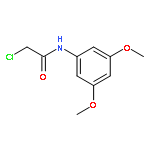
Co-reporter:Chris J. Vickers;Gonzalo E. González-Páez;Jeffrey C. Umotoy;Charmagne Cayanan-Garrett;Dr. Steven J. Brown; Dr. Dennis W. Wolan
ChemBioChem 2013 Volume 14( Issue 12) pp:1419-1422
Publication Date(Web):
DOI:10.1002/cbic.201300315
.jpg)



![1-(chloroacetyl)-4-[(2-fluorophenyl)sulfonyl]piperazine](http://img.cochemist.com/ccimg/923300/923204-90-4.png)
![1-(chloroacetyl)-4-[(2-fluorophenyl)sulfonyl]piperazine](http://img.cochemist.com/ccimg/923300/923204-90-4_b.png)
![N-[5-(aminosulfonyl)-2-(diethylamino)phenyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/923200/923168-91-6.png)
![N-[5-(aminosulfonyl)-2-(diethylamino)phenyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/923200/923168-91-6_b.png)
![Ethenesulfonamide,N-methyl-2-[1-(phenylmethyl)-1H-indol-5-yl]-, (1E)-](http://img.cochemist.com/ccimg/894400/894351-84-9.png)
![Ethenesulfonamide,N-methyl-2-[1-(phenylmethyl)-1H-indol-5-yl]-, (1E)-](http://img.cochemist.com/ccimg/894400/894351-84-9_b.png)
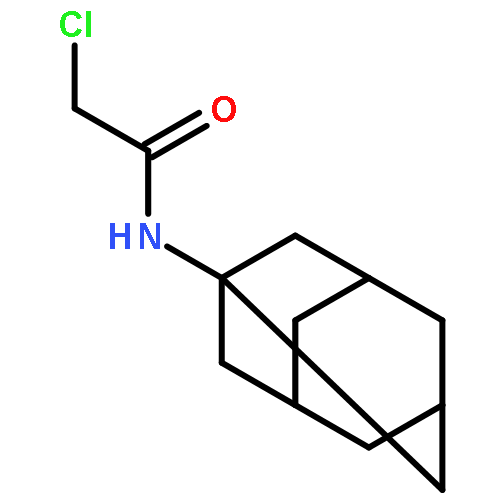
![N-[(1-adamantylamino)carbonyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/725800/725711-03-5.png)
![N-[(1-adamantylamino)carbonyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/725800/725711-03-5_b.png)
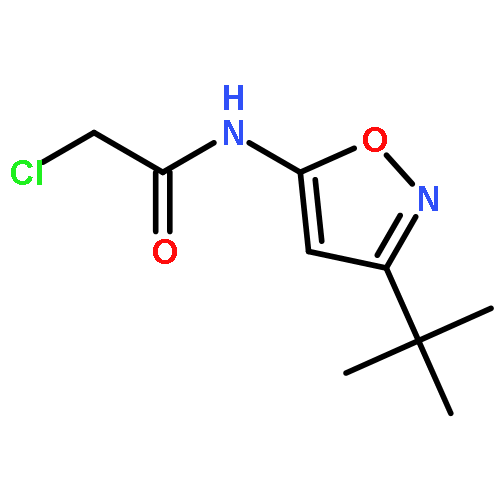
![N-[3,5-bis(trifluoromethyl)phenyl]-2-bromopropanamide](http://img.cochemist.com/ccimg/646500/646497-42-9.png)
![N-[3,5-bis(trifluoromethyl)phenyl]-2-bromopropanamide](http://img.cochemist.com/ccimg/646500/646497-42-9_b.png)
![Pentanal, 5-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/564500/564469-81-4.png)
![Pentanal, 5-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/564500/564469-81-4_b.png)


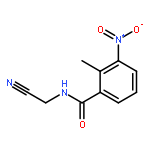
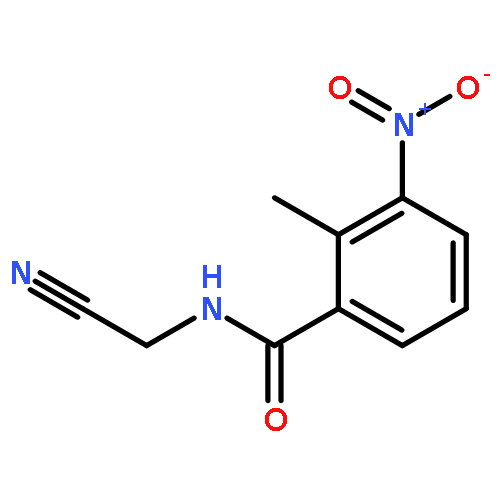
![N-Benzo[1,3]dioxol-5-yl-2-chloro-acetamide](http://img.cochemist.com/ccimg/227200/227199-07-7.png)
![N-Benzo[1,3]dioxol-5-yl-2-chloro-acetamide](http://img.cochemist.com/ccimg/227200/227199-07-7_b.png)
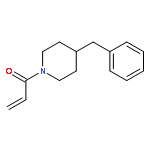












![Carbamic acid, [(1S)-1-formyl-3-methylbutyl]-, 9H-fluoren-9-ylmethylester](http://img.cochemist.com/ccimg/146900/146803-42-1.png)
![Carbamic acid, [(1S)-1-formyl-3-methylbutyl]-, 9H-fluoren-9-ylmethylester](http://img.cochemist.com/ccimg/146900/146803-42-1_b.png)
















![2-[(chloroacetyl)amino]-2-deoxy-D-glucose](http://img.cochemist.com/ccimg/59900/59817-28-6.png)
![2-[(chloroacetyl)amino]-2-deoxy-D-glucose](http://img.cochemist.com/ccimg/59900/59817-28-6_b.png)




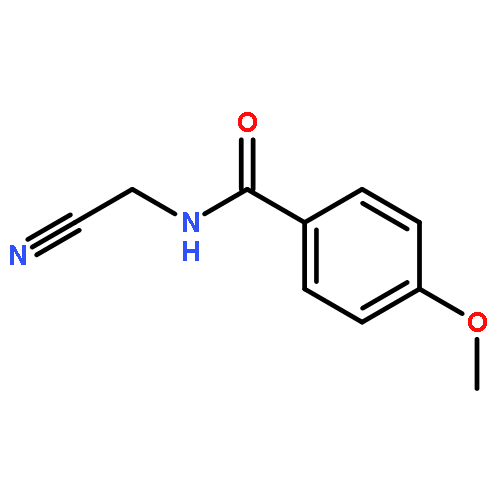

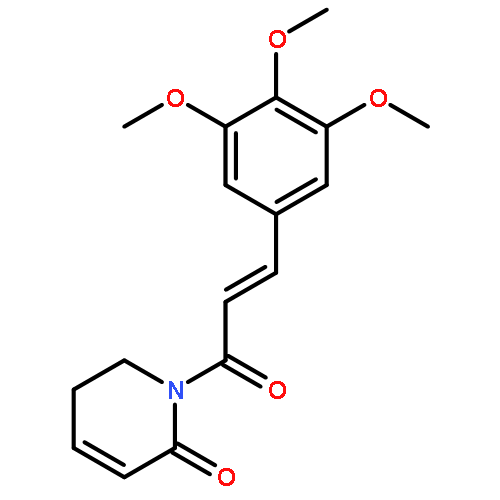
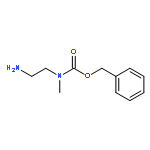
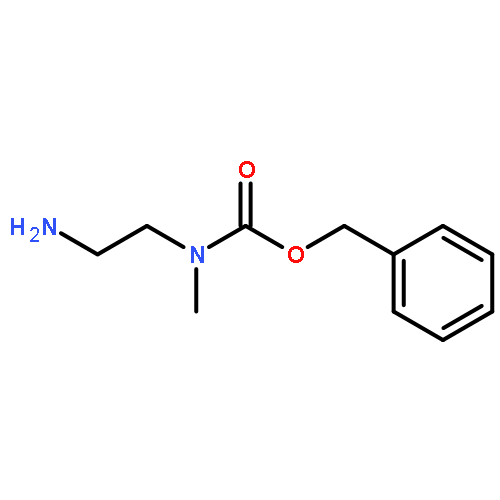











![N-(3-CHLORO-4-FLUOROPHENYL)-N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-1<WBR />,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/3300/3289-77-8.png)
![N-(3-CHLORO-4-FLUOROPHENYL)-N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-1<WBR />,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/3300/3289-77-8_b.png)
![Ethanone,2-chloro-1-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-](http://img.cochemist.com/ccimg/200/143-85-1.png)
![Ethanone,2-chloro-1-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-](http://img.cochemist.com/ccimg/200/143-85-1_b.png)
![Pentanoic acid,5-chloro-3-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-4-oxo-, (3S)-](http://img.cochemist.com/ccimg/260500/260434-72-8.png)
![Pentanoic acid,5-chloro-3-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-4-oxo-, (3S)-](http://img.cochemist.com/ccimg/260500/260434-72-8_b.png)




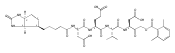
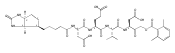






![L-Valinamide,N-acetyl-L-a-aspartyl-L-a-glutamyl-N-[(1S)-2-carboxy-1-formylethyl]-](http://img.cochemist.com/ccimg/169400/169332-60-9.png)
![L-Valinamide,N-acetyl-L-a-aspartyl-L-a-glutamyl-N-[(1S)-2-carboxy-1-formylethyl]-](http://img.cochemist.com/ccimg/169400/169332-60-9_b.png)





