Co-reporter:Priya Murria, Caleb K. Miskin, Robert Boyne, Laurance T. Cain, Ravikiran Yerabolu, Ruihong Zhang, Evan C. Wegener, Jeffrey T. Miller, Hilkka I. Kenttämaa, and Rakesh Agrawal
Inorganic Chemistry December 4, 2017 Volume 56(Issue 23) pp:14396-14396
Publication Date(Web):November 13, 2017
DOI:10.1021/acs.inorgchem.7b01359
Thiol-amine mixtures are an attractive medium for the solution processing of semiconducting thin films because of their remarkable ability to dissolve a variety of metals, metal chalcogenides, metal salts, and chalcogens. However, very little is known about their dissolution chemistry. Electrospray ionization high-resolution tandem mass spectrometry and X-ray absorption spectroscopy were employed to identify the species formed upon dissolution of CuCl and CuCl2 in 1-propanethiol and n-butylamine. Copper was found to be present exclusively in the 1+ oxidation state for both solutions. The copper complexes detected include copper chlorides, copper thiolates, and copper thiolate chlorides. No complexes of copper with amines were observed. Additionally, alkylammonium ions and alkylammonium chloride adducts were observed. These findings suggest that the dissolution is initiated by proton transfer from the thiol to the amine, followed by coordination of the thiolate anions with copper cations. Interestingly, the mass and X-ray absorption spectra of the solutions of CuCl and CuCl2 in thiol-amine were essentially identical. However, dialkyl disulfides were identified by Raman spectroscopy as an oxidation product only for the copper(II) solution, wherein copper(II) had been reduced to copper(I). Analysis of several thiol-amine pairs suggested that the dissolution mechanism is quite general. Finally, analysis of thin films prepared from these solutions revealed persistent chlorine impurities, in agreement with previous studies. These impurities are explained by the mass spectrometric finding that chloride ligands are not completely displaced by thiolates upon dissolution. These results suggest that precursors other than chlorides will likely be preferred for the generation of high-efficiency copper chalcogenide films, despite the reasonable efficiencies that have been obtained for films generated from chloride precursors in the past.
Co-reporter:Hanyu Zhu;Tiffany M. Jarrell
Journal of The American Society for Mass Spectrometry 2017 Volume 28( Issue 2) pp:393-393
Publication Date(Web):05 December 2016
DOI:10.1007/s13361-016-1561-3
Erratum to: J. Am. Soc. Mass Spectrom (2016)DOI: 10.1007/s13361-016-1442-9Several authors were inadvertently excluded from the list of authors. The list is correct as displayed above.
Co-reporter:Chunfen Jin, Jyrki Viidanoja, Mingzhe Li, Yuyang Zhang, Elias Ikonen, Andrew Root, Mark Romanczyk, Jeremy Manheim, Eric Dziekonski, and Hilkka I. Kenttämaa
Analytical Chemistry 2016 Volume 88(Issue 21) pp:10592
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.analchem.6b02789
Direct infusion atmospheric pressure chemical ionization mass spectrometry (APCI-MS) was compared to field ionization mass spectrometry (FI-MS) for the determination of hydrocarbon class distributions in lubricant base oils. When positive ion mode APCI with oxygen as the ion source gas was employed to ionize saturated hydrocarbon model compounds (M) in hexane, only stable [M – H]+ ions were produced. Ion–molecule reaction studies performed in a linear quadrupole ion trap suggested that fragment ions of ionized hexane can ionize saturated hydrocarbons via hydride abstraction with minimal fragmentation. Hence, APCI-MS shows potential as an alternative of FI-MS in lubricant base oil analysis. Indeed, the APCI-MS method gave similar average molecular weights and hydrocarbon class distributions as FI-MS for three lubricant base oils. However, the reproducibility of APCI-MS method was found to be substantially better than for FI-MS. The paraffinic content determined using the APCI-MS and FI-MS methods for the base oils was similar. The average number of carbons in paraffinic chains followed the same increasing trend from low viscosity to high viscosity base oils for the two methods.
Co-reporter:James S. Riedeman, Naveen Reddy Kadasala, Alexander Wei, and Hilkka I. Kenttämaa
Energy & Fuels 2016 Volume 30(Issue 2) pp:805-809
Publication Date(Web):January 6, 2016
DOI:10.1021/acs.energyfuels.5b02002
Crude oil deposition in oil transfer pipelines and bore wells afflicts many oil reservoirs. Asphaltenes play a major role in this process because of their tendency to precipitate in pipelines upon changes in temperature and/or pressure. Asphaltenes are defined by their lack of solubility in n-alkane solvents, which means that they likely contain many compounds that do not actively contribute to the deposition of crude oil in pipelines. The preponderance of studies in the literature have focused on asphaltenes derived from crude oil, whereas far fewer investigations have focused on asphaltenes derived from oil deposits. In this study, structural parameters of oil-deposit asphaltenes were examined using Raman spectroscopy and tandem mass spectrometry and compared to results reported previously for petroleum asphaltenes. On the basis of D1 and G band intensities in the Raman spectrum of oil-deposit asphaltenes, the average aromatic sheet size of these molecules was 21.0 Å, slightly larger than earlier values reported for petroleum asphaltenes (15.2–18.8 Å). Mass spectrometric experiments of oil-deposit asphaltenes ionized via atmospheric pressure chemical ionization (APCI) using CS2 solvent were used to measure the molecular weight distribution (MWD), saturated carbon content, and the number of fused aromatic rings in the cores of the asphaltene molecules. The MWD was found to be 150–1050 Da with an average molecular weight (average MW) of 497 Da, which are significantly lower than those reported previously for petroleum asphaltenes (200–1500 Da and 570–700 Da, respectively). Aromatic core sizes were estimated to contain 8 fused rings on average for the most abundant species in oil-deposit asphaltenes, with 5–15 carbons in their alkyl side chains, as compared to averages of 3–7 aromatic rings and 17–41 alkyl carbons for petroleum asphaltenes.
Co-reporter:Dr. Nelson R. Vinueza;Dr. Bart&x142;omiej J. Jankiewicz;Vanessa A. Gallardo;Gregory Z. LaFavers;Dane DeSutter;Dr. John J. Nash; Hilkka I. Kenttämaa
Chemistry - A European Journal 2016 Volume 22( Issue 2) pp:809-815
Publication Date(Web):
DOI:10.1002/chem.201502502
Abstract
The chemical properties of the 4,5,8-tridehydroisoquinolinium ion (doublet ground state) and related mono- and biradicals were examined in the gas phase in a dual-cell Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer. The triradical abstracted three hydrogen atoms in a consecutive manner from tetrahydrofuran (THF) and cyclohexane molecules; this demonstrates the presence of three reactive radical sites in this molecule. The high (calculated) electron affinity (EA=6.06 eV) at the radical sites makes the triradical more reactive than two related monoradicals, the 5- and 8-dehydroisoquinolinium ions (EA=4.87 and 5.06 eV, respectively), the reactivity of which is controlled predominantly by polar effects. Calculated triradical stabilization energies predict that the most reactive radical site in the triradical is not position C4, as expected based on the high EA of this radical site, but instead position C5. The latter radical site actually destabilizes the 4,8-biradical moiety, which is singlet coupled. Indeed, experimental reactivity studies show that the radical site at C5 reacts first. This explains why the triradical is not more reactive than the 4-dehydroisoquinolinium ion because the C5 site is the intrinsically least reactive of the three radical sites due to its low EA. Although both EA and spin–spin coupling play major roles in controlling the overall reactivity of the triradical, spin–spin coupling determines the relative reactivity of the three radical sites.
Co-reporter:Hanyu Zhu;Joann P. Max
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 11) pp:1813-1823
Publication Date(Web):2016 November
DOI:10.1007/s13361-016-1442-9
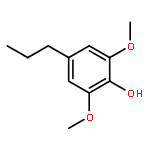
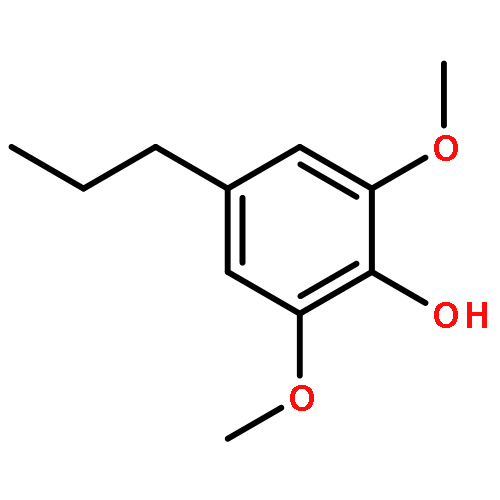
Conversion of lignin into smaller molecules provides a promising alternate and sustainable source for the valuable chemicals currently derived from crude oil. Better understanding of the chemical composition of the resulting product mixtures is essential for the optimization of such conversion processes. However, these mixtures are complex and contain isomeric molecules with a wide variety of functionalities, which makes their characterization challenging. Tandem mass spectrometry based on ion–molecule reactions has proven to be a powerful tool in functional group identification and isomer differentiation for previously unknown compounds. This study demonstrates that the identification of the phenol functionality, the most commonly observed functionality in lignin degradation products, can be achieved via ion–molecule reactions between diethylmethoxyborane (DEMB) and the deprotonated analyte in the absence of strongly electron-withdrawing substituents in the ortho- and para-positions. Either a stable DEMB adduct or an adduct that has lost a methanol molecule (DEMB adduct-MeOH) is formed for these ions. Deprotonated phenols with an adjacent phenol or hydroxymethyl functionality or a conjugated carboxylic acid functionality can be identified based on the formation of DEMB adduct-MeOH. Deprotonated compounds not containing the phenol functionality and phenols containing an electron-withdrawing ortho- or para-substituent were found to be unreactive toward diethylmethoxyborane. Hence, certain deprotonated isomeric compounds with phenol and carboxylic acid, aldehyde, carboxylic acid ester, or nitro functionalities can be differentiated via these reactions. The above mass spectrometry method was successfully coupled with high-performance liquid chromatography for the analysis of a complex biomass degradation mixture.
Co-reporter:Huaming Sheng, Weijuan Tang, Ravikiran Yerabolu, Joann Max, Raghavendhar R. Kotha, James S. Riedeman, John J. Nash, Minli Zhang, and Hilkka. I. Kenttämaa
The Journal of Organic Chemistry 2016 Volume 81(Issue 2) pp:575-586
Publication Date(Web):December 11, 2015
DOI:10.1021/acs.joc.5b02409
The in vivo oxidation of sulfur and nitrogen atoms in many drugs into sulfoxide and N-oxide functionalities is a common biotransformation process. Unfortunately, the unambiguous identification of these metabolites can be challenging. In the present study, ion–molecule reactions of tris(dimethylamino)borane followed by collisionally activated dissociation (CAD) in an ion trap mass spectrometer are demonstrated to allow the identification of N-oxide and sulfoxide functionalities in protonated polyfunctional drug metabolites. Only ions with N-oxide or sulfoxide functionality formed diagnostic adducts that had lost dimethyl amine (DMA). This was demonstrated even for an analyte that contains a substantially more basic functionality than the functional group of interest. CAD of the diagnostic product ions (M) resulted mainly in type A (M – DMA) and B fragment ions (M – HO–B(N(CH3)2)2) for N-oxides, but sulfoxides also formed diagnostic C ions (M – O═BN(CH3)2), thus allowing differentiation of the functionalities. Some protonated analytes yielded abundant TDMAB adducts that had lost two DMA molecules instead of just one. This provides information on the environment of the N-oxide and sulfoxide functionalities. Quantum chemical calculations were performed to explore the mechanisms of the above-mentioned reactions. The method can be implemented on HPLC for real drug analysis.
Co-reporter:Weijuan Tang, Matthew R. Hurt, Huaming Sheng, James S. Riedeman, David J. Borton, Peter Slater, and Hilkka I. Kenttämaa
Energy & Fuels 2015 Volume 29(Issue 3) pp:1309-1314
Publication Date(Web):February 3, 2015
DOI:10.1021/ef501242k
In this work, six petroleum asphaltene samples of different geographical origins were studied using atmospheric pressure chemical ionization (APCI) in positive-ion mode in a linear quadrupole ion trap mass spectrometer (LQIT). APCI doped with carbon disulfide reagent was selected as the ionization method because it has been previously demonstrated to generate stable molecular ions with no fragmentation for asphaltene molecules. The mass spectra measured using this approach revealed the apparent molecular weights (MWs) of the molecules in the asphaltene samples. The results show that petroleum asphaltenes from the American continent, Europe, and China have similar apparent molecular weight distributions, ranging from 200 to 1450 Da, with slightly different apparent average MWs ranging from 570 to 700 Da. Further, molecular ions with eight randomly selected mass-to-charge ratios (m/z) ranging from m/z 500 to 808 were isolated for each asphaltene sample and subjected to collisionally activated dissociation (CAD) at the same collision energy to examine their structures. The CAD mass spectra (MS2 experiment) provided information on the maximum total number of carbons in the alkyl chains and the smallest possible size of the aromatic cores in the ionized molecules. Additionally, MS3 experiments were performed to investigate the fragmentation patterns of the fragment ions generated in the MS2 experiments. The results obtained support the island structural model for these asphaltenes. Moreover, molecules of greater MWs are shown to have more carbons in alkyl chains (ranging from 17 to 41), but the minimum core size is fairly constant.
Co-reporter:Tiffany M. Jarrell, James S. Riedeman, Hilkka Kenttämaa
International Journal of Mass Spectrometry 2015 Volume 378() pp:206-211
Publication Date(Web):15 February 2015
DOI:10.1016/j.ijms.2014.08.011
•ClMn2+ chemical ionization reagent ion efficiently ionizes various types of analytes, both polar and nonpolar, to exclusively form ClMn+ adduct ions.•Above chemical ionization method appears to have no bias toward specific analytes.•Above characteristics make this method especially suitable for mixture analysis.A chemical ionization reagent ion, ClMn2+, has been identified for the analysis of mixtures of organic compounds since it allows ionization of both polar and nonpolar analytes so that only one product ion, ClMn+ adduct of the analyte, is generated without fragmentation. The reagent ion is formed upon ionization of ClMn(CO)5 via corona discharge in an atmospheric pressure chemical ionization source of a linear quadrupole ion trap mass spectrometer. Volatile analytes were introduced into the ion trap via a reagent mixing manifold. Nonvolatile analytes were deposited on a titanium foil and desorbed using laser-induced acoustic desorption (LIAD). Formation of a ClMn+ adduct with no accompanying fragmentation was observed for all analytes, including branched saturated hydrocarbons. Calculations indicate that ClMn+ binds to saturated hydrocarbons via an agostic interaction involving the manganese center and a CH bond of the analyte. The reagent ion Mn+ was also investigated. This ion forms a stable adduct with most analytes studied. It binds fairly strongly to saturated hydrocarbons via two agostic interactions with two CH bonds instead of insertion into a CH bond. However, it was found to cause fragmentation for alcohols. ClMn(H2O)+ has been previously shown to ionize most compounds by ClMn+ adduct formation but also to yield molecular ions for amines due to their low ionization energies (<8.3 eV). However, electron transfer was not observed upon ionization of amines with the reagents reported here. The CpCo+ ion has been reported earlier to ionize most saturated hydrocarbons without fragmentation. However, it induces CC bond cleavages for highly branched alkanes upon ionization. This was not observed for the reagent ions studied here. ClMn2+ ion is a more universal ionization reagent than Mn+ and the previously reported reagent ions due to its ability to ionize analytes with low ionization energies without electron transfer and due to the complete lack of fragmentation for all analytes. Furthermore, based on the examination of six very different analytes, it has no significant bias toward specific types of analytes.
Co-reporter:Ashley M. Wittrig, Enada F. Archibold, Huaming Sheng, John J. Nash, Hilkka I. Kenttämaa
International Journal of Mass Spectrometry 2015 Volume 377() pp:39-43
Publication Date(Web):1 February 2015
DOI:10.1016/j.ijms.2014.04.017
The gas-phase reactivity of charged para-benzynes is entirely unexplored as they and/or their precursors tend to undergo ring-opening upon their generation. We report here a gas-phase reactivity study of two such benzynes, the 2,5-didehydropyridinium and 5,8-didehydroisoquinolinium cations, generated in a modified dual-linear quadrupole ion trap (DLQIT) mass spectrometer. Both biradicals were found to form diagnostic products with organic molecules, indicating the presence of two radical sites. As opposed to earlier predictions that the singlet–triplet (S–T) splitting controls the radical reactivity of such species, the 2,5-didehydropyridinium cation reacts much faster in spite of its larger S–T splitting. Calculated vertical electron affinities of the radical sites of the para-benzynes, a parameter related to the polarity of the transition states of their reactions, appears to be the most important reactivity controlling factor.
Co-reporter:John C. Degenstein, Priya Murria, Mckay Easton, Huaming Sheng, Matt Hurt, Alex R. Dow, Jinshan Gao, John J. Nash, Rakesh Agrawal, W. Nicholas Delgass, Fabio H. Ribeiro, and Hilkka I. Kenttämaa
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1909-1914
Publication Date(Web):January 6, 2015
DOI:10.1021/jo5025255
A fast-pyrolysis probe/tandem mass spectrometer combination was utilized to determine the initial fast-pyrolysis products for four different selectively 13C-labeled cellobiose molecules. Several products are shown to result entirely from fragmentation of the reducing end of cellobiose, leaving the nonreducing end intact in these products. These findings are in disagreement with mechanisms proposed previously. Quantum chemical calculations were used to identify feasible low-energy pathways for several products. These results provide insights into the mechanisms of fast pyrolysis of cellulose.
Co-reporter:Tiffany M. Jarrell, Christopher L. Marcum, Huaming Sheng, Benjamin C. Owen, C. J. O'Lenick, Hagen Maraun, Joseph J. Bozell and Hilkka I. Kenttämaa
Green Chemistry 2014 vol. 16(Issue 5) pp:2713-2727
Publication Date(Web):26 Feb 2014
DOI:10.1039/C3GC42355G
Lignin is an aromatic biopolymer that may yield valuable chemicals currently obtained solely from petroleum. However, extraction of lignin by using traditional methods, such as organosolv extraction, produces very complex mixtures. Molecular level characterization of the major components is essential to be able to rationally tailor methodology for the conversion of these mixtures to transportation fuel and valuable chemicals. In this study, high performance liquid chromatography/high resolution tandem mass spectrometry (HPLC/MSn) was used to obtain molecular weight, elemental composition and structural information for the major components in an organosolv lignin sample. HPLC/MSn coupled with hydroxide-doped electrospray ionization was used to identify the structures of the major components by using a Thermo Scientific linear quadrupole ion trap-Fourier transform ion cyclotron resonance hybrid mass spectrometer (LQIT/FT-ICR). The results reported here demonstrate that the major products of organosolv extraction are low molecular weight compounds, including monomeric and dimeric lignin units, with various functionalities.
Co-reporter:Jinshan Gao, Bartłomiej J. Jankiewicz, Jennifer Reece, Huaming Sheng, Christopher J. Cramer, John J. Nash and Hilkka I. Kenttämaa
Chemical Science 2014 vol. 5(Issue 6) pp:2205-2215
Publication Date(Web):27 Feb 2014
DOI:10.1039/C4SC00194J
The reactivities of eleven 3,5-didehydropyridinium and six 2,4-didehydropyridinium cations toward cyclohexane were examined in the gas phase by using Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometry as well as high-level quantum chemical calculations. The results unequivocally demonstrate that the reactivity of meta-benzyne analogs can be “tuned” from more radical-like to less radical-like by changing the type and position of substituents. For example, σ-acceptor substituents at the 4-position and π-donor substituents at the 2-position in 3,5-didehydropyridinium cations partially decouple the biradical electrons, which results in lower energy transition states, and faster radical reactions. In contrast, σ-acceptors at the 2-position and π-donors at the 4-position in 3,5-didehydropyridinium cations cause stronger coupling between the biradical electrons, which results in lower radical reactivity. Three main factors are found to control the reactivity of these biradicals: (1) the energy required to distort the minimum energy dehydrocarbon atom separation to the separation of the transition state, (2) the S–T splitting at the separation of the transition state, and (3) the electron affinity at the separation of the transition state.
Co-reporter:Tiffany Jarrell, James Riedeman, Mark Carlsen, Randall Replogle, Tim Selby, and Hilkka Kenttämaa
Analytical Chemistry 2014 Volume 86(Issue 13) pp:6533
Publication Date(Web):June 4, 2014
DOI:10.1021/ac501034v
Ion–molecule reactions provide a powerful tool for structural elucidation of ionized pharmaceutical analytes in tandem mass spectrometry. However, all previous interfaces for the introduction of reagents for ion–molecule reactions have utilized a single reagent approach. In this study, a multiported pulsed valve system was designed and characterized for rapid introduction of three neutral reagents into a linear quadrupole ion trap. Additionally, automatic triggering was used to allow for the introduction of the reagents on a chromatographic time scale. This system enables automatic, high throughput screening of complex mixtures by using at least three different ion–molecule reactions. Further, rapid testing of new neutral reagents is also possible.
Co-reporter:Huaming Sheng, Peggy E. Williams, Weijuan Tang, Minli Zhang and Hilkka I. Kenttämaa
Analyst 2014 vol. 139(Issue 17) pp:4296-4302
Publication Date(Web):05 Jun 2014
DOI:10.1039/C4AN00677A
A mass spectrometric method utilizing gas-phase ion/molecule reactions of 2-methoxypropene (MOP) has been developed for the identification of the sulfoxide functionality in protonated analytes in a LQIT mass spectrometer. Protonated sulfoxide analytes react with MOP to yield an abundant addition product (corresponding to 37–99% of the product ions), which is accompanied by a much slower proton transfer. The total efficiency (percent of gas-phase collisions leading to products) of the reaction is moderate (3–14%). A variety of compounds with different functional groups, including sulfone, hydroxylamino, N-oxide, aniline, phenol, keto, ester, amino and hydroxy, were examined to probe the selectivity of this reaction. Most of the protonated compounds with proton affinities lower than that of MOP react mainly via proton transfer to MOP. The formation of adduct-MeOH ions was found to be characteristic for secondary N-hydroxylamines. N-Oxides formed abundant MOP adducts just like sulfoxides, but sulfoxides can be differentiated from N-oxides based on their high reaction efficiencies. The reaction was tested by using the anti-inflammatory drug sulindac (a sulfoxide) and its metabolite sulindac sulfone. The presence of a sulfoxide functionality in the drug but a sulfone functionality in the metabolite was readily demonstrated. The presence of other functionalities in addition to sulfoxide in the analytes was found not to influence the diagnostic reactivity.
Co-reporter:Huaming Sheng, Peggy E. Williams, Weijuan Tang, James S. Riedeman, Minli Zhang, and Hilkka I. Kenttämaa
The Journal of Organic Chemistry 2014 Volume 79(Issue 7) pp:2883-2889
Publication Date(Web):February 26, 2014
DOI:10.1021/jo402645a
A tandem mass spectrometric method is presented for the rapid identification of drug metabolites that contain the sulfone functional group. This method is based on a gas-phase ion/molecule reaction of protonated sulfone analytes with trimethyl borate (TMB) that yields a diagnostic product ion, adduct-Me2O, at high reaction efficiency. A variety of compounds with different functional groups, such as sulfoxides, hydroxylamines, N-oxides, anilines, phenol, an aliphatic amine, and an aliphatic alcohol, were examined to probe the selectivity of this reaction. Except for protonated sulfones, most of the protonated compounds react very slowly or not at all with TMB. Most importantly, none of them give the adduct-Me2O product. A mechanism that explains the observed selectivity is proposed for the diagnostic reaction and is supported by quantum chemical calculations. The reaction was tested with the anti-inflammatory drug sulindac and its metabolite, sulindac sulfone, which were readily distinguished. The presence of other functionalities in addition to sulfone was found not to influence the diagnostic reactivity.
Co-reporter:Tiffany M. Jarrell, Chunfen Jin, James S. Riedeman, Benjamin C. Owen, Xiaoli Tan, Alexander Scherer, Rik R. Tykwinski, Murray R. Gray, Peter Slater, Hilkka I. Kenttämaa
Fuel 2014 Volume 133() pp:106-114
Publication Date(Web):1 October 2014
DOI:10.1016/j.fuel.2014.04.040
•Methodology will facilitate the elucidation of the structures of asphaltenes.•Unique fragmentation shown for island and archipelago structures.•Distinguishable characteristics: alkyl chain lengths and sizes of aromatic cores.•Fragmentation influenced by presence of nitrogen.•No influence from the presence of sulfur.Despite extensive studies of the asphaltene fraction of petroleum, the molecular structures of asphaltenes remain a highly debated topic. Tandem mass spectrometry is the only technique that allows the examination of the structures of individual asphaltene molecules due to the extreme complexity of asphaltenes. Recently, atmospheric pressure chemical ionization (APCI) using CS2 as the reagent was demonstrated to produce abundant and stable molecular ions for polyaromatic hydrocarbons with long alkyl chains. Hence, coupling APCI/CS2 with tandem mass spectrometry appears to be a promising method for the examination of the structures of molecules in asphaltenes. However, the fragmentation pathways of the molecular ions of large alkyl aromatic compounds are not well understood. In order to address this issue, a detailed examination of the collision-activated dissociation reactions of the molecular ions and several of their fragment ions (MSn experiments) was carried out for several model compounds of asphaltenes. The results show that information on various structural aspects of asphaltenes can be obtained from these experiments, such as alkyl chain lengths and sizes of aromatic cores. Based on these results, MS2 experiments may provide enough information to determine approximate core sizes for molecules with archipelago structures. However, the number of ion isolation and collision-activated dissociation (CAD) experiments needed to elucidate maximum structural information for molecules with island structures depends on the number of carbon chains on the aromatic core.
Co-reporter:Peggy E. Williams, Bartłomiej J. Jankiewicz, Linan Yang, and Hilkka I. Kenttämaa
Chemical Reviews 2013 Volume 113(Issue 9) pp:6949
Publication Date(Web):August 29, 2013
DOI:10.1021/cr400121w
Co-reporter:David J. Borton, Lucas M. Amundson, Matthew R. Hurt, Alex Dow, Jeremy T. Madden, Garth J. Simpson, and Hilkka I. Kenttämaa
Analytical Chemistry 2013 Volume 85(Issue 12) pp:5720
Publication Date(Web):May 16, 2013
DOI:10.1021/ac4000333
Laser-induced acoustic desorption (LIAD) was recently coupled to atmospheric pressure chemical ionization (APCI) and shown to be of great utility for the analysis of a variety of thermally labile nonpolar analytes that are not amenable to ionization via electrospray ionization, such as nonvolatile hydrocarbons. Despite these advancements, LIAD still suffered from several limitations, including only being able to sample a small fraction of the analyte molecules deposited on a Ti foil for desorption, poor reproducibility, as well as limited laser power throughput to the backside of the foil. These limitations severely hinder the analysis of especially challenging analytes, such as asphaltenes. To address these issues, a novel high-throughput LIAD probe and an assembly for raster sampling of a LIAD foil were designed, constructed, and tested. The new probe design allows 98% of the initial laser power to be realized at the backside of the foil over the 25% achieved previously, thus improving reproducibility and allowing for the analysis of large nonvolatile analytes, including asphaltenes. The raster assembly provided a 5.7 fold increase in the surface area of a LIAD foil that could be sampled and improved reproducibility and sensitivity for LIAD experiments. The raster assembly can also improve throughput as foils containing multiple analytes can be prepared and analyzed.
Co-reporter:Matthew R. Hurt, John C. Degenstein, Piotr Gawecki, David J. Borton II, Nelson R. Vinueza, Linan Yang, Rakesh Agrawal, W. Nicholas Delgass, Fabio H. Ribeiro, and Hilkka I. Kenttämaa
Analytical Chemistry 2013 Volume 85(Issue 22) pp:10927
Publication Date(Web):October 7, 2013
DOI:10.1021/ac402380h
Mass spectrometric methodology was developed for the determination and manipulation of the primary products of fast pyrolysis of carbohydrates. To determine the true primary pyrolysis products, a very fast heating pyroprobe was coupled to a linear quadrupole ion trap mass spectrometer through a custom-built adaptor. A home-built flow tube that simulates pyrolysis reactor conditions was used to examine the secondary reactions of the primary products. Depending on the experiment, the pyrolysis products were either evaporated and quenched or allowed to react for a period of time. The quenched products were ionized in an atmospheric pressure chemical ionization (APCI) source infused with one of two ionization reagents, chloroform or ammonium hydroxide, to aid in ionization. During APCI in negative ion mode, chloroform produces chloride anions that are known to readily add to carbohydrates with little bias and little to no fragmentation. On the other hand, in positive ion mode APCI, ammonium hydroxide forms ammonium adducts with carbohydrates with little to no fragmentation. The latter method ionizes compounds that are not readily ionized upon negative ion mode APCI, such as furan derivatives. Six model compounds were studied to verify the ability of the ionization methods to ionize known pyrolysis products: glycolaldehyde, hydroxyacetone, furfural, 5-hydroxymethylfurfural, levoglucosan, and cellobiosan. The method was then used to examine fast pyrolysis of cellobiose. The primary fast pyrolysis products were determined to consist of only a handful of compounds that quickly polymerize to form anhydro-oligosaccharides when allowed to react at high temperatures for an extended period of time.
Co-reporter:Benjamin C. Owen, Tiffany M. Jarrell, Jae C. Schwartz, Rob Oglesbee, Mark Carlsen, Enada F. Archibold, and Hilkka I. Kenttämaa
Analytical Chemistry 2013 Volume 85(Issue 23) pp:11284
Publication Date(Web):October 31, 2013
DOI:10.1021/ac401956f
A novel differentially pumped dual linear quadrupole ion trap (DLQIT) mass spectrometer was designed and built to facilitate tandem MS experiments free from interfering reactions. The instrument consists of two differentially pumped Thermo Scientific linear quadrupole ion trap (LQIT) systems that have been connected via an ion transfer octupole encased in a machined manifold. Tandem MS experiments can be performed in the front trap and then the resulting product ions can be transferred via axial ejection into the back trap for further, independent tandem MS experiments in a differentially pumped area. This approach allows the examination of consecutive collision-activated dissociation (CAD) and ion–molecule reactions without unwanted side reactions that often occur when CAD and ion–molecule reactions are examined in the same space. Hence, it greatly facilitates investigations of ion structures. In addition, the overall lower pressure of the DLQIT, as compared to commercial LQIT instruments, results in a reduction of unwanted side reactions with atmospheric contaminants, such as water and oxygen, in CAD and ion–molecule experiments.
Co-reporter:Matthew R. Hurt, David J. Borton, Heewon J. Choi, and Hilkka I. Kenttämaa
Energy & Fuels 2013 Volume 27(Issue 7) pp:3653-3658
Publication Date(Web):May 29, 2013
DOI:10.1021/ef302024z
Asphaltenes are a complex mixture of aromatic compounds that are present in petroleum and liquefied coal and have unknown molecular structures. Determination of the molecular structures of asphaltenes is greatly hindered by the extreme complexity of the mixtures. The average molecular weight of coal asphaltenes is known to be roughly half that of petroleum asphaltenes. In this study, tandem mass spectrometry experiments were used to compare the fragmentation patterns of molecular ions of the same mass-to-charge (m/z) value derived from coal and petroleum asphaltenes. The fragmentation pathways of the asphaltene ions were used to determine structural differences between the two types of asphaltenes. This type of analysis is new to the field of asphaltenes research and has not yet been utilized to compare coal and petroleum asphaltenes. Thus, this study was performed to both confirm its viability as well as further corroborate previous data gathered using different techniques. The results reported here demonstrate that the molecules in coal asphaltenes have shorter alkyl chains and larger aromatic cores than their petroleum-derived counterparts with the same molecular weight (MW). Furthermore, they have a higher aromatic-to-alkane carbon ratio, independent of the MW. However, the typical petroleum and coal asphaltenes (when considering the most abundant molecules in each) have a similarly sized aromatic core (∼8 rings), but very different total lengths of the alkyl side chains (∼4 for coal asphaltenes, ∼22 for petroleum asphaltenes).
Co-reporter:Bart&x142;omiej J. Jankiewicz;Nelson R. Vinueza;Lindsey M. Kirkpatrick;Vanessa A. Gallardo;Guannan Li;John J. Nash ;Hilkka I. Kenttämaa
Journal of Physical Organic Chemistry 2013 Volume 26( Issue 9) pp:707-714
Publication Date(Web):
DOI:10.1002/poc.3120
Reactive intermediates are key species involved in many chemical and biochemical processes. For example, carbon-centered aromatic σ,σ-biradicals formed in biological systems from naturally occurring enediyne antitumor antibiotics are responsible for the irreversible cleavage of double-stranded DNA caused by these prodrugs. However, because of their high reactivity, it is very difficult or impossible to isolate and investigate these biradicals. The aromatic σ,σ-biradical, 2,6-didehydropyridine, has been speculated for many years to be formed in certain organic reactions; however, no definitive proof of its generation has been obtained. We report here the successful generation of protonated 2,6-didehydropyridine and the examination of its chemical properties in the gas phase by using a Fourier transform ion cyclotron resonance mass spectrometer. The results suggest that a mixture of singlet (ground) state and triplet (excited) state 2,6-didehydropyridinium cations was generated. The two different states show qualitatively different reactivity, with the triplet state showing greater Brønsted acidity than that of the singlet state. The triplet state also shows much greater radical reactivity than that of the singlet state, as expected because of the coupling of the nonbonding electrons in the singlet state. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Dr. Lindsey M. Kirkpatrick;Dr. Nelson R. Vinueza;Dr. Bart&x142;omiej J. Jankiewicz;Vanessa A. Gallardo;Dr. Enada F. Archibold;Dr. John J. Nash; Hilkka I. Kenttämaa
Chemistry - A European Journal 2013 Volume 19( Issue 27) pp:9022-9033
Publication Date(Web):
DOI:10.1002/chem.201203264
Abstract
Experimental and computational studies on the formation of three gaseous, positively-charged para-benzyne analogues in a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer are reported. The structures of the cations were examined by isolating them and allowing them to react with various neutral reagents whose reactions with aromatic carbon-centered σ-type mono- and biradicals are well understood. Cleavage of two iodine–carbon bonds in N-deuterated 1,4-diiodoisoquinolinium cation by collision-activated dissociation (CAD) produced a long-lived cation that showed nonradical reactivity, which was unexpected for a para-benzyne. However, the reactivity closely resembles that of an isomeric enediyne, N-deuterated 2-ethynylbenzonitrilium cation. A theoretical study on possible rearrangement reactions occurring during CAD revealed that the cation formed upon the first iodine atom loss undergoes ring-opening before the second iodine atom loss to form an enediyne instead of a para-benzyne. Similar results were obtained for the 5,8-didehydroisoquinolinium cation and the 2,5-didehydropyridinium cation. The findings for the 5,8-didehydroisoquinolinium cation are in contradiction with an earlier report on this cation. The cation described in the literature was regenerated by using the literature method and demonstrated to be the isomeric 5,7-didehydro-isoquinolinium cation and not the expected 5,8-isomer.
Co-reporter:Linan Yang;Tefsit Bekele;Mark A. Lipton
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 4) pp:563-572
Publication Date(Web):2013 April
DOI:10.1007/s13361-012-0567-8
A negatively charged biradical intermediate was successfully generated in the gas phase via cyclization of the deprotonated bicyclo[8.3.0]trideca-12-ene-2,7-diyn-1-one precursor. The inherent negative charge of this biradical allows its characterization via collision-activated dissociation and reactions with a variety of neutral substrates in an FT-ICR mass spectrometer. Although the biradical is unreactive toward reagents that usually react rapidly with positively charged biradicals, such as dimethyl disulfide, it reacts with the halogen-containing substrates carbon tetrachloride, carbon tetrabromide, and bromotrichloromethane via bromine or chlorine atom abstraction, which supports its biradical structure. The results presented in this study indicate that cyclizations commonly used in solution to form biradical intermediates from enediyne compounds may also occur in the gas phase.
Co-reporter:Fanny Widjaja;Zhicheng Jin;John J. Nash
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 4) pp:469-480
Publication Date(Web):2013 April
DOI:10.1007/s13361-012-0543-3
The reactivity of the three distonic isomers of the pyridine radical cation toward tetrahydrofuran is compared in solution and in the gas phase. In solution, the distonic ions were generated by UV photolysis at 300 nm from iodo-precursors in acidic 50:50 tetrahydrofuran/water solutions. In the gas phase, the ions were generated by collisionally activated dissociation (CAD) of protonated iodo-precursors in an FT-ICR mass spectrometer, as described in the literature. The same major reaction, hydrogen atom abstraction, was observed in solution and in the gas phase. Attempts to cleave the iodine atom from the 2-iodopyridinium cation in the gas phase and in solution yielded the 2-pyridyl cation in addition to the desired 2-dehydropyridinium cation. In the gas phase, this ion was ejected prior to the examination of the desired ion’s chemical properties. This was not possible in solution. This study suggests that solvation effects are not significant for radical reactions of charged radicals. On the other hand, the even-electron ion studied, the 2-pyridyl cation, shows substantial solvation effects. For example, in solution, the 2-pyridyl cation forms a stable adduct with tetrahydrofuran, whereas in the gas phase, only addition/elimination reactions were observed.
Co-reporter:Nelson R. Vinueza, Vanessa A. Gallardo, John F. Klimek, Nicholas C. Carpita, Hilkka I. Kenttämaa
Fuel 2013 Volume 105() pp:235-246
Publication Date(Web):March 2013
DOI:10.1016/j.fuel.2012.08.012
The ability to characterize oligosaccharides directly in complex mixtures would greatly benefit many research efforts focused on the development of biofuels from lignocellulosic biomass. We report here on the utility of chloride anion attachment in atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI) tandem mass spectrometry for MW determination and structural elucidation of several mono-, di- and oligosaccharides directly without sample pretreatment or derivatization. Chloride anion forms stable adducts with these sugars upon ESI or APCI, which can be readily identified due to the chlorine isotope pattern. Fragment anions or other product anions are usually not observed, which is beneficial for mixture analysis. Upon collisional activation in MS2 experiments, these adducts readily lose HCl, which helps verify the molecular weight of each analyte. Isolating the resulting anion and subjecting it to further collision-activated dissociation experiments (MSn; n = 3–4) until no ion signal remains yields useful structural information. Examination of equimolar mixtures of mono-, di- and oligosaccharides and mixtures of enzymatically digested biomass demonstrates the ability of this methodology to ionize all the components of the mixtures without bias and with nearly equal efficiency.Highlights► We have developed a method for the analysis of cellulose degradation products. ► Chloride attachment in atmospheric pressure gently ionizes oligosaccharides. ► Tandem mass spectrometry provides valuable structural information.
Co-reporter:Vanessa A. Gallardo ; Bartłomiej J. Jankiewicz ; Nelson R. Vinueza ; John J. Nash ;Hilkka I. Kenttämaa
Journal of the American Chemical Society 2012 Volume 134(Issue 4) pp:1926-1929
Publication Date(Web):January 2, 2012
DOI:10.1021/ja209068z
The 2,4,6-tridehydropyridine radical cation, an analogue of the elusive 1,2,3,5-tetradehydrobenzene, was generated in the gas phase and its reactivity examined. Surprisingly, the tetraradical was found not to undergo radical reactions. This behavior is rationalized by resonance structures hindering fast radical reactions. This makes the cation highly electrophilic, and it rapidly reacts with many nucleophiles by quenching the N–C ortho-benzyne moiety, thereby generating a relatively unreactive meta-benzyne analogue.
Co-reporter:Benjamin C. Owen, Laura J. Haupert, Tiffany M. Jarrell, Christopher L. Marcum, Trenton H. Parsell, Mahdi M. Abu-Omar, Joseph J. Bozell, Stuart K. Black, and Hilkka I. Kenttämaa
Analytical Chemistry 2012 Volume 84(Issue 14) pp:6000
Publication Date(Web):June 13, 2012
DOI:10.1021/ac300762y
In the search for a replacement for fossil fuel and the valuable chemicals currently obtained from crude oil, lignocellulosic biomass has become a promising candidate as an alternative biorenewable source for crude oil. Hence, many research efforts focus on the extraction, degradation, and catalytic transformation of lignin, hemicellulose, and cellulose. Unfortunately, these processes result in the production of very complex mixtures. Further, while methods have been developed for the analysis of mixtures of oligosaccharides, this is not true for the complex mixtures generated upon degradation of lignin. For example, high-performance liquid chromatography/multiple stage tandem mass spectrometry (HPLC/MSn), a tool proven to be invaluable in the analysis of complex mixtures derived from many other biopolymers, such as proteins and DNA, has not been implemented for lignin degradation products. In this study, we have developed an HPLC separation method for lignin degradation products that is amenable to negative-ion-mode electrospray ionization (ESI doped with NaOH), the best method identified thus far for ionization of lignin-related model compounds without fragmentation. The separated and ionized compounds are then analyzed by MS3 experiments to obtain detailed structural information while simultaneously performing high-resolution measurements to determine their elemental compositions in the two parts of a commercial linear quadrupole ion trap/Fourier-transform ion cyclotron resonance mass spectrometer. A lignin degradation product mixture was analyzed using this method, and molecular structures were proposed for some components. This methodology significantly improves the ability to analyze complex product mixtures that result from degraded lignin.
Co-reporter:Mingkun Fu, Penggao Duan, Jinshan Gao and Hilkka I. Kenttämaa
Analyst 2012 vol. 137(Issue 24) pp:5720-5722
Publication Date(Web):12 Oct 2012
DOI:10.1039/C2AN35986C
A mass spectrometric method utilizing gas-phase ion–molecule reactions of 1-butanethiol and di-tert-butyl peroxide has been developed for the differentiation of primary, secondary and tertiary hydroxyl functionalities in protonated analytes in a FT-ICR mass spectrometer.
Co-reporter:Thomas N. Loegel, Neil D. Danielson, David J. Borton, Matthew R. Hurt, and Hilkka I. Kenttämaa
Energy & Fuels 2012 Volume 26(Issue 5) pp:2850-2857
Publication Date(Web):April 23, 2012
DOI:10.1021/ef201919x
The use of a 4.6 × 250 mm, 5 μm cyanopropyl column is effective for the liquid chromatography (LC) separation of asphaltenes with sequential ultraviolet (UV) and florescence detection. The mobile-phase composition is an optimized gradient from acetonitrile (MeCN) and water to N-methyl-2-pyrrolidone (NMP) and tetrahydrofuran (THF). A low flow rate of 0.5 mL min–1 is used to maintain lower operating pressure to minimize aggregate formation. Using a 0.02 g L–1 asphaltene sample for preliminary optimization, three peaks, with two partially resolved, are evident in the fluorescence chromatogram. The UV chromatogram revealed an extra weakly retained peak, suggesting aggregates that quench fluorescence. Aggregation of asphaltenes increases with time up to about 10 h and is dependent upon the choice of sample solvent. On the basis of the reversed-phase mobile-phase gradient, the relative polarity of the peaks from least to most retained can be estimated over the polarity index (P′) range from about 6.3–4.3 on a scale of 0.1 for hexane (least polar) to 10.6 for water (most polar). The sample concentration is increased to 1 g L–1 for separation and collection of 12 fractions. Selected fractions are subjected to characterization using atmospheric pressure chemical ionization mass spectrometry (APCI–MS) using a linear quadrupole ion trap (LQIT). The variation of the molecular-weight distribution of the asphaltenes for the 12 fractions is fairly constant, indicating that the retention mechanism is not controlled by size exclusion but likely a partitioning/adsorption mechanism.
Co-reporter:Lucas M. Amundson, Vanessa A. Gallardo, Nelson R. Vinueza, Benjamin C. Owen, Jennifer N. Reece, Steven C. Habicht, Mingkun Fu, Ryan C. Shea, Allen B. Mossman, and Hilkka I. Kenttämaa
Energy & Fuels 2012 Volume 26(Issue 5) pp:2975-2989
Publication Date(Web):March 26, 2012
DOI:10.1021/ef2019098
A tandem mass spectrometric method using a commercial linear quadrupole ion trap (LQIT) mass spectrometer and another LQIT coupled with a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer is described for the identification and counting of different oxygen-containing functionalities and alkyl groups in unknown aromatic analytes. A total of 64 aromatic model compounds were evaporated and ionized via positive-mode atmospheric pressure chemical ionization (APCI). Ionization of the model compounds primarily results in the formation of protonated molecules, [M + H]+. In some cases, the molecular radical cation, [M]+ •, and/or a fragment ion, [M – H]+, are formed instead. Only in one case, no ions were observed near the m/z value of the molecular ion, and the ion with the greatest m/z value is a fragment ion, [M + H – H2O]+. Once ionized, the ions were subjected to multiple isolation and collision-activated dissociation (CAD) events until no more fragmentation was observed (up to MS5). In most cases, all functionalities were sequentially cleaved, one or more at a time, by the CAD events. The type of neutral molecule cleaved and the number of times that it was cleaved facilitate the identification and counting of the functionalities. The method was successfully used in concert with high-performance liquid chromatography (HPLC). The HPLC retention times offer further structural information for the analytes. This methodology benefits the chemical, pharmaceutical, and biofuels industries by facilitating the identification of previously unknown compounds directly in complex mixtures, such as crude products of chemical processes, drug metabolites, and lignin degradation products.
Co-reporter:Bartłomiej J. Jankiewicz, Jinshan Gao, Jennifer N. Reece, Nelson R. Vinueza, Padmaja Narra, John J. Nash, and Hilkka I. Kenttämaa
The Journal of Physical Chemistry A 2012 Volume 116(Issue 12) pp:3089-3093
Publication Date(Web):February 21, 2012
DOI:10.1021/jp2101557
Recent studies have shown that the reactivity of the 4-dehydropyridinium cation significantly differs from the reactivities of its isomers toward tetrahydrofuran. While only hydrogen atom abstraction was observed for the 2- and 3-dehydropyridinium cations, nonradical reactions were observed for the 4-isomer. In order to learn more about these reactions, the gas-phase reactivities of the 4-dehydropyridinium cation and several of its derivatives toward tetrahydrofuran were investigated in a Fourier transform ion electron resonance mass spectrometer. Both radical and nonradical reactions were observed for most of these positively charged radicals. The major parameter determining whether nonradical reactions occur was found to be the electron affinity of the radicals—only those with relatively high electron affinities underwent nonradical reactions. The reactivities of the monoradicals are also affected by hydrogen bonding and steric effects.
Co-reporter:Dr. Nelson R. Vinueza;Enada F. Archibold;Dr. Bart&x142;omiej J. Jankiewicz;Vanessa A. Gallardo;Dr. Steven C. Habicht;Mohammad Sabir Aqueel;Dr. John J. Nash; Hilkka I. Kenttämaa
Chemistry - A European Journal 2012 Volume 18( Issue 28) pp:8692-8698
Publication Date(Web):
DOI:10.1002/chem.201103628
Abstract
The chemical properties of a 1,8-didehydronaphthalene derivative, the 4,5-didehydroisoquinolinium cation, were examined in the gas phase in a dual-cell Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer. This is an interesting biradical because it has two radical sites in close proximity, yet their coupling is very weak. In fact, the biradical is calculated to have approximately degenerate singlet and triplet states. This biradical was found to exclusively undergo radical reactions, as opposed to other related biradicals with nearby radical sites. The first bond formation occurs at the radical site in the 4-position, followed by that in the 5-position. The proximity of the radical sites leads to reactions that have not been observed for related mono- or biradicals. Interestingly, some ortho-benzynes have been found to yield similar products. Since ortho-benzynes do not react via radical mechanisms, these products must be especially favorable thermodynamically.
Co-reporter:Dr. Bart&x142;omiej J. Jankiewicz;Dr. Nelson R. Vinueza;Dr. Jennifer N. Reece;Young C. Lee;Peggy Williams;Dr. John J. Nash; Hilkka I. Kenttämaa
Chemistry - A European Journal 2012 Volume 18( Issue 3) pp:969-974
Publication Date(Web):
DOI:10.1002/chem.201102641
Abstract
The reactivity of 3-hydroxy-2,4,6-tridehydropyridinium cation was found to be drastically different from the reactivity of 2,4,6-tridehydropyridinium cation. While the latter triradical reacts with tetrahydrofuran, dimethyl disulfide and ally iodide via three consecutive atom or group abstractions, the former triradical exhibits this behavior only with tetrahydrofuran. Only a single atom or group abstraction was observed for the 3-hydroxy-2,4,6-tridehydropyridinium cation upon interaction with dimethyl disulfide and allyl iodide. This change in reactivity is caused by the hydroxyl group that strengthens the interactions between the two radical sites adjacent to it, thus reducing their reactivity. This explanation is supported by the observation of similar behavior for related biradicals.
Co-reporter:Ryan J. Eismin;Mingkun Fu;Sonoeun Yem
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 1) pp:12-22
Publication Date(Web):2012 January
DOI:10.1007/s13361-011-0249-y
A mass spectrometric method has been delineated for the identification of the epoxide functionalities in unknown monofunctional analytes. This method utilizes gas-phase ion/molecule reactions of protonated analytes with neutral trimethyl borate (TMB) followed by collision-activated dissociation (CAD) in an ion trapping mass spectrometer (tested for a Fourier-transform ion cyclotron resonance and a linear quadrupole ion trap). The ion/molecule reaction involves proton transfer from the protonated analyte to TMB, followed by addition of the analyte to TMB and elimination of methanol. Based on literature, this reaction allows the general identification of oxygen-containing analytes. Vinyl and phenyl epoxides can be differentiated from other oxygen-containing analytes, including other epoxides, based on the loss of a second methanol molecule upon CAD of the addition/methanol elimination product. The only other analytes found to undergo this elimination are some amides but they also lose O = B-R (R = group bound to carbonyl), which allows their identification. On the other hand, other epoxides can be differentiated from vinyl and phenyl epoxides and from other monofunctional analytes based on the loss of (CH3O)2BOH or formation of protonated (CH3O)2BOH upon CAD of the addition/methanol elimination product. For propylene oxide and 2,3-dimethyloxirane, the (CH3O)2BOH fragment is more basic than the hydrocarbon fragment, and the diagnostic ion (CH3O)2BOH2+ is formed. These reactions involve opening of the epoxide ring. The only other analytes found to undergo (CH3O)2BOH elimination are carboxylic acids, but they can be differentiated from the rest based on several published ion/molecule reaction methods. Similar results were obtained in the Fourier-transform ion cyclotron resonance and linear quadrupole ion trap mass spectrometer.
Co-reporter:Jinshan Gao;Benjamin C. Owen
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 5) pp:816-822
Publication Date(Web):2012 May
DOI:10.1007/s13361-012-0347-5
Saturated and unsaturated, linear, branched, and cyclic hydrocarbons, as well as polyaromatic and heteroaromatic hydrocarbons, were successfully ionized by atmospheric pressure chemical ionization (APCI) using small hydrocarbons as reagents in a linear quadrupole ion trap (LQIT) mass spectrometer. Pentane was proved to be the best reagent among the hydrocarbon reagents studied. This ionization method generated different types of abundant ions (i.e., [M + H]+, M+•, [M – H]+ and [M – 2H]+ •), with little or no fragmentation. The radical cations can be differentiated from the even-electron ions by using dimethyl disulfide, thus facilitating molecular weight (MW) determination. While some steroids and lignin monomer model compounds, such as androsterone and 4-hydroxy-3-methoxybenzaldehyde, also formed abundant M+• and [M + H]+ ions, this was not true for all of them. Analysis of two known mixtures as well as a base oil sample demonstrated that each component of the known mixtures could be observed and that a correct MW distribution was obtained for the base oil. The feasibility of using this ionization method on the chromatographic time scale was demonstrated by using high-performance liquid chromatography (HPLC) with hexane as the mobile phase (and APCI reagent) to separate an artificial mixture prior to mass spectrometric analysis.
Co-reporter:George O. Pates ; Leonard Guler ; John J. Nash ;Hilkka I. Kenttämaa
Journal of the American Chemical Society 2011 Volume 133(Issue 24) pp:9331-9342
Publication Date(Web):May 25, 2011
DOI:10.1021/ja111280t
The reactivity of 10 charged phenyl radicals toward several amino acids was examined in the gas phase in a dual-cell Fourier transform ion cyclotron resonance mass spectrometer. All radicals abstract a hydrogen atom from the amino acids, as expected. The most electrophilic radicals (with the greatest calculated vertical electron affinities (EA) at the radical site) also react with these amino acids via NH2 abstraction (a nonradical nucleophilic addition–elimination reaction). Both the radical (hydrogen atom abstraction) and nonradical (NH2 abstraction) reaction efficiencies were found to increase with the electrophilicity (EA) of the radical. However, NH2 abstraction is more strongly influenced by EA. In contrast to an earlier report, the ionization energies of the amino acids do not appear to play a general reactivity-controlling role. Studies using several partially deuterium-labeled amino acids revealed that abstraction of a hydrogen atom from the α-carbon is only preferred for glycine; for the other amino acids, a hydrogen atom is preferentially abstracted from the side chain. The electrophilicity of the radicals does not appear to have a major influence on the site from which the hydrogen atom is abstracted. Hence, the regioselectivity of hydrogen atom abstraction appears to be independent of the structure of the radical but dependent on the structure of the amino acid. Surprisingly, abstraction of two hydrogen atoms was observed for the N-(3-nitro-5-dehydrophenyl)pyridinium radical, indicating that substituents on the radical not only influence the EA of the radical but also can be involved in the reaction. In disagreement with an earlier report, proline was found to display several unprecedented reaction pathways that likely do not proceed via a radical mechanism but rather by a nucleophilic addition–elimination mechanism. Both NH2 and 15NH2 groups were abstracted from lysine labeled with 15N on the side chain, indicating that NH2 abstraction occurs both from the amino terminus and from the side chain. Quantum chemical calculations were employed to obtain insights into some of the reaction mechanisms.
Co-reporter:Fanny Widjaja ; Zhicheng Jin ; John J. Nash ;Hilkka I. Kenttämaa
Journal of the American Chemical Society 2011 Volume 134(Issue 4) pp:2085-2093
Publication Date(Web):December 7, 2011
DOI:10.1021/ja207899j
To directly compare the reactivity of positively charged carbon-centered aromatic σ-radicals toward methanol in solution and in the gas phase, the 2-, 3-, and 4-dehydropyridinium cations (distonic isomers of the pyridine radical cation) were generated by ultraviolet photolysis of the corresponding iodo precursors in a mixture of water and methanol at varying pH. The reaction mixtures were analyzed by using liquid chromatography/mass spectrometry. Hydrogen atom abstraction was the only reaction observed for the 3- and 4-dehydropyridinium cations (and pyridines) in solution. This also was the major reaction observed earlier in the gas phase. Depending on the pH, the hydrogen atom can be abstracted from different molecules (i.e., methanol or water) and from different sites (in methanol) by the 3- and 4-dehydropyridinium cations/pyridines in solution. In the pH range 1–4, the methyl group of methanol is the main hydrogen atom donor site for both 3- and 4-dehydropyridinium cations (just like in the gas phase). At higher pH, the hydroxyl groups of water and methanol also act as hydrogen atom donors. This finding is rationalized by a greater abundance of the unprotonated radicals that preferentially abstract hydrogen atoms from the polar hydroxyl groups. The percentage yield of hydrogen atom abstraction by these radicals was found to increase with lowering the pH in the pH range 1.0–3.2. This pH effect is rationalized by polar effects: the lower the pH, the greater the fraction of protonated (more polar) radicals in the solution. This finding is consistent with previous results obtained in the gas phase and suggests that gas-phase studies can be used to predict solution reactivity, but only as long as the same reactive species is studied in both experiments. This was found not to be the case for the 2-iodopyridinium cation. Photolysis of this precursor in solution resulted in the formation of two major addition products, 2-hydroxy- and 2-methoxypyridinium cations, in addition to the hydrogen atom abstraction product. These addition products were not observed in the earlier gas-phase studies on 2-dehydropyridinium cation. Their observation in solution is explained by the formation of another reactive intermediate, the 2-pyridylcation, upon photolysis of 2-iodopyridinium cation (and 2-iodopyridine). The same intermediate was observed in the gas phase but it was removed before examining the reactions of the desired radical, 2-dehydropyridinium cation (which cannot be done in solution).
Co-reporter:Lucas M. Amundson, Ryan J. Eismin, Jennifer N. Reece, Mingkun Fu, Steven C. Habicht, Allan B. Mossman, Ryan C. Shea, and Hilkka I. Kenttämaa
Energy & Fuels 2011 Volume 25(Issue 7) pp:3212-3222
Publication Date(Web):April 15, 2011
DOI:10.1021/ef200141w
Identification and counting of different oxygen-containing functional groups in 40 small aromatic analytes, including a lignin monomer, was explored using a linear quadrupole ion trap (LQIT) mass spectrometer. The analytes were evaporated and ionized by negative-mode electrospray ionization (ESI). In an effort to cleave off all of the functionalities, one at a time, the deprotonated analytes were then subjected to multiple consecutive collision-activated dissociation (CAD) events until no more fragmentation was observed (up to MS5). In most cases, the number and types of functionalities could be determined. This approach was demonstrated to be feasible on the high-performance liquid chromatographic (HPLC) time scale. Hence, valuable structural information can be obtained for previously unknown aromatic analytes directly in complex mixtures, such as lignin degradation products.
Co-reporter:Zhicheng Jin, Shivani Daiya, Hilkka I. Kenttämaa
International Journal of Mass Spectrometry 2011 Volume 301(1–3) pp:234-239
Publication Date(Web):30 March 2011
DOI:10.1016/j.ijms.2010.11.001
Laser-induced acoustic desorption (LIAD) combined with ClMn(H2O)+ chemical ionization (CI) was tested for the analysis of nonpolar lipids and selected steroids in a Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR). The nonpolar lipids studied, cholesterol, 5α-cholestane, cholesta-3,5-diene, squalene, and β-carotene, were found to solely form the desired water replacement product (adduct-H2O) upon reaction with the ClMn(H2O)+ ions. The steroids, androsterone, dehydroepiandrosterone (DHEA), estrone, estradiol, and estriol, also form abundant adduct-H2O ions, but less abundant adduct-2H2O ions were also observed. Neither (+)APCI nor (+)ESI can ionize the saturated hydrocarbon lipid, cholestane. APCI successfully ionizes the unsaturated hydrocarbon lipids to form exclusively the intact protonated analytes. However, it causes extensive fragmentation for cholesterol and the steroids. The worst case is cholesterol that does not produce any stable protonated molecules. On the other hand, ESI cannot ionize any of the hydrocarbon analytes, saturated or unsaturated. However, ESI can be used to protonate the oxygen-containing analytes with substantially less fragmentation than for APCI in all cases except for cholesterol and estrone. In conclusion, LIAD/ClMn(H2O)+ chemical ionization is superior over APCI and ESI for the mass spectrometric characterization of underivatized nonpolar lipids and steroids.Graphical abstractResearch highlights▶ Different ionization methods were compared for the mass spectrometric analysis of nonpolar lipids. ▶ LIAD/CI was found to be superior to ESI and APCI. ▶ A stable pseudo-molecular ion was formed for all analytes, including saturated hydrocarbons.
Co-reporter:Jayalakshmi Somuramasami;Penggao Duan
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 6) pp:1040-1051
Publication Date(Web):2011 June
DOI:10.1007/s13361-011-0099-7
Several lignin model compounds were examined to test whether gas-phase ion–molecule reactions of trimethylborate (TMB) in a FTICR can be used to differentiate the ortho-, meta-, and para-isomers of protonated aromatic compounds, such as those formed during degradation of lignin. All three regioisomers could be differentiated for methoxyphenols and hydroxyphenols. However, only the differentiation of the ortho-isomer from the meta- and para-isomers was possible for hydroxyacetophenones and hydroxybenzoic acids. Consideration of the previously reported proton affinities at all basic sites in the isomeric hydroxyphenols, and the calculated proton affinities at all basic sites in the three methoxyphenol isomers, revealed that the proton affinities of the analytes relative to that of TMB play an important role in determining whether and how they react with TMB. The loss of two methanol molecules (instead of one) from the adducts formed with TMB either during ion–molecule reactions, or during sustained-off resonance irradiated collision-activated dissociation of the ion–molecule reaction products, revealed the presence of two functionalities in almost all the isomers. This finding supports earlier results suggesting that TMB can be used to count the functionalities in unknown oxygen-containing analytes.
Co-reporter:Jinshan Gao;David J. Borton II
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 3) pp:531-538
Publication Date(Web):2011 March
DOI:10.1007/s13361-010-0048-x
Laser-induced acoustic desorption (LIAD) was successfully coupled to a conventional atmospheric pressure chemical ionization (APCI) source in a commercial linear quadrupole ion trap mass spectrometer (LQIT). Model compounds representing a wide variety of different types, including basic nitrogen and oxygen compounds, aromatic and aliphatic compounds, as well as unsaturated and saturated hydrocarbons, were tested separately and as a mixture. These model compounds were successfully evaporated into the gas phase by using LIAD and then ionized by using APCI with different reagents. From the four APCI reagent systems tested, neat carbon disulfide provided the best results. The mixture of methanol and water produced primarily protonated molecules, as expected. However, only the most basic compounds yielded ions under these conditions. In sharp contrast, using APCI with either neat benzene or neat carbon disulfide as the reagent resulted in the ionization of all the analytes studied to predominantly yield stable molecular ions. Benzene yielded a larger fraction of protonated molecules than carbon disulfide, which is a disadvantage. A similar but minor amount of fragmentation was observed for these two reagents. When the experiment was performed without a liquid reagent (nitrogen gas was the reagent), more fragmentation was observed. Analysis of a known mixture as well as a petroleum cut was also carried out. In summary, the new experiment presented here allows the evaporation of thermally labile compounds, both polar and nonpolar, without dissociation or aggregation, and their ionization to predominantly form stable molecular ions.
Co-reporter:Steve C. Habicht;Nelson R. Vinueza
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 3) pp:520-530
Publication Date(Web):2011 March
DOI:10.1007/s13361-010-0050-3
We report here a comparison of the use of diagnostic ion–molecule reactions for the identification of oxygen-containing functional groups in Fourier-transform ion cyclotron resonance (FTICR) and linear quadrupole ion trap (LQIT) mass spectrometers. The ultimate goal of this research is to be able to identify functionalities in previously unknown analytes by using many different types of mass spectrometers. Previous work has focused on the reactions of various boron reagents with protonated oxygen-containing analytes in FTICR mass spectrometers. By using a LQIT modified to allow the introduction of neutral reagents into the helium buffer gas, this methodology has been successfully implemented to this type of an ion trap instrument. The products obtained from the reactions of trimethyl borate (TMB) with various protonated analytes are compared for the two instruments. Finally, the ability to integrate these reactions into LC-MS experiments on the LQIT is demonstrated.
Co-reporter:Lucas M. Amundson;Benjamin C. Owen
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 4) pp:670-682
Publication Date(Web):2011 April
DOI:10.1007/s13361-011-0079-y
Positive-mode atmospheric pressure chemical ionization tandem mass spectrometry (APCI-MSn) was tested for the differentiation of regioisomeric aromatic ketocarboxylic acids. Each analyte forms exclusively an abundant protonated molecule upon ionization via positive-mode APCI in a commercial linear quadrupole ion trap (LQIT) mass spectrometer. Energy-resolved collision-activated dissociation (CAD) experiments carried out on the protonated analytes revealed fragmentation patterns that varied based on the location of the functional groups. Unambiguous differentiation between the regioisomers was achieved in each case by observing different fragmentation patterns, different relative abundances of ion-molecule reaction products, or different relative abundances of fragment ions formed at different collision energies. The mechanisms of some of the reactions were examined by H/D exchange reactions and molecular orbital calculations.
Co-reporter:Steven C. Habicht, Lucas M. Amundson, Penggao Duan, Nelson R. Vinueza and Hilkka I. Kenttämaa
Analytical Chemistry 2010 Volume 82(Issue 2) pp:608
Publication Date(Web):December 15, 2009
DOI:10.1021/ac901943k
In recent years, laser-induced acoustic desorption (LIAD) coupled with a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer has been demonstrated to provide a valuable technique for the analysis of a wide variety of nonvolatile, thermally labile compounds, including analytes that could not previously be analyzed by mass spectrometry. Although FT-ICR instruments are very powerful, they are also large and expensive and, hence, mainly used as research instruments. In contrast, linear quadrupole ion trap (LQIT) mass spectrometers are common due to several qualities that make these instruments attractive for both academic and industrial settings, such as high sensitivity, large dynamic range, and experimental versatility. Further, the relatively small size of the instruments, comparatively low cost, and the lack of a magnetic field provide some distinct advantages over FT-ICR instruments. Hence, we have coupled the LIAD technique with a commercial LQIT, the Thermo Fischer Scientific LTQ mass spectrometer. The LQIT was modified for a LIAD probe by outfitting the removable back plate of the instrument with a 6 in. ConFlat flange (CFF) port, gate valve, and sample lock. Reagent ions were created using the LQIT’s atmospheric pressure ionization source and trapped in the mass analyzer for up to 10 s to allow chemical ionization reactions with the neutral molecules desorbed via LIAD. These initial experiments focused on demonstrating the feasibility of performing LIAD in the LQIT. Hence, the results are compared to those obtained using an FT-ICR mass spectrometer. Despite the lower efficiency in the transfer of desorbed neutral molecules into the ion trap, and the smaller maximum number of available laser pulses, the intrinsically higher sensitivity of the LQIT resulted in a higher sensitivity relative to the FT-ICR.
Co-reporter:David S. Pinkston, Penggao Duan, Vanessa A. Gallardo, Mingkun Fu, Steven C. Habicht and Hilkka I. Kenttämaa
Energy & Fuels 2010 Volume 24(Issue 5) pp:3119-3124
Publication Date(Web):May 5, 2010
DOI:10.1021/ef100244y
Chemical ionization (CI) using the [ClMn(H2O)]+ reagent ion combined with collision-activated dissociation is demonstrated to allow the differentiation of isomeric alkanes (C8H18 and C9H20) and alkenes (C8H16) in a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. The [ClMn(H2O)]+ ion reacts with each isomeric hydrocarbon molecule to produce a water replacement product [hence providing molecular weight (MW) information], with no accompanying fragmentation. Differentiation of the isomers is achieved on the basis of the different fragmentation patterns observed upon sustained off-resonance irradiation collision-activated dissociation (SORI−CAD) of the isolated water replacement products. Upon activation, the weak bond of the hydrocarbon to ClMn+ is broken. ClMn+ is the major fragment ion for linear alkanes. However, for branched alkanes and all alkenes, the leaving ClMn+ abstracts a weakly bound hydride, an alkyl anion, or an alkene from the hydrocarbon to generate diagnostic carbocation fragment ions. Because the experiment involves isolation of the ionized analytes prior to structural characterization (MS/MS), it is well-suited for direct analysis of hydrocarbon mixtures.
Co-reporter:David Borton II, David S. Pinkston, Matthew R. Hurt, Xiaoli Tan, Khalid Azyat, Alexander Scherer, Rik Tykwinski, Murray Gray, Kuangnan Qian, and Hilkka I. Kenttämaa
Energy & Fuels 2010 Volume 24(Issue 10) pp:5548-5559
Publication Date(Web):September 20, 2010
DOI:10.1021/ef1007819
We report here an examination of the mass spectrometric fragmentation behavior of molecular ions generated (and excited) by electron ionization (EI) from several asphaltene model compounds simulating both the island and archipelago structural models. This behavior is compared to that of protonated molecules generated from the same compounds by atmospheric pressure chemical ionization (APCI) and excited by collision-activated dissociation (CAD). The fragmentation behavior of the protonated molecules and molecular ions is surprisingly similar. Both types of ions yielded distinct fragmentation patterns for both types of model compounds. Ions derived from the island-type model compounds fragment predominantly by losing their alkyl chains (with either all carbons or all but one), one after another, which allows for the identification of the chain lengths and counting the number of chains. Increasing the length of the alkyl chains reduces the extent of spontaneous fragmentation occurring upon EI, likely because of more efficient cooling of the fragmenting ions via emission of infrared (IR) light made possible by the reduced fragmentation rates of the longer chains. Ions derived from the archipelago model compounds with ethylene bridges connecting two or three aromatic cores (without alkyl side chains) readily undergo cleavages in these bridges. Increasing the length of the alkyl chain between the aromatic cores reduces the extent of fragmentation caused by EI. Similarly, the addition of long external alkyl chains to archipelago model compounds with an ethylene bridging two aromatic cores greatly hinders fragmentation upon EI. When these molecules are protonated and subjected to high-energy CAD, they appear to fragment almost randomly but, nevertheless, indicating some preference for cleavages of the bonds in the chain connecting the aromatic cores. A comparison of these findings to the fragmentation patterns observed for protonated asphaltenes indicates that the asphaltene molecules studied are likely composed of many isomeric and isobaric molecules. Each may contain several aromatic rings and a distribution of mostly aliphatic alkyl chains (and possibly naphthenic rings) ranging in size from 1 to at least 14 carbons, several containing methyl branching at the α carbons. The results do not allow for the unambiguous differentiation between island- and archipelago-type structures, although they are in a better agreement with the island model.
Co-reporter:Steven C. Habicht, Penggao Duan, Nelson R. Vinueza, Mingkun Fu, Hilkka I. Kenttämaa
Journal of Pharmaceutical and Biomedical Analysis 2010 51(4) pp: 805-811
Publication Date(Web):
DOI:10.1016/j.jpba.2009.09.047
Co-reporter:Anthony Adeuya, John J. Nash, and Hilkka I. Kenttämaa
The Journal of Physical Chemistry A 2010 Volume 114(Issue 49) pp:12851-12857
Publication Date(Web):November 16, 2010
DOI:10.1021/jp107254k
The reactions of several substituted, positively charged dehydropyridinium cations with cyclohexane, methanol, and tetrahydrofuran have been examined in a Fourier-transform ion cyclotron resonance mass spectrometer. All of the charged monoradicals react with the neutral reagents exclusively via hydrogen atom abstraction. For cyclohexane, there is a good correlation between the reaction efficiencies and the calculated electron affinities at the radical sites; that is, the greater the electron affinity of the charged monoradical at the radical site, the faster the reaction. The reaction efficiencies with methanol and tetrahydrofuran, however, do not correlate with the calculated electron affinities. Density functional theory (DFT) calculations indicate that for these reagents a stabilizing hydrogen bonding interaction exists in the hydrogen atom abstraction transition states for some of the charged monoradicals but not for others. At both the MPW1K and G3MP2B3 levels of theory, there is a good correlation between the calculated activation enthalpies and the observed reaction efficiencies, although the G3MP2B3 method provides a slightly better correlation than the MPW1K method. The extent of enhancement in the reaction efficiencies caused by the hydrogen bonding interactions parallels the calculated hydrogen bond lengths in the transition states.
Co-reporter:Jayalakshmi Somuramasami;Brian E. Winger
Journal of The American Society for Mass Spectrometry 2010 Volume 21( Issue 5) pp:773-784
Publication Date(Web):2010 May
DOI:10.1016/j.jasms.2009.11.008
A mass spectrometric method has been developed for the identification of carbonyl and hydroxyl functional groups, as well as for counting the functional groups, in previously unknown protonated bifunctional oxygen-containing analytes. This method utilizes solution reduction before mass spectrometric analysis to convert the carbonyl groups to hydroxyl groups. Gas-phase ion-molecule reactions of the protonated reduced analytes with neutral trimethylborate (TMB) in a FT-ICR mass spectrometer give diagnostic product ions. The reaction sequence likely involves three consecutive steps, proton abstraction from the protonated analyte by TMB, addition of the neutral analyte to the boron reagent, and elimination of a neutral methanol molecule. The number of methanol molecules eliminated upon reactions with TMB reveals the number of hydroxyl groups in the analyte. Comparison of the reactions of the original and reduced analytes reveals the presence and number of carbonyl and hydroxyl groups in the analyte.
Co-reporter:Mingkun Fu;Sen Li;Enada Archibold
Journal of The American Society for Mass Spectrometry 2010 Volume 21( Issue 10) pp:1737-1752
Publication Date(Web):2010 October
DOI:10.1016/j.jasms.2010.06.010
Gas-phase reactivity of a positively charged aromatic σ,σ-biradical (N-methyl-6,8-didehydroquinolinium) was examined toward six aliphatic amino acids and 15 dipeptides by using Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR) and laser-induced acoustic desorption (LIAD). While previous studies have revealed that H-atom and NH2 abstractions dominate the reactions of related monoradicals with aliphatic amino acids and small peptides, several additional, unprecedented reaction pathways were observed for the reactions of the biradical. For amino acids, these are 2H-atom abstraction, H2O abstraction, addition — CO2, addition — HCOOH, and formation of a stable adduct. The biradical reacts with aliphatic dipeptides similarly as with aliphatic amino acids, but undergoes also one additional reaction pathway, addition/C-terminal amino acid elimination (addition — CO — NHCHRC). These reactions are initiated by H-atom abstraction by the biradical from the amino acid or peptide, or nucleophilic addition of an NH2 or a HO group of the amino acid or peptide at the radical site at C-6 in the biradical. Reactions of the unquenched C-8 radical site then yield the products not observed for related monoradicals. The biradical reacts with aromatic dipeptides with an aromatic ring in N-terminus (i.e., Tyr-Leu, Phe-Val, and Phe-Pro) similarly as with aliphatic dipeptides. However, for those aromatic dipeptides that contain an aromatic ring in the C-terminus (i.e., Leu-Tyr and Ala-Phe), one additional pathway, addition/N-terminal amino acid elimination (addition — CO — NHCHRN), was observed. This reaction is likely initiated by radical addition of the biradical at the aromatic ring in the C-terminus. Related monoradicals add to aromatic amino acids and small peptides, which is followed by Cα-Cβ bond cleavage, resulting in side-chain abstraction by the radical. For biradicals, with one unquenched radical site after the initial addition, the reaction ultimately results in the loss of the N-terminal amino acid. Similar to monoradicals, the C-S bond in amino acids and dipeptides was found to be especially susceptible to biradical attack.
Co-reporter:Steven C. Habicht;Nelson R. Vinueza
Journal of The American Society for Mass Spectrometry 2010 Volume 21( Issue 4) pp:559-563
Publication Date(Web):2010 April
DOI:10.1016/j.jasms.2009.12.015
We report here an automated method for the identification of N-oxide functional groups in drug metabolites by using the combination of liquid chromatography/tandem mass spectrometry (LC/MSn) based on ion-molecule reactions and collision-activated dissociation (CAD). Data-dependent acquisition, which has been readily utilized for metabolite characterization using CAD-based methods, is adapted for use with ion-molecule reaction-based tandem mass spectrometry by careful choice of select experimental parameters. Two different experiments utilizing ion-molecule reactions are demonstrated, data-dependent neutral gain MS3 and data-dependent neutral gain pseudo-MS3, both of which generate functional group selective mass spectral data in a single experiment and facilitate increased throughput in structural elucidation of unknown mixture components. Initial results have been generated by using an LC/MSn method based on ion-molecule reactions developed earlier for the identification of the N-oxide functional group in pharmaceutical samples, a notoriously difficult functional group to identify via CAD alone. Since commercial software and straightforward, external instrument modification are used, these experiments are readily adaptable to the industrial pharmaceutical laboratory.
Co-reporter:David S. Pinkston, Penggao Duan, Vanessa A. Gallardo, Steven C. Habicht, Xiaoli Tan, Kuangnan Qian, Murray Gray, Klaus Müllen and Hilkka I. Kenttämaa
Energy & Fuels 2009 Volume 23(Issue 11) pp:5564
Publication Date(Web):September 16, 2009
DOI:10.1021/ef9006005
Laser-induced acoustic desorption (LIAD)/electron ionization (EI) was used to study asphaltene model compounds and asphaltenes derived from North American crude oil in a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (MS). Successful desorption by LIAD of all model compounds (including a polyphenylated vanadoyl porphyrin) as intact neutral molecules into the mass spectrometer indicates that this method allows the evaporation of most if not all components of asphaltenes into mass spectrometers for further characterization. Electron ionization is a universal ionization method that ionizes all organic compounds. Hence, it is not surprising that all the model compounds studied were successfully ionized by using this method. Furthermore, this method yielded stable molecular ions for all model compounds studied. Because LIAD/EIMS provides MW information for these model compounds, this is almost certainly also true for all components of asphaltenes. Examination of asphaltene samples derived from North American crude oil by using this technique yielded a MW distribution of about 350−1050 Da and provided structural information for asphaltene components.
Co-reporter:Michael J. Yurkovich, Penggao Duan, Ryan C. Shea, Michael A. Watkins, Sarah M. Mandell, Eric M. Tippmann, Sen Jason Li, Matthew S. Platz, Hilkka I. Kenttämaa
International Journal of Mass Spectrometry 2009 Volume 287(1–3) pp:16-20
Publication Date(Web):15 October 2009
DOI:10.1016/j.ijms.2009.03.011
The reactivity of two metallated nitrenium ions toward various substrates was examined in the gas phase. The nitrenium ions were generated by a reaction of benzoyl azide with laser-ablated Mg+ or Cu+ in a Fourier transform ion cyclotron resonance mass spectrometer. The two nitrenium ions show drastically different reactivity. While the Mg-nitrenium ion reacts by radical mechanisms (e.g., H atom abstraction), the Cu-nitrenium ion follows non-radical pathways (e.g., metal ion transfer).Two metallated nitrenium ions show drastically different reactivity. While the Mg-nitrenium ion reacts by radical mechanisms, the Cu-nitrenium ion follows non-radical pathways.
Co-reporter:Mingkun Fu, Ryan J. Eismin, Penggao Duan, Sen Li, Hilkka I. Kenttämaa
International Journal of Mass Spectrometry 2009 Volume 282(Issue 3) pp:77-84
Publication Date(Web):1 May 2009
DOI:10.1016/j.ijms.2009.01.018
A mass spectrometric method is presented that facilitates the identification and differentiation of primary, secondary and tertiary amino functionalities in protonated monofunctional analytes. This method utilizes gas-phase ion–molecule reactions of protonated analytes with neutral hexamethylphosphoramide (HMPA) and diethylmethylphosphonate (DEMP) in a Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR). A variety of protonated analytes containing different functional groups, namely, amino, amido, N-oxide and various oxygen-containing functional groups, were examined to demonstrate that protonated primary and secondary amines can be identified and differentiated by reactions with HMPA and DEMP. However, differentiation of tertiary amines from some N-oxides requires additional experiments. First, protonated secondary and tertiary amines can be differentiated from protonated primary amines, amides and oxygen-containing functionalities, as well as from each other (but not from protonated N-oxides), by using HMPA. Protonated primary amines, amides, some N-oxides and oxygen-containing analytes, most with a proton affinity (PA) < 229 kcal/mol, only transfer a proton to HMPA (PA = 229 kcal/mol). In contrast, protonated secondary amines also form two stable hydrogen-bound adducts (MH+ + HMPA, MH+ + 2HMPA; M: amine), and tertiary amines and some N-oxides (with PA ≥ 222 kcal/mol) react with HMPA by forming just one stable hydrogen-bound adduct (MH+ + HMPA). Further, ion–molecule reactions with the other reagent, DEMP, allow the differentiation of protonated primary and secondary amines from tertiary amines and N-oxides and from protonated oxygen-containing analytes and amides. Protonated primary amines and secondary amines (most with PA ≥ 220 kcal/mol) react with DEMP (PA = 219 kcal/mol) by forming two stable hydrogen-bound adducts (MH+ + DEMP and MH+ + 2DEMP), while protonated oxygen-containing analytes and amides (with PA ≤ 221 kcal/mol) solely transfer a proton to DEMP. Protonated tertiary amines and N-oxides react yet differently, and yield only one stable hydrogen-bound adduct (MH+ + DEMP) with DEMP. Protonated N-oxides can be differentiated from protonated tertiary amines by using previously reported methods based on diagnostic ion–molecule reactions of protonated N-oxides with 2-methoxypropene, dimethyl disulfide and/or tri(dimethylamine)borane.Differentiation of a protonated primary and tertiary amine (M) based on their reactions with hexamethylphosphoramide (HMPA).
Co-reporter:Sen Li, Mingkun Fu, Steven C. Habicht, George O. Pates, John J. Nash and Hilkka I. Kenttämaa
The Journal of Organic Chemistry 2009 Volume 74(Issue 20) pp:7724-7732
Publication Date(Web):September 22, 2009
DOI:10.1021/jo901470f
Laser-induced acoustic desorption (LIAD) incorporated with Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR) has been utilized to investigate phenyl radical-induced damage to dipeptides in the gas phase. On the basis of the product branching ratios measured for the reactions of two different positively charged phenyl radicals with 17 different dipeptides, the overall order of susceptibility to attack of the different sites in the dipeptides was determined to be heteroaromatic side chain ≈ S atom in SCH3 group > H atom in SH group > H atom in CH group > aromatic side chain > S atom in SH group > NH2 in side chain > N-terminal NH2 > COOH in side chain ≈ C-terminal COOH. The amino acid sequence also influences the selectivity of these reactions. As expected, the ability of a phenyl radical to damage dipeptides increases as the electrophilicity of the phenyl radical increases.
Co-reporter:Anthony Adeuya, Jason M. Price, Bartłomiej J. Jankiewicz, John J. Nash and Hilkka I. Kenttämaa
The Journal of Physical Chemistry A 2009 Volume 113(Issue 49) pp:13663-13674
Publication Date(Web):November 10, 2009
DOI:10.1021/jp901380y
To explore the effects of the electronic nature of charged phenyl radicals on their reactivity, reactions of the three distonic isomers of n-dehydropyridinium cation (n = 2, 3, or 4) have been investigated in the gas phase by using Fourier-transform ion cyclotron resonance mass spectrometry. All three isomers react with cyclohexane, methanol, ethanol, and 1-pentanol exclusively via hydrogen atom abstraction and with allyl iodide mainly via iodine atom abstraction, with a reaction efficiency ordering of 2 > 3 > 4. The observed reactivity ordering correlates well with the calculated vertical electron affinities of the charged radicals (i.e., the higher the vertical electron affinity, the faster the reaction). Charged radicals 2 and 3 also react with tetrahydrofuran exclusively via hydrogen atom abstraction, but the reaction of 4 with tetrahydrofuran yields products arising from nonradical reactivity. The unusual reactivity of 4 is likely to result from the contribution of an ionized carbene-type resonance structure that facilitates nucleophilic addition to the most electrophilic carbon atom (C-4) in this charged radical. The influence of such a resonance structure on the reactivity of 2 is not obvious, and this may be due to stabilizing hydrogen-bonding interactions in the transition states for this molecule. Charged radicals 2 and 3 abstract a hydrogen atom from the substituent in both phenol and toluene, but 4 abstracts a hydrogen atom from the phenyl ring, a reaction that is unprecedented for phenyl radicals. Charged radical 4 reacts with tert-butyl isocyanide mainly by hydrogen cyanide (HCN) abstraction, whereas CN abstraction is the principal reaction for 2 and 3. The different reactivity observed for 4 (as compared to 2 and 3) is likely to result from different charge and spin distributions of the reaction intermediates for these charged radicals.
Co-reporter:Mingkun Fu;Penggao Duan;Sen Li
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 7) pp:1251-1262
Publication Date(Web):2009 July
DOI:10.1016/j.jasms.2009.02.020
A mass spectrometric method is presented for the identification of analytes with two basic functionalities and PA between 222 and 245 kcal/mol, including diamines. This method utilizes gas-phase ion-molecule reactions of protonated analytes with neutral 1,1-diethoxyethene (DEE) in a Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR). A variety of protonated mono-, bi-, and trifunctional analytes containing different functional groups, namely, amido, amino, N-oxide, hydroxy, carboxylic acid, keto, thio, thioether, alkene, phosphite, and phosphonate, were tested in the FT-ICR. The results demonstrate that basic protonated bifunctional compounds (PA between 222 and 245 kcal/mol) react selectively with DEE by forming a specific addition/elimination product ion (adduct - EtOH) (this product was also observed for lysine with three functionalities). The diagnostic reaction sequence involves proton transfer from the protonated analyte to the basic vinyl group in DEE, followed by addition of one of the functional groups of the analyte to the electrophilic α-carbon in protonated DEE. The next step involves proton transfer from this functionality to the other analyte functionality, followed by proton transfer to DEE and elimination of ethanol. Since the mechanism involves proton transfer between two functional groups of the analyte, the reaction does not occur for analytes where the two functionalities cannot be in close proximity (i.e., meta-phenylenediamine), and where no proton is available (i.e., dimethylaminoketone).
Co-reporter:Mingkun Fu, Penggao Duan, Sen Li, Steven C. Habicht, David S. Pinkston, Nelson R. Vinueza and Hilkka I. Kenttämaa
Analyst 2008 vol. 133(Issue 4) pp:452-454
Publication Date(Web):25 Feb 2008
DOI:10.1039/B801961D
A mass spectrometric method utilizing regioselective ion–molecule reactions has been developed for the differentiation of protonatedisomericaromatic diamines in FT-ICR, linear quadrupole ion trap and triple quadrupole mass spectrometers.
Co-reporter:Bart&x142;omiejJ. Jankiewicz;JenniferN. Reece;NelsonR. Vinueza;JohnJ. Nash Dr. ;HilkkaI. Kenttämaa
Angewandte Chemie International Edition 2008 Volume 47( Issue 51) pp:9860-9865
Publication Date(Web):
DOI:10.1002/anie.200802714
Co-reporter:Jayalakshmi Somuramasami, Penggao Duan, Michael A. Watkins, Brian E. Winger, Hilkka I. Kenttamaa
International Journal of Mass Spectrometry 2007 Volume 265(2–3) pp:359-371
Publication Date(Web):1 September 2007
DOI:10.1016/j.ijms.2007.06.005
A mass spectrometric method has been developed for the identification of functional groups in unknown bifunctional oxygen-containing compounds and for the identification and counting of the hydroxyl groups in polyols. This method utilizes gas-phase ion–molecule reactions of protonated analytes with neutral trimethylborate (TMB) in a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. The diagnostic reaction sequence involves proton abstraction from the protonated analyte by TMB, followed by the addition of the analyte to TMB and elimination of methanol. The functional groups in bifunctional oxygen-containing compounds are identified based on the total number of TMB molecules that have added to the protonated analyte and/or the number of methanol molecules lost, or by sustained off-resonance irradiation collision-activated dissociation (SORI-CAD) of the reaction products. The number of hydroxyl groups in polyols is revealed by the number of methanol molecules lost during their reactions with TMB. Reactions of protonated carboxylic acids with TMB also lead to elimination of a methanol molecule. However, carboxylic acids can be differentiated from the isomeric hydroxyketones based on the loss of HO-B(OCH3)2 upon SORI-CAD of the reaction product. Reactions of protonated amides with TMB also lead to elimination of a methanol molecule. However, amides can be differentiated from bifunctional oxygen-containing compounds based on the loss of OBR from the reaction product upon SORI-CAD. Protonated amines do not react with TMB.
Co-reporter:Bartłomiej J. Jankiewicz;Anthony Adeuya Dr.;Michael J. Yurkovich;Nelson R. Vinueza;Samuel J. Gardner III;Meng Zhou;John J. Nash Dr.;Hilkka I. Kenttämaa
Angewandte Chemie 2007 Volume 119(Issue 48) pp:
Publication Date(Web):24 SEP 2007
DOI:10.1002/ange.200701732
Tri-, Di- und Monoradikale: Die Reaktivität des σ,σ,σ-Triradikals 2,4,6-Tridehydropyridinium wurde mit derjenigen verwandter Mono- und Diradikale in einem Fourier-Transformations-Ionenzyklotronresonanz-Massenspektrometer verglichen. Das Triradikal hat einen Dublett-Grundzustand und geht an drei Positionen Radikalreaktionen ein. Bezüglich der Reaktivität steht es verwandten Monoradikalen näher als Diradikalen.
Co-reporter:Bartłomiej J. Jankiewicz;Anthony Adeuya Dr.;Michael J. Yurkovich;Nelson R. Vinueza;Samuel J. Gardner III;Meng Zhou;John J. Nash Dr.;Hilkka I. Kenttämaa
Angewandte Chemie International Edition 2007 Volume 46(Issue 48) pp:
Publication Date(Web):24 SEP 2007
DOI:10.1002/anie.200701732
Tri-, bi-, and monoradicals: The reactivity of a σ,σ,σ-triradical, 2,4,6-tridehydropyridinium cation, was compared with that of related mono- and biradicals in a Fourier transform ion cyclotron resonance mass spectrometer. The triradical has a doublet ground state and contains three interacting radical sites. The reactivity of the triradical more closely resembles that of related monoradicals than related biradicals.
Co-reporter:Jayalakshmi Somuramasami
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 3) pp:525-540
Publication Date(Web):2007 March
DOI:10.1016/j.jasms.2006.10.009
A novel mass spectrometric method has been developed for obtaining sequence information on small peptides. The peptides are desorbed as intact neutral molecules into a Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR) by means of laser-induced acoustic desorption (LIAD). Reactions of the neutral peptides with the dimethoxyphosphenium ion, P(OCH3)2+, occur predominantly by addition of the peptide to P(OCH3)2+ followed by the loss of two methanol molecules, thus yielding product ions with the composition (peptide+P−2H)+. Upon sustained off-resonance irradiation for collision-activated dissociation (SORI-CAD), the (peptide+P−2H)+ ions undergo successive losses of CO and NH=CHR or H2O, CO, and NH=CHR to yield sequence-related fragment ions in addition to the regular an- and bn-type ions. Under the same conditions, SORI-CAD of the analogous protonated peptides predominantly yields the regular an- and bn-type ions. The mechanisms of the reactions of peptides with P(OCH3)2+ and the dissociation of the (peptide+P−2H)+ ions were examined by using model peptides and molecular orbital calculations.
Co-reporter:Anthony Adeuya, Linan Yang, F. Sedinam Amegayibor, John J. Nash, Hilkka I. Kenttämaa
Journal of the American Society for Mass Spectrometry 2006 Volume 17(Issue 10) pp:1325-1334
Publication Date(Web):October 2006
DOI:10.1016/j.jasms.2006.07.015
The gas-phase reactions of sugars with aromatic, carbon-centered σ,σ-biradicals with varying polarities [as reflected by their calculated electron affinities (EA)] and extent of spin-spin coupling [as reflected by their calculated singlet-triplet (S-T) gaps] have been studied. The biradicals are positively charged, which allows them to be manipulated and their reactions to be studied in a Fourier-transform ion cyclotron resonance mass spectrometer. Hydrogen atom abstraction from sugars was found to be the dominant reaction for the biradicals with large EA values, while the biradicals with large S-T gaps tend to form addition/elimination products instead. Hence, not all σ,σ-biradicals may be able to damage DNA by hydrogen atom abstraction. The overall reaction efficiencies of the biradicals towards a given substrate were found to be directly related to the magnitude of their EA values, and inversely related to their S-T gaps. The EA of a biradical appears to be a very important rate-controlling factor, and it may even counterbalance the reduced radical reactivity characteristic of singlet biradicals that have large S-T gaps.
Co-reporter:Chris Petucci, Leo Guler, Hilkka I Kenttämaa
Journal of the American Society for Mass Spectrometry 2002 Volume 13(Issue 4) pp:362-370
Publication Date(Web):April 2002
DOI:10.1016/S1044-0305(02)00344-6
A chemical ionization method is reported for distinction of diastereomeric hydroxysteroids by using Fourier-transform ion cyclotron mass spectrometry (FT-ICR). Certain phosphenium ions are demonstrated to react with stereoisomeric steroids to yield qualitatively different product ions. For example, 1,3,5(10)-estratriene-3,16β,17β-triol (cis-estriol) reacts with the dimethoxy phosphenium ion to form a diagnostic product ion (not formed for the trans-estriol) through addition followed by the loss of two molecules of methanol. In an analogous manner, the 1,3-dioxolan-2-phosphenium ion produces a diagnostic product ion through the loss of ethylene glycol from the adduct of cis-estriol only. The 1,3,5(10)-estratriene-3,16α,17β-triol (trans-estriol), on the other hand, reacts with each phosphenium ion only via hydroxide abstraction-initiated pathways that indicate the presence of at least two hydroxyl groups in the molecule. These specific reactions take place for all hydroxysteroids examined, independent of their stereochemistry. Another isomer pair, cholestan-3α,5α-diol (cis-cholestandiol) and cholestan-3β,5α-diol (trans-cholestandiol), is differentiated based on selective elimination of water only from the adduct of the cis-isomer. However, the method does not allow distinction between the stereoisomeric 5β-pregnane-3α,17α,20α-triol and 5β-pregnane-3α,17α,20β-triol. The different reactivities of the three pairs of steroid isomers and of each diastereomeric compound pair are rationalized by reaction enthalpies and steric effects based on straightforward and predictable reaction mechanisms.
Co-reporter:Christopher J Petzold, Luis E Ramírez-Arizmendi, Jenny L Heidbrink, James Pérez, Hilkka I Kenttämaa
Journal of the American Society for Mass Spectrometry 2002 Volume 13(Issue 2) pp:192-194
Publication Date(Web):February 2002
DOI:10.1016/S1044-0305(01)00344-0
A generally applicable method for the study of phenyl radicals’ reactions with neutral biomolecules in the gas phase is demonstrated. Neutral biomolecules were evaporated into a Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR) by means of laser-induced acoustic desorption (LIAD) and subsequently reacted with trapped charged phenyl radicals. The structural integrity of the evaporated alanylalanine molecules was verified by reaction with dichlorophosphenium ions. Examination of the reactions of charged phenyl radicals with alanylalanine and thymidine evaporated via LIAD revealed hydrogen atom abstraction for both alanylalanine and thymidine as well as an addition/elimination product for the reaction with thymidine. These reactions are consistent with the results obtained by others in solution. Further, a previously unstudied reaction of the nucleotide of thymine (T1) with charged phenyl radical was found to yield analogous products as the reaction with thymidine.
Co-reporter:James Pérez, Christopher J. Petzold, Michael A. Watkins, Weldon E. Vaughn, Hilkka I. Kenttämaa
Journal of the American Society for Mass Spectrometry 1999 Volume 10(Issue 11) pp:1105-1110
Publication Date(Web):November 1999
DOI:10.1016/S1044-0305(99)00084-7
We report here the first application of laser desorption (LD) in transmission geometry (backside irradiation of the sample through a transparent support) inside a Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR). A probe-mounted fiber optic assembly was used to simplify the implementation of this LD technique. This setup requires little or no instrument modifications, has minimum maintenance requirements, and is relatively inexpensive to build. The performance of the probe was tested by determining the molecular weight of a commercial polystyrene standard from its matrix-assisted laser desorption/ionization (MALDI) spectrum. The measured average molecular weight is comparable to that obtained for the same sample by MALDI in the conventional top-illumination arrangement (reflection geometry) and by the manufacturer of the sample by gel permeation chromatography. The average velocities measured for ions evaporated by transmission mode LD of several neat samples are about half the velocity of those obtained by using the reflection geometry. Therefore, transmission mode irradiation of the sample holds promise to desorb ions that are easier to trap in an ICR cell. An oscillating capillary nebulizer was adapted for the deposition of analytes to improve sampling reproducibility.
Co-reporter:M.C Nyman, J Pérez, E.R Blatchley III, H.I Kenttämaa
Journal of the American Society for Mass Spectrometry 1999 Volume 10(Issue 11) pp:1152-1156
Publication Date(Web):November 1999
DOI:10.1016/S1044-0305(99)00080-X
3,3′-Dichlorobenzidine (DCB) and its degradation products, 3-chlorobenzidine (MCB) and benzidine, are of environmental concern because of their carcinogenic nature. The suitability of a small Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer for the analysis of these environmental contaminants in different types of matrices was explored. All the measurements were carried out by depositing the sample solution directly on a disk that was introduced into the mass spectrometer. This approach is very fast and simple because it requires no prior chromatographic separation or derivatization. Calibration curves determined by collecting 70-eV electron ionization mass spectra of neat samples yielded lower limits of detection of 29 and 17 pg (total amount on the solids probe) for DCB and benzidine, respectively (based on a signal to noise ratio of ≥2:1), while chemical ionization with ammonia resulted in lower limits of detection of 21 pg for DCB and 9 pg for benzidine (total amount on the solids probe). FT-ICR analysis of sediments collected from Lake Macatawa (Holland, MI) verified the presence of DCB in this complex, environmentally significant sample matrix. Laboratory experiments designed to probe biodegradation and photodegradation pathways showed that DCB undergoes sequential dehalogenation to yield MCB and then benzidine under exposure to microorganisms and under simulated tropospheric solar radiation. The ability of the FT-ICR to determine elemental compositions of compounds introduced as described above was demonstrated for one of the degradation products.
Co-reporter:Mingkun Fu, Sen Li, Enada Archibold, Michael J. Yurkovich, John J. Nash, Hilkka I. Kenttämaa
Journal of the American Society for Mass Spectrometry (October 2010) Volume 21(Issue 10) pp:1737-1752
Publication Date(Web):1 October 2010
DOI:10.1016/j.jasms.2010.06.010
Gas-phase reactivity of a positively charged aromatic σ,σ-biradical (N-methyl-6,8-didehydroquinolinium) was examined toward six aliphatic amino acids and 15 dipeptides by using Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR) and laser-induced acoustic desorption (LIAD). While previous studies have revealed that H-atom and NH2 abstractions dominate the reactions of related monoradicals with aliphatic amino acids and small peptides, several additional, unprecedented reaction pathways were observed for the reactions of the biradical. For amino acids, these are 2H-atom abstraction, H2O abstraction, addition – CO2, addition – HCOOH, and formation of a stable adduct. The biradical reacts with aliphatic dipeptides similarly as with aliphatic amino acids, but undergoes also one additional reaction pathway, addition/C-terminal amino acid elimination (addition – CO – NHCHRC). These reactions are initiated by H-atom abstraction by the biradical from the amino acid or peptide, or nucleophilic addition of an NH2 or a HO group of the amino acid or peptide at the radical site at C-6 in the biradical. Reactions of the unquenched C-8 radical site then yield the products not observed for related monoradicals. The biradical reacts with aromatic dipeptides with an aromatic ring in N-terminus (i.e., Tyr-Leu, Phe-Val, and Phe-Pro) similarly as with aliphatic dipeptides. However, for those aromatic dipeptides that contain an aromatic ring in the C-terminus (i.e., Leu-Tyr and Ala-Phe), one additional pathway, addition/N-terminal amino acid elimination (addition – CO – NHCHRN), was observed. This reaction is likely initiated by radical addition of the biradical at the aromatic ring in the C-terminus. Related monoradicals add to aromatic amino acids and small peptides, which is followed by Cα–Cβ bond cleavage, resulting in side-chain abstraction by the radical. For biradicals, with one unquenched radical site after the initial addition, the reaction ultimately results in the loss of the N-terminal amino acid. Similar to monoradicals, the C–S bond in amino acids and dipeptides was found to be especially susceptible to biradical attack.A mass spectrum measured for the reaction of the biradical (m/z 142) with Phe-Val (MW 264) in FT-ICR.Download high-res image (83KB)Download full-size image
Co-reporter:Jayalakshmi Somuramasami, Brian E. Winger, Todd A. Gillespie, Hilkka I. Kenttämaa
Journal of the American Society for Mass Spectrometry (May 2010) Volume 21(Issue 5) pp:773-784
Publication Date(Web):1 May 2010
DOI:10.1016/j.jasms.2009.11.008
A mass spectrometric method has been developed for the identification of carbonyl and hydroxyl functional groups, as well as for counting the functional groups, in previously unknown protonated bifunctional oxygen-containing analytes. This method utilizes solution reduction before mass spectrometric analysis to convert the carbonyl groups to hydroxyl groups. Gas-phase ion-molecule reactions of the protonated reduced analytes with neutral trimethylborate (TMB) in a FT-ICR mass spectrometer give diagnostic product ions. The reaction sequence likely involves three consecutive steps, proton abstraction from the protonated analyte by TMB, addition of the neutral analyte to the boron reagent, and elimination of a neutral methanol molecule. The number of methanol molecules eliminated upon reactions with TMB reveals the number of hydroxyl groups in the analyte. Comparison of the reactions of the original and reduced analytes reveals the presence and number of carbonyl and hydroxyl groups in the analyte.A mass spectrometric method was developed for the identification and counting of carbonyl and hydroxyl functional groups in previously unknown protonated bifunctional oxygen containing analytes.Download high-res image (39KB)Download full-size image
Co-reporter:Mingkun Fu, Penggao Duan, Sen Li, Ryan J. Eismin, Hilkka I. Kenttämaa
Journal of the American Society for Mass Spectrometry (July 2009) Volume 20(Issue 7) pp:1251-1262
Publication Date(Web):1 July 2009
DOI:10.1016/j.jasms.2009.02.020
A mass spectrometric method is presented for the identification of analytes with two basic functionalities and PA between 222 and 245 kcal/mol, including diamines. This method utilizes gas-phase ion–molecule reactions of protonated analytes with neutral 1,1-diethoxyethene (DEE) in a Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR). A variety of protonated mono-, bi-, and trifunctional analytes containing different functional groups, namely, amido, amino, N-oxide, hydroxy, carboxylic acid, keto, thio, thioether, alkene, phosphite, and phosphonate, were tested in the FT-ICR. The results demonstrate that basic protonated bifunctional compounds (PA between 222 and 245 kcal/mol) react selectively with DEE by forming a specific addition/elimination product ion (adduct − EtOH) (this product was also observed for lysine with three functionalities). The diagnostic reaction sequence involves proton transfer from the protonated analyte to the basic vinyl group in DEE, followed by addition of one of the functional groups of the analyte to the electrophilic α-carbon in protonated DEE. The next step involves proton transfer from this functionality to the other analyte functionality, followed by proton transfer to DEE and elimination of ethanol. Since the mechanism involves proton transfer between two functional groups of the analyte, the reaction does not occur for analytes where the two functionalities cannot be in close proximity (i.e., meta-phenylenediamine), and where no proton is available (i.e., dimethylaminoketone).A mass spectrometric method is presented for the identification of analytes with two basic functionalities and PA between 222 and 245 kcal/mol.Download high-res image (77KB)Download full-size image
Co-reporter:Steven C. Habicht, Nelson R. Vinueza, Penggao Duan, Mingkun Fu, Hilkka I. Kenttämaa
Journal of the American Society for Mass Spectrometry (April 2010) Volume 21(Issue 4) pp:559-563
Publication Date(Web):1 April 2010
DOI:10.1016/j.jasms.2009.12.015
We report here an automated method for the identification of N-oxide functional groups in drug metabolites by using the combination of liquid chromatography/tandem mass spectrometry (LC/MSn) based on ion-molecule reactions and collision-activated dissociation (CAD). Data-dependent acquisition, which has been readily utilized for metabolite characterization using CAD-based methods, is adapted for use with ion-molecule reaction-based tandem mass spectrometry by careful choice of select experimental parameters. Two different experiments utilizing ion-molecule reactions are demonstrated, data-dependent neutral gain MS3 and data-dependent neutral gain pseudo-MS3, both of which generate functional group selective mass spectral data in a single experiment and facilitate increased throughput in structural elucidation of unknown mixture components. Initial results have been generated by using an LC/MSn method based on ion-molecule reactions developed earlier for the identification of the N-oxide functional group in pharmaceutical samples, a notoriously difficult functional group to identify via CAD alone. Since commercial software and straightforward, external instrument modification are used, these experiments are readily adaptable to the industrial pharmaceutical laboratory.A data-dependent tandem MS method based on ion-molecule reactions has been developed for the identification of the N-oxide functionality in drug metabolites.Download high-res image (152KB)Download full-size image
Co-reporter:Jinshan Gao, Bartłomiej J. Jankiewicz, Jennifer Reece, Huaming Sheng, Christopher J. Cramer, John J. Nash and Hilkka I. Kenttämaa
Chemical Science (2010-Present) 2014 - vol. 5(Issue 6) pp:NaN2215-2215
Publication Date(Web):2014/02/27
DOI:10.1039/C4SC00194J
The reactivities of eleven 3,5-didehydropyridinium and six 2,4-didehydropyridinium cations toward cyclohexane were examined in the gas phase by using Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometry as well as high-level quantum chemical calculations. The results unequivocally demonstrate that the reactivity of meta-benzyne analogs can be “tuned” from more radical-like to less radical-like by changing the type and position of substituents. For example, σ-acceptor substituents at the 4-position and π-donor substituents at the 2-position in 3,5-didehydropyridinium cations partially decouple the biradical electrons, which results in lower energy transition states, and faster radical reactions. In contrast, σ-acceptors at the 2-position and π-donors at the 4-position in 3,5-didehydropyridinium cations cause stronger coupling between the biradical electrons, which results in lower radical reactivity. Three main factors are found to control the reactivity of these biradicals: (1) the energy required to distort the minimum energy dehydrocarbon atom separation to the separation of the transition state, (2) the S–T splitting at the separation of the transition state, and (3) the electron affinity at the separation of the transition state.

![Acetamide,N-[2-hydroxy-4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/110500/110479-60-2.png)
![Acetamide,N-[2-hydroxy-4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/110500/110479-60-2_b.png)




![L-Cysteine,N-acetyl-S-[5-(acetylamino)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/52400/52372-86-8.png)
![L-Cysteine,N-acetyl-S-[5-(acetylamino)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/52400/52372-86-8_b.png)

![Acetamide, N-[2-hydroxy-5-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/37600/37596-92-2.png)
![Acetamide, N-[2-hydroxy-5-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/37600/37596-92-2_b.png)

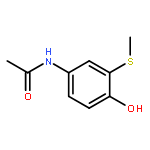



![Benzo[a]fluorene](http://img.cochemist.com/ccimg/30800/30777-18-5.png)
![Benzo[a]fluorene](http://img.cochemist.com/ccimg/30800/30777-18-5_b.png)




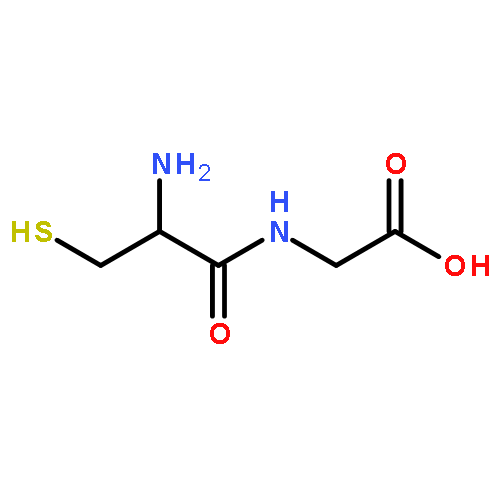











![Butanoic acid,2-amino-4-[(2-amino-2-carboxyethyl)dithio]-](http://img.cochemist.com/ccimg/5000/4985-47-1.png)
![Butanoic acid,2-amino-4-[(2-amino-2-carboxyethyl)dithio]-](http://img.cochemist.com/ccimg/5000/4985-47-1_b.png)
![6,7-Isoquinolinediol,1-[(3,4-dihydroxyphenyl)methyl]-1,2,3,4-tetrahydro-](http://img.cochemist.com/ccimg/4800/4747-99-3.png)
![6,7-Isoquinolinediol,1-[(3,4-dihydroxyphenyl)methyl]-1,2,3,4-tetrahydro-](http://img.cochemist.com/ccimg/4800/4747-99-3_b.png)




![ACETAMIDE,N-(4'-AMINO[1,1'-BIPHENYL]-4-YL)-](http://img.cochemist.com/ccimg/3400/3366-61-8.png)
![ACETAMIDE,N-(4'-AMINO[1,1'-BIPHENYL]-4-YL)-](http://img.cochemist.com/ccimg/3400/3366-61-8_b.png)


![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)

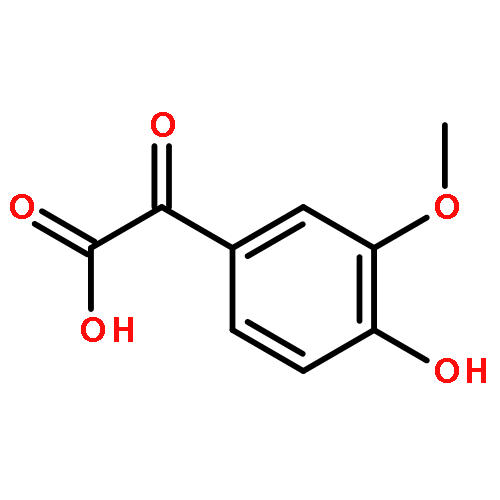





![Acetamide,N,N'-[1,1'-biphenyl]-4,4'-diylbis-](http://img.cochemist.com/ccimg/700/613-35-4.png)
![Acetamide,N,N'-[1,1'-biphenyl]-4,4'-diylbis-](http://img.cochemist.com/ccimg/700/613-35-4_b.png)


![Pyrrolo[2,3-b]indol-5-ol,1,2,3,3a,8,8a-hexahydro-1,3a,8-trimethyl-, (3aS,8aR)-](http://img.cochemist.com/ccimg/500/469-22-7.png)
![Pyrrolo[2,3-b]indol-5-ol,1,2,3,3a,8,8a-hexahydro-1,3a,8-trimethyl-, (3aS,8aR)-](http://img.cochemist.com/ccimg/500/469-22-7_b.png)



![Dibenzo[def,mno]chrysene](http://img.cochemist.com/ccimg/200/191-26-4.png)
![Dibenzo[def,mno]chrysene](http://img.cochemist.com/ccimg/200/191-26-4_b.png)
![Benzo[rst]pentaphene](http://img.cochemist.com/ccimg/200/189-55-9.png)
![Benzo[rst]pentaphene](http://img.cochemist.com/ccimg/200/189-55-9_b.png)










![N-[4-Hydroxy-3-(sulfooxy)phenyl]acetamide Sodium Salt](http://img.cochemist.com/ccimg/60700/60603-11-4.png)
![N-[4-Hydroxy-3-(sulfooxy)phenyl]acetamide Sodium Salt](http://img.cochemist.com/ccimg/60700/60603-11-4_b.png)
![L-Cysteine,S-[5-(acetylamino)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/53500/53446-10-9.png)
![L-Cysteine,S-[5-(acetylamino)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/53500/53446-10-9_b.png)
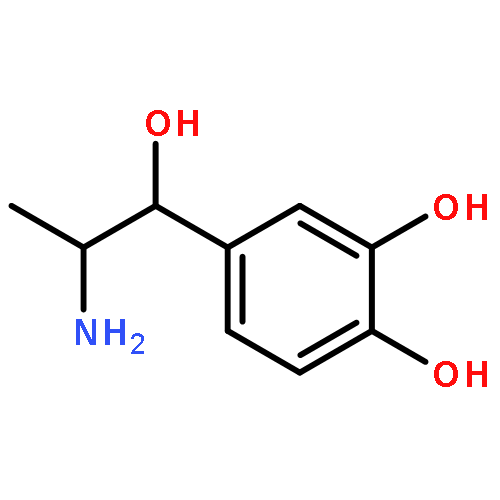


![4(3H)-Pteridinone,2-amino-6-[(1S,2S)-1,2,3-trihydroxypropyl]-](http://img.cochemist.com/ccimg/2300/2277-42-1.png)
![4(3H)-Pteridinone,2-amino-6-[(1S,2S)-1,2,3-trihydroxypropyl]-](http://img.cochemist.com/ccimg/2300/2277-42-1_b.png)
![4(3H)-Pteridinone,2-amino-7,8-dihydro-6-[(1S,2R)-1,2,3-trihydroxypropyl]-](http://img.cochemist.com/ccimg/1300/1218-98-0.png)
![4(3H)-Pteridinone,2-amino-7,8-dihydro-6-[(1S,2R)-1,2,3-trihydroxypropyl]-](http://img.cochemist.com/ccimg/1300/1218-98-0_b.png)
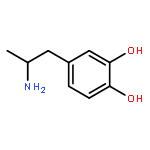
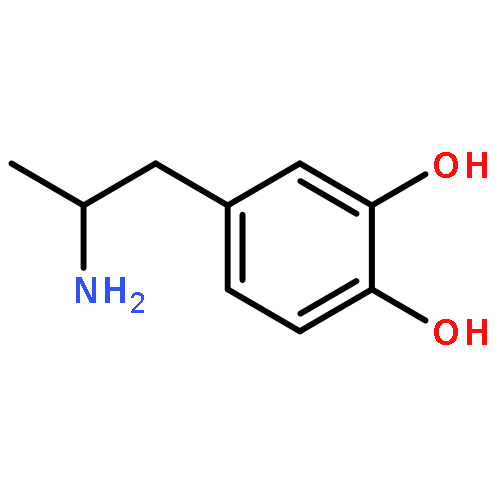
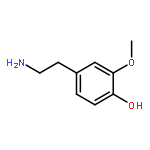

![1,2-Benzenediol,4-[2-(methylamino)ethyl]-](http://img.cochemist.com/ccimg/600/501-15-5.png)
![1,2-Benzenediol,4-[2-(methylamino)ethyl]-](http://img.cochemist.com/ccimg/600/501-15-5_b.png)

![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6.png)
![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6_b.png)



![4(3H)-Pteridinone,2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-7,8-dihydro-](http://img.cochemist.com/ccimg/6800/6779-87-9.png)
![4(3H)-Pteridinone,2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-7,8-dihydro-](http://img.cochemist.com/ccimg/6800/6779-87-9_b.png)


![4(3H)-Pteridinone,2-amino-6-[(1S,2R)-1,2,3-trihydroxypropyl]-](http://img.cochemist.com/ccimg/2100/2009-64-5.png)
![4(3H)-Pteridinone,2-amino-6-[(1S,2R)-1,2,3-trihydroxypropyl]-](http://img.cochemist.com/ccimg/2100/2009-64-5_b.png)





![Benzenemethanol,4-hydroxy-3-methoxy-a-[(methylamino)methyl]-](http://img.cochemist.com/ccimg/5100/5001-33-2.png)
![Benzenemethanol,4-hydroxy-3-methoxy-a-[(methylamino)methyl]-](http://img.cochemist.com/ccimg/5100/5001-33-2_b.png)


![1-hydroxy-1-[1-(1-oxo-1λ4-benzothiophen-2-yl)ethyl]urea](http://img.cochemist.com/ccimg/1147600/1147524-83-1.png)
![1-hydroxy-1-[1-(1-oxo-1λ4-benzothiophen-2-yl)ethyl]urea](http://img.cochemist.com/ccimg/1147600/1147524-83-1_b.png)

![2,2'-Bipyridine, 4,4'-bis[2-(9-anthracenyl)ethyl]-](http://img.cochemist.com/ccimg/500700/500615-72-5.png)
![2,2'-Bipyridine, 4,4'-bis[2-(9-anthracenyl)ethyl]-](http://img.cochemist.com/ccimg/500700/500615-72-5_b.png)
![2,2'-Bipyridine, 4-methyl-4'-[2-(1-pyrenyl)ethyl]-](http://img.cochemist.com/ccimg/500700/500615-70-3.png)
![2,2'-Bipyridine, 4-methyl-4'-[2-(1-pyrenyl)ethyl]-](http://img.cochemist.com/ccimg/500700/500615-70-3_b.png)
![Ethanol,2-[2-[4-(5-oxidodibenzo[b,f][1,4]thiazepin-11-yl)-1-piperazinyl]ethoxy]-](http://img.cochemist.com/ccimg/329300/329216-63-9.png)
![Ethanol,2-[2-[4-(5-oxidodibenzo[b,f][1,4]thiazepin-11-yl)-1-piperazinyl]ethoxy]-](http://img.cochemist.com/ccimg/329300/329216-63-9_b.png)
![10H-Thieno[2,3-b][1,5]benzodiazepine,2-methyl-4-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/174800/174794-02-6.png)
![10H-Thieno[2,3-b][1,5]benzodiazepine,2-methyl-4-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/174800/174794-02-6_b.png)
![10H-Thieno[2,3-b][1,5]benzodiazepine-2-methanol,4-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/174800/174756-45-7.png)
![10H-Thieno[2,3-b][1,5]benzodiazepine-2-methanol,4-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/174800/174756-45-7_b.png)
![D-[5-13C]GLUCOSE](http://img.cochemist.com/ccimg/120400/120388-24-1.png)
![D-[5-13C]GLUCOSE](http://img.cochemist.com/ccimg/120400/120388-24-1_b.png)





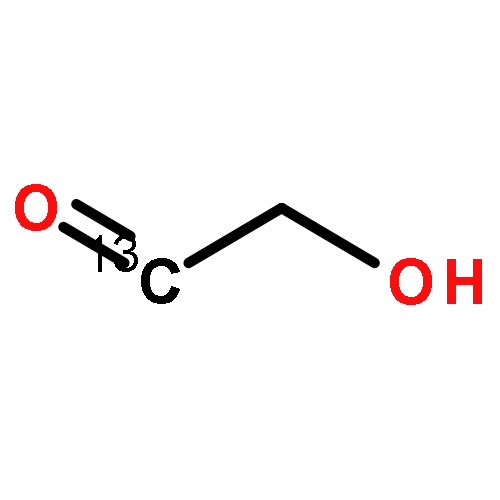
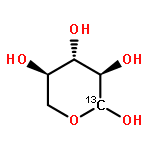
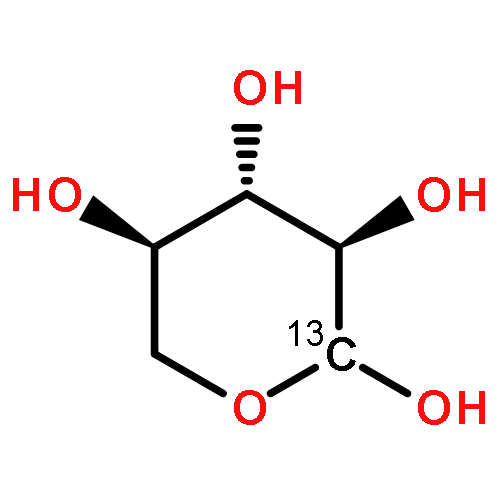
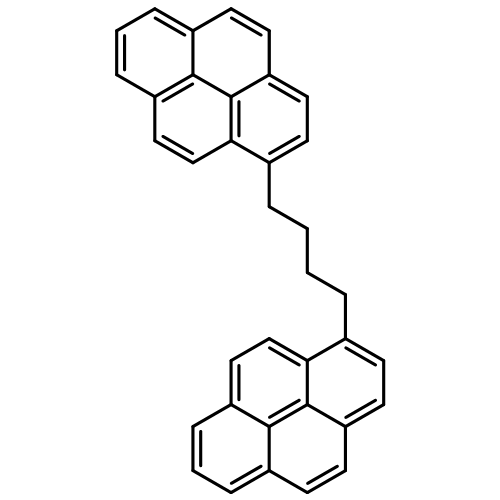
![1H-Indene-3-aceticacid, 5-fluoro-2-methyl-1-[[4-(methylsulfonyl)phenyl]methylene]-](http://img.cochemist.com/ccimg/59900/59864-04-9.png)
![1H-Indene-3-aceticacid, 5-fluoro-2-methyl-1-[[4-(methylsulfonyl)phenyl]methylene]-](http://img.cochemist.com/ccimg/59900/59864-04-9_b.png)









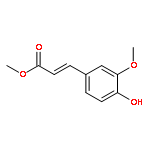

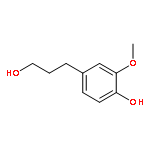
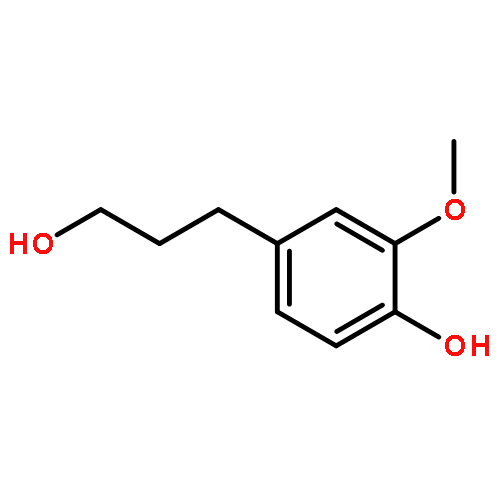
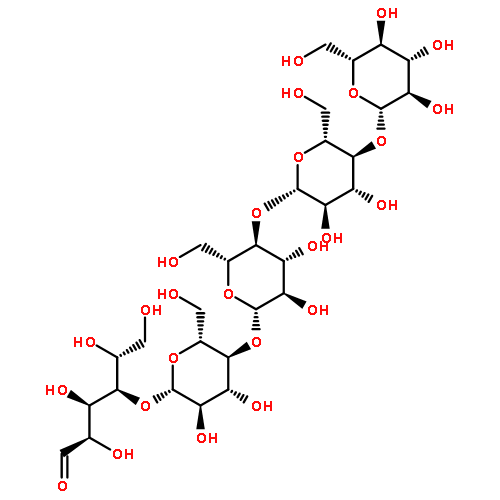




![1H-Indene-3-aceticacid, 5-fluoro-2-methyl-1-[[4-(methylsulfonyl)phenyl]methylene]-, (1Z)-](http://img.cochemist.com/ccimg/60000/59973-80-7.png)
![1H-Indene-3-aceticacid, 5-fluoro-2-methyl-1-[[4-(methylsulfonyl)phenyl]methylene]-, (1Z)-](http://img.cochemist.com/ccimg/60000/59973-80-7_b.png)