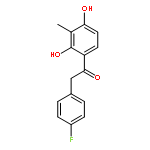
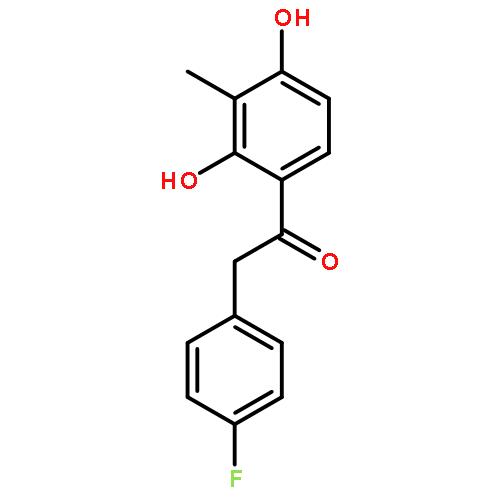
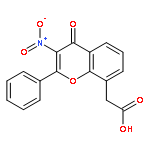
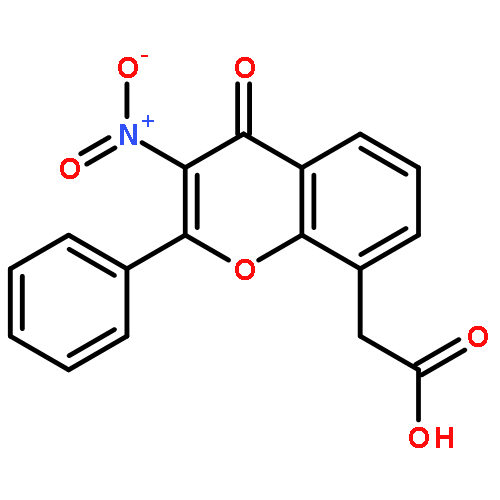
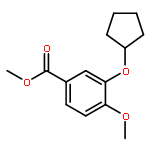
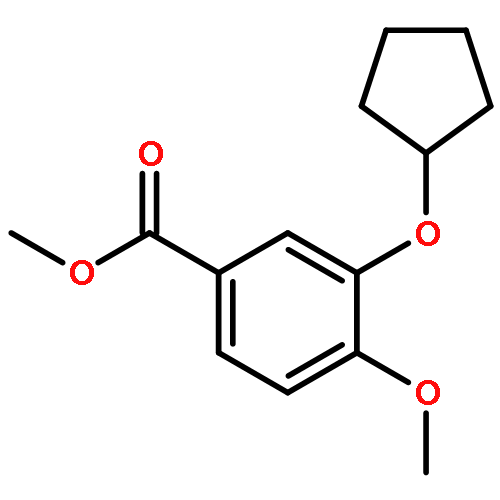
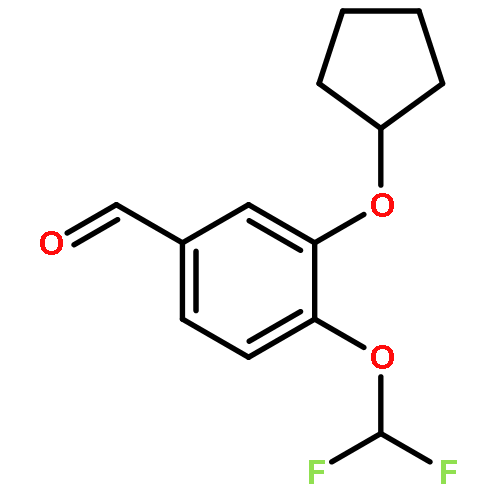
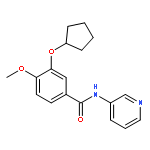
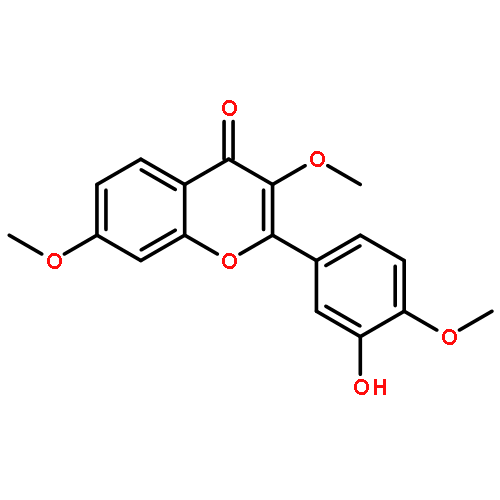
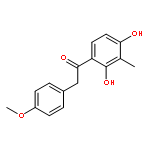
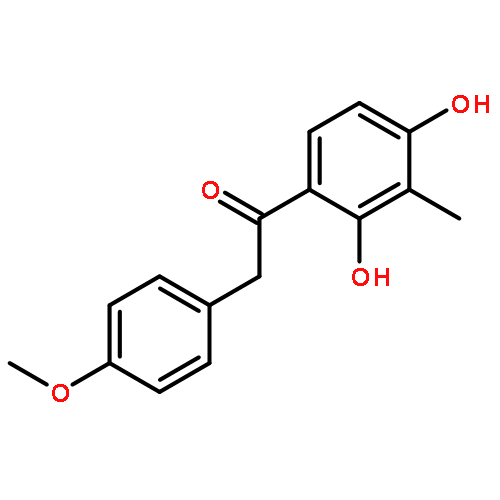
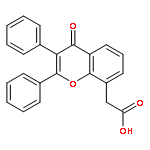
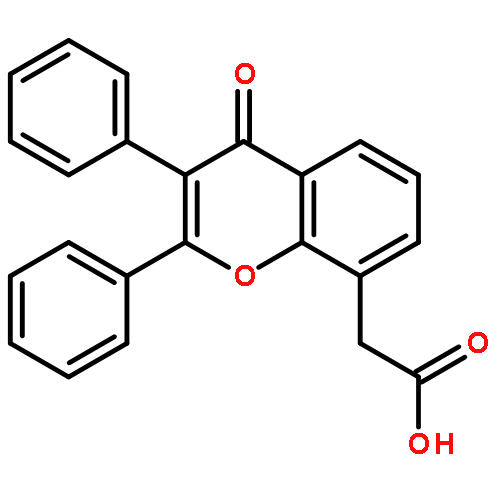
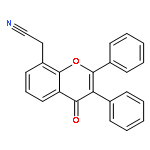
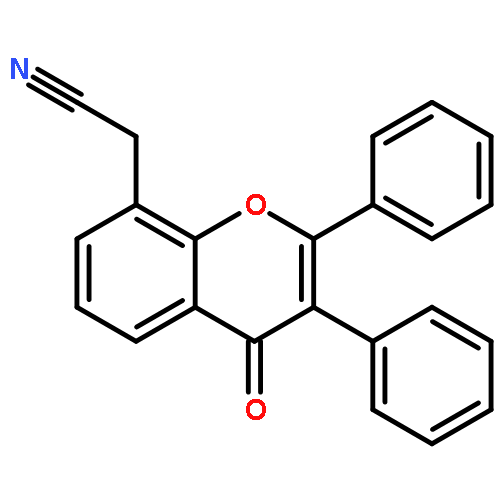
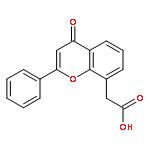
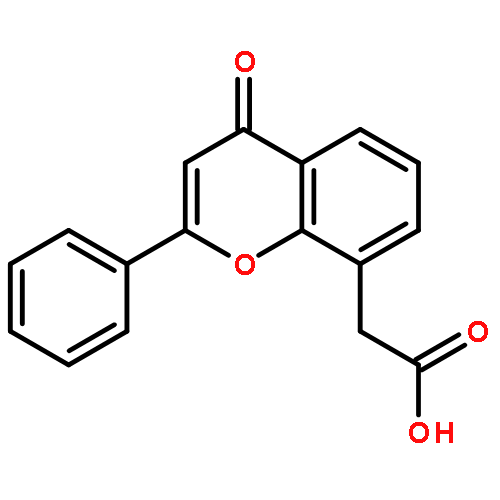
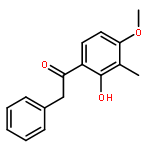
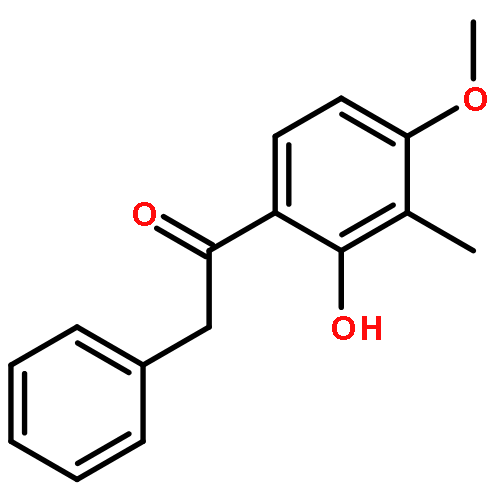
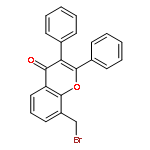
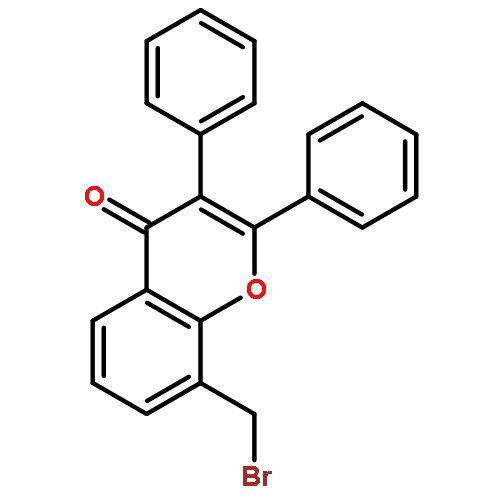
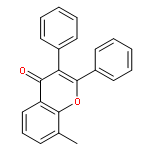
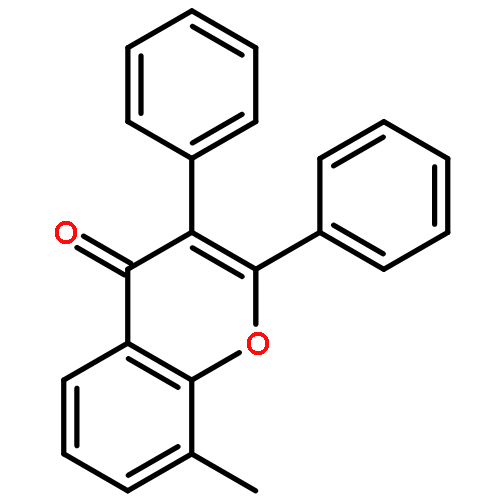
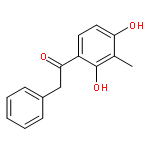
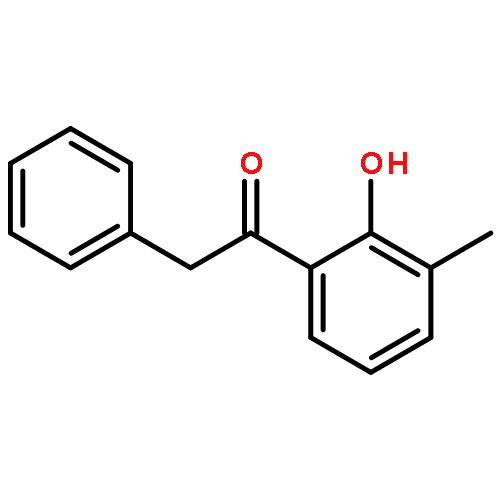
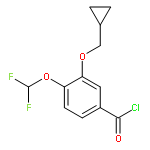
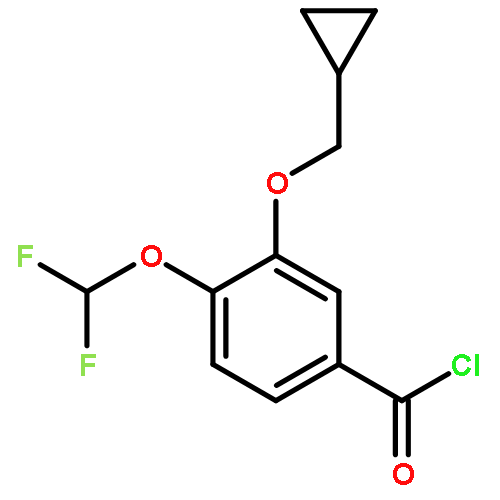
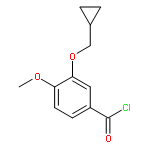
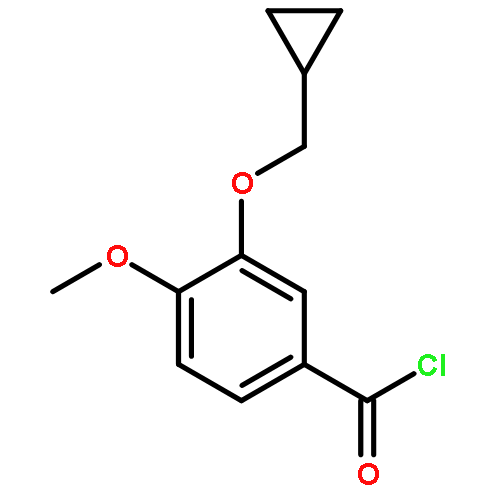
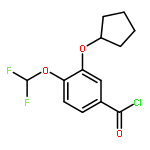
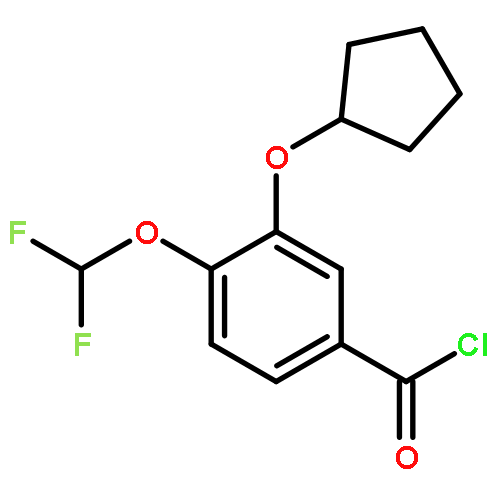
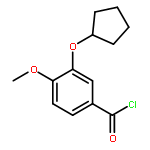
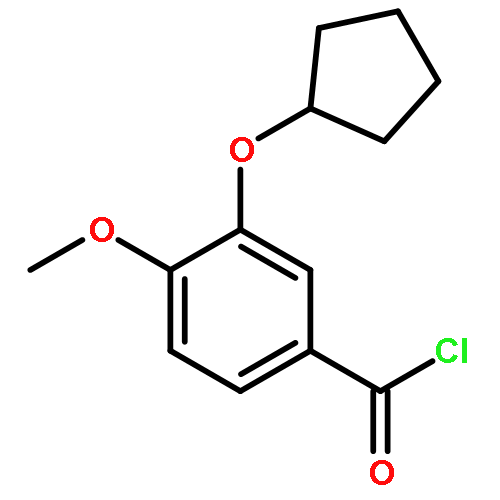
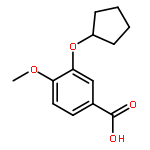
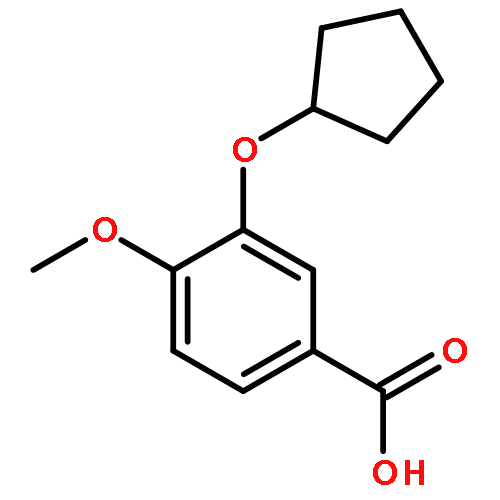
Co-reporter:Yi Liu, Gang Nie, Zhongzhen Zhou, Lihui Jia, and Yunfeng Chen
The Journal of Organic Chemistry September 1, 2017 Volume 82(Issue 17) pp:9198-9198
Publication Date(Web):July 27, 2017
DOI:10.1021/acs.joc.7b01429
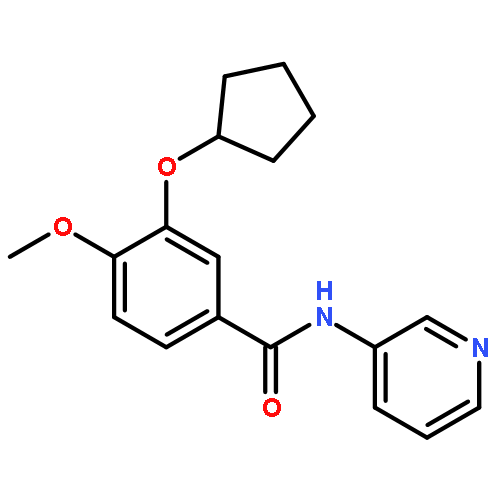
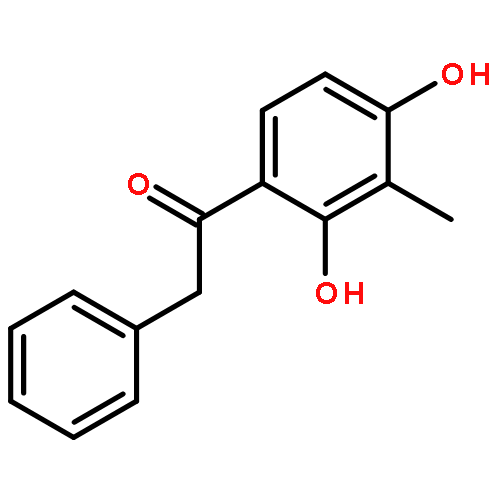
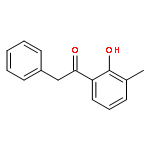
A copper-catalyzed three-component reaction of methyl ketones, organic azides, and various one-carbon (C1) donors was developed that provides 4-acyl-1,2,3-triazoles in moderate to good yields. While DMF, DMA, TMEDA, or DMSO can serve as the C1 donor, best yields were obtained using DMF. The transformation is proposed to proceed via an oxidative C–H/C–H cross-dehydrogenative coupling followed by an oxidative 1,3-dipolar cycloaddition.
Co-reporter:Zhong-Zhen Zhou, Yu-Fang Cheng, Zheng-Qiang Zou, Bing-Chen Ge, Hui Yu, Cang Huang, Hai-Tao Wang, Xue-Mei Yang, and Jiang-Ping Xu
ACS Chemical Neuroscience 2017 Volume 8(Issue 1) pp:
Publication Date(Web):October 3, 2016
DOI:10.1021/acschemneuro.6b00271
Depression involving neuroinflammation is one of the most common disabling and life-threatening psychiatric disorders. Phosphodiesterase 4 (PDE4) inhibitors produce potent antidepressant-like and cognition-enhancing effects. However, their clinical utility is limited by their major side effect of emesis. To obtain more selective PDE4 inhibitors with antidepressant and anti-neuroinflammation potential and less emesis, we designed and synthesized a series of N-alkyl catecholamides by modifying the 4-methoxybenzyl group of our hit compound, FCPE07, with an alkyl side chain. Among these compounds, 10 compounds displayed submicromolar IC50 values in the mid- to low-nanomolar range. Moreover, 4-difluoromethoxybenzamides 10g and 10j, bearing isopropyl groups, exhibited the highest PDE4 inhibitory activities, with IC50 values in the low-nanomolar range and with higher selectivities for PDE4 (approximately 5000-fold and 2100-fold over other PDEs, respectively). Furthermore, compound 10j displayed anti-neuroinflammation potential, promising antidepressant-like effects, and a zero incidence rate of emesis at 0.8 mg/kg within 180 min.Keywords: anti-neuroinflammation; antidepressant-like effects; less side-effect; N-Alkyl catecholamide; selective phosphodiesterase-4 inhibitors; structure−activity relationships;
Co-reporter:Zhong-Zhen Zhou, Xiu-Dong Shi, Hong-Fang Feng, Yu-Fang Cheng, Hai-Tao Wang, Jiang-Ping Xu
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.07.054
•Two series of 9H-purin-6-amines and 6-dichloro-9H-purines were designed and synthesized.•Compounds 9d and 11e-h exhibited low-micromole GI50 values against all test cell lines.•Compound 9d induced apoptosis at G2/M phase arrest in the human AGS cells.•Compounds 10b and 10d showed selective antiproliferative activities towards SH-SY5Y cells.•Compounds 10b and 10d displayed sub-micromole GI50 values against SH-SY5Y cells.Two series of N-(4-methoxyphenyl)-N-methyl-9H-purin-6-amines (9a-d and 10a-h) and 9-substituted benzyl-6-chloro-9H-purines (11a-h) were designed and synthesized. Their antiproliferative activities against human myelogenous leukemia (K562), human neuroblastoma (SH-SY5Y) and gastric cancer (AGS) cell lines were evaluated using the MTT assay. The preliminary results indicated that compounds 9d and 11e-h displayed low-micromole GI50 values against all tested cell lines. In addition, compounds 10b and 10d showed wonderful antiproliferative activities towards SH-SY5Y cells with selectivity of >230-fold over K562 and AGS cells. Among them, compounds 9d, 10b, 10d and 11g with good antitumor activities exhibited high selectivity for tumor cell lines over immortalized mouse hippocampal (HT22) cell line. Moreover, compound 9d with sub-micromole GI50 values toward AGS cells exhibited moderate tubulin polymerization inhibitory activity, and induced apoptosis at G2/M phase arrest with a dose-dependent manner in the human AGS cells.Download high-res image (179KB)Download full-size image
Co-reporter:Bing-Chen Ge, Hong-Fang Feng, Yu-Fang Cheng, Hai-Tao Wang, Bao-Ming Xi, Xue-Mei Yang, Jiang-Ping Xu, Zhong-Zhen Zhou
European Journal of Medicinal Chemistry 2017 Volume 141(Volume 141) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.ejmech.2017.09.077
•Twelve substituted aminopyridazin-3(2H)-ones derivatives were synthesized.•Compounds 8a-b exhibited potent antitumor activities with low-micromole GI50 value.•Compounds 8a exhibited comparable activities against all test cells with fluorouracil.•Compound 8b exhibited more potent activities against SH-SY5Y cells than fluorouracil.•Compounds 8a-b arrests cell cycle in G0/G1-phas phase and induces apoptosis in SH-SY5Y cells.A series of aminopyridazin-3(2H)-one derivatives has been designed and synthesized. Their antiproliferative activities were evaluated against three human cancer cell lines (SH-SY5Y human neuroblastoma, K562 human myelogenous leukemia and AGS gastric cancer cell lines) using the MTT assay. The preliminary activity test displayed that compound 8a exhibited comparable activities against all test cells with the positive control fluorouracil. Meanwhile compounds 8b, 8e and 9c-e displayed selective antiproliferative activities for SH-SY5Y cells. Furthermore, compounds 8a-b with low-micromole GI50 value for SH-SY5Y cells induced apoptosis with cell cycle arrest at G0/G1 phase in SH-SY5Y cells in a dose-dependent manner.In this paper, Novel (2-(2-methoxyphenoxy)ethyl)aminopyridazin-3(2H)-one derivatives bearing substituted benzyl groups were designed and synthesized. Among these compounds, compounds 8a-b exhibited wonderful antiproliferative activities against SH-SY5Y cells, and induced apoptosis and G0/G1-phase cell cycle arrest in human SH-SY5Y cells in a dose-dependent manner.Download high-res image (154KB)Download full-size image
Co-reporter:Zhong-Zhen Zhou, Bing-Chen Ge, Qiu-Ping Zhong, Chang Huang, Yu-Fang Cheng, Xue-Mei Yang, Hai-Tao Wang, Jiang-Ping Xu
European Journal of Medicinal Chemistry 2016 Volume 124() pp:372-379
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.08.052
•A series of catecholamides bearing different aromatic rings was synthesized.•Eight compounds displayed submicromolar IC50 values (30–360 nM).•Compounds 7i and 7j exhibited significant selectivity for PDE4.•The replacement of 4-methoxy group with difluoromethoxy group improve activity.•Compound 7j inhibited anti-neuroinflammation potential in microglia.In this study, catecholamides (7a–l) bearing different aromatic rings (such as pyridine-2-yl, pyridine-3-yl, phenyl, and 2-chlorophenyl groups) were synthesized as potent phosphodiesterase (PDE) 4 inhibitors. The inhibitory activities of these compounds were evaluated against the core catalytic domains of human PDE4 (PDE4CAT), full-length PDE4A4, PDE4B1, PDE4C1, and PDE4D7 enzymes, and other PDE family members. Eight of the synthesized compounds were identified as having submicromolar IC50 values in the mid-to low-nanomolar range. Careful analysis on the structure-activity relationship of compounds 7a-l revealed that the replacement of the 4-methoxy group with the difluoromethoxy group improved inhibitory activities. More interesting, 4-difluoromethoxybenzamides 7i and 7j exhibited preference for PDE4 with higher selectivities of about 3333 and 1111-fold over other PDEs, respectively. In addition, compound 7j with wonderful PDE4D7 inhibitory activities inhibited LPS-induced TNF-α production in microglia.In this paper, a series of catecholamides bearing aromatic rings was designed and synthesized as PDE4 inhibitors. Among these compounds, compound 7j with anti-neuroinflammation activity showed preference for PDE4 with higher selectivity over other PDEs.
Co-reporter:Zhong-Zhen Zhou, Bing-Chen Ge, Yu-Fang Chen, Xiu-Dong Shi, Xue-Mei Yang, Jiang-Ping Xu
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 22) pp:7332-7339
Publication Date(Web):15 November 2015
DOI:10.1016/j.bmc.2015.10.033
In this study, a series of catechol-based amides (8a–n) with different amide linkers linking the catecholic moiety to the terminal phenyl ring was designed and synthesized as potent phosphodiesterase (PDE) 4D inhibitors. The inhibitory activities of these compounds were evaluated against the core catalytic domains of human PDE4 (PDE4CAT), full-length PDE4B1 and PDE4D7 enzymes, and other PDE family members. The results indicated the majority of compounds 8a–n displayed moderate to good inhibitory activities against PDE4CAT. Among these compounds, compound 8j with a short amide linker (CONHCH2) displayed comparable PDE4CAT inhibitory activity (IC50 = 410 nM) with rolipram. More interestingly, compound 8g, a potent and selective PDE4D inhibitor (IC50 = 94 nM), exhibited a 10-fold selectivity over the PDE4B subtypes and an over 1000-fold selectivity against other PDE family members. Docking simulations suggested that 8g forms three extra H-bonds with the NH of residue Asn487 and two water molecules.
Co-reporter:Guang-Hua Yan, Xiao-Fang Li, Bing-Chen Ge, Xiu-Dong Shi, Yu-Fang Chen, Xue-Mei Yang, Jiang-Ping Xu, Shu-Wen Liu, Pei-Liang Zhao, Zhong-Zhen Zhou, Chun-Qiong Zhou, Wen-Hua Chen
European Journal of Medicinal Chemistry 2015 90() pp: 251-257
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.11.030