Co-reporter:Zhe Qiang, Yanfeng Xia, Xuhui Xia, and Bryan D. Vogt
Chemistry of Materials December 12, 2017 Volume 29(Issue 23) pp:10178-10178
Publication Date(Web):November 20, 2017
DOI:10.1021/acs.chemmater.7b04061
High concentrations of heteroatom can be doped into ordered mesoporous carbon by infiltration of molten dopants into silica-reinforced mesoporous cross-linked polymer (resol) and subsequent carbonization. The high concentration of dopants relative to polymer enables a high probability of heteroatoms to be dynamically integrated into the framework through carbonization, while the silica in the framework prevents loss of the ordered structure. This method is demonstrated to generate ordered mesoporous carbons with high heteroatom content (up to 26 atom % N, 15 atom % B, 7 atom % P, or 4 atom % S) for a wide variety of elements through melt infusion of the appropriate dopant (melamine, boric anhydride, ammonium dihydrogen phosphate, or dibenzyl sulfide). The ratio of the solid dopants to mesoporous silica–resol in a physical mixture during the melt infusion provides a simple methodology to precisely tune the doping content in the carbonized material. Etching of the silica postcarbonization generates additional micropores to produce porous doped carbons with high surface areas that can exceed 2000 m2/g. Increasing the doping of the mesoporous carbon leads to a decrease in the surface area as the framework is swollen during the incorporation of the heteroatoms that leads to an increase in the d-spacing of the mesostructure, but the ordered structure is maintained with well-defined mesopores. With the advantages of high surface area, well-defined pore size, and tunable doping concentration through a straightforward methodology, it is expected that this family of mesoporous carbons will provide model materials for fundamental studies in diverse applications from energy storage to catalysis to adsorption for separations.
Co-reporter:Zhiyang Zhao, Fang Peng, Kevin A. Cavicchi, Mukerrem Cakmak, R. A. Weiss, and Bryan D. Vogt
ACS Applied Materials & Interfaces August 16, 2017 Volume 9(Issue 32) pp:27239-27239
Publication Date(Web):July 25, 2017
DOI:10.1021/acsami.7b07816
Three-dimensional printing enables the net shape manufacturing of objects with minimal material waste and low tooling costs, but the functionality is generally limited by available materials, especially for extrusion-based printing, such as fused deposition modeling (FDM). Here, we demonstrate shape memory behavior of 3D printed objects with FDM using a commercially available olefin ionomer, Surlyn 9520, which is zinc-neutralized poly(ethylene-co-methacrylic acid). The initial fixity for 3D printed and compression-molded samples was similar, but the initial recovery was much lower for the 3D printed sample (R = 58%) than that for the compression-molded sample (R = 83%). The poor recovery in the first cycle is attributed to polyethylene crystals formed during programming that act to resist the permanent network recovery. This effect is magnified in the 3D printed part due to the higher strain (lower modulus in the 3D printed part) at a fixed programming stress. The fixity and recovery in subsequent shape memory cycles are greater for the 3D printed part than for the compression-molded part. Moreover, the programmed strain can be systematically modulated by inclusion of porosity in the printed part without adversely impacting the fixity or recovery. These characteristics enable the direct formation of complex shapes of thermoplastic shape memory polymers that can be recovered in three dimensions with the appropriate trigger, such as heat, through the use of FDM as a 3D printing technology.Keywords: 4D printing; additive manufacturing; fused deposition modeling; shape memory polymer; surlyn;
Co-reporter:Zhe Qiang, Xinye Liu, Feng Zou, Kevin A. Cavicchi, Yu Zhu, and Bryan D. Vogt
The Journal of Physical Chemistry C August 10, 2017 Volume 121(Issue 31) pp:16702-16702
Publication Date(Web):July 17, 2017
DOI:10.1021/acs.jpcc.7b03795
Bimodal porous carbon-silica (BP-CS) nanocomposites exhibit advantageous properties from a design perspective for low-cost lithium-ion battery anodes. The BP-CS nanocomposites were fabricated using cooperative self-assembly of phenolic resin, tetraethylorthosilicate, and Pluronic F127 via a scalable roll-to-roll method. An etching reaction between molten KOH and silica at high temperature (∼700 °C) introduces micropores and increases the surface area from 446 m2/g to 1718 m2/g without the loss of the ordered mesostructure. This large surface area after etching is generally advantageous for electrochemical energy storage. The carbon framework not only provides electrical conductivity but also constrains the volumetric changes of SiO2 during Li+ insertion and extraction to improve the capacity stability on charge–discharge cycling. The bimodal pores of BP-CS facilitate lithium-ion diffusion (mesopores) while maximizing the contact area between the electrolyte and electrode (micropores) as well as providing stress relief from Li+ insertion. These characteristics lead to a discharge capacity of 611 mAh g–1 after 200 cycles at 200 mA g–1 with over 99.5% Coulombic efficiency for all discharge cycles. Even when increasing the current rate to 3 A g–1, a capacity of 313 mAh g–1 is retained after 1500 cycles, corresponding to <0.005% fade in the capacity per cycle. The combination of a high rate performance, a good cycle stability at a high rate, and a scalable synthesis route with low-cost precursors makes BP-CS a promising inexpensive, carbon/SiO2-based anode material for long lifetime batteries.
Co-reporter:Chao Wang;Yipin Duan;Nicole S. Zacharia
Soft Matter (2005-Present) 2017 vol. 13(Issue 6) pp:1161-1170
Publication Date(Web):2017/02/08
DOI:10.1039/C6SM02439D
Composite hydrogels containing graphene oxide (GO) offer advantageous mechanical properties, but tuning these properties generally requires the synthesis of new hydrogels or if the hydrogel is thermally responsive, utilization of a chemistry determined temperature window. Here, we demonstrate a simple route to generate a family of GO-based hydrogels from aqueous solution based assembly of GO with polycationic poly(ethylenimine), PEI, without any secondary chemical crosslinking. Tuning the ratio of GO : PEI during the assembly produces a family of hydrogels that responds to mechanical compression by irreversibly altering their equilibrium water content and mechanical properties in a controllable manner. Despite the lack of chemical crosslinks, the hydrogels are stable when stored in an excess of water or NaCl solutions (up to 1 M) and exhibit a tunable swelling ratio (mass hydrogel : mass solid) between 44 and 162 based on both composition and compression history. Consequently, the storage modulus from shear rheology can be increased by more than 3 orders of magnitude from this irreversible mechanical compression of the hydrogel. This stiffening of the hydrogels in response to mechanical stimuli enables the prior compression loading of the hydrogel to be determined. We demonstrate that this strategy is generalizable to other anionic 2D materials such as clay (cloisite). This family of mechanically adaptive hydrogels enables facile fabrication and tuning of physical properties that could be advantageous for sensing, energy dissipation, and other applications.
Co-reporter:Changhuai Ye;Chao Wang;Jing Wang;Clinton G. Wiener;Xuhui Xia;Stephen Z. D. Cheng;Ruipeng Li;Kevin G. Yager;Masafumi Fukuto
Soft Matter (2005-Present) 2017 vol. 13(Issue 39) pp:7074-7084
Publication Date(Web):2017/10/11
DOI:10.1039/C7SM01366C
Crystal orientation in semi-crystalline polymers tends to enhance their performance, such as increased yield strength and modulus, along the orientation direction. Zone annealing (ZA) orients the crystal lamellae through a sharp temperature gradient that effectively directs the crystal growth, but the sweep rate (VZA) of this gradient significantly impacts the extent of crystal orientation. Here, we demonstrate rotational zone annealing (RZA) as an efficient method to elucidate the influence of VZA on the crystal morphology of thin films in a single experiment using isotactic poly(1-butene), PB-1, as a model semi-crystalline polymer. These RZA results are confirmed using standard, serial linear ZA to tune the structure from an almost unidirectional oriented morphology to weakly oriented spherulites. The overall crystallinity is only modestly changed in comparison to isothermal crystallization (maximum of 55% from ZA vs. 48% for isothermal crystallization). However, the average grain size increases and the spherulites become anisotropic from ZA. Due to these structural changes, the Young's modulus of the oriented films, both parallel and perpendicular to the spherulite orientation direction, is significantly increased by ZA. The modulus does become anisotropic after ZA due to the directionality in the crystal structure, with more than a threefold increase in the modulus parallel to the orientation direction for the highest oriented film in comparison to the modulus from isothermal crystallization. RZA enables rapid identification of conditions to maximize orientation of crystals in thin polymer films, which could find utility in determining conditions to improve crystallinity and performance in organic electronics.
Co-reporter:Yanfeng Xia;Zhe Qiang;Byeongdu Lee;Matthew L. Becker
CrystEngComm (1999-Present) 2017 vol. 19(Issue 30) pp:4294-4303
Publication Date(Web):2017/07/31
DOI:10.1039/C7CE00900C
Soft templating using block copolymers provides a generalized synthetic strategy to fabricate mesoporous materials, but it is generally challenging to obtain highly crystalline frameworks without significant deformation or loss of the mesoporous structure. Here, we demonstrate a simple route to generate ordered mesoporous crystalline manganese oxide films by block copolymer templating through microwave processing to convert carbonate precursors to oxides, remove the polymeric template and crystallize the Mn3O4, all within 1 min. The microwave heating in this case is driven primarily by the high microwave cross-section of the substrate (silicon wafer), but manganese oxide also absorbs microwaves to provide energy locally for promoting nucleation/crystallization. Conversely, conventional heating in a muffle furnace at an analogous surface temperature leads to either significant residual copolymer or nanostructure collapse with low crystallinity. This difference in the behavior is attributed to the rapid and local heating of the manganese oxide by microwaves to crystallize the oxide. Microwaves rapidly generate the crystals as evidenced by the invariance in the refractive index of the films after 45 s on further microwave heating. Additionally, the microwave processing leads to nearly twice the specific surface area for the films than that of mesoporous films fabricated by calcination in the furnace. Microwave energy appears to be an attractive alternative to enable the fabrication of a highly crystalline framework in soft-templated ordered mesoporous materials when the microwaves can be absorbed by the framework of interest.
Co-reporter:Clinton G. Wiener, Chao WangYun Liu, R. A. Weiss, Bryan D. Vogt
Macromolecules 2017 Volume 50(Issue 4) pp:
Publication Date(Web):February 9, 2017
DOI:10.1021/acs.macromol.6b02680
The nanostructure changes associated with stress dissipation in a tough, supramolecular hydrogel were determined by small-angle neutron scattering (SANS) and compared with stress-relaxation measurements to understand the molecular origin of the toughness. The hydrogels were formed from random copolymers of N,N-dimethylacrylamide (DMA) and 2-(N-ethylperfluorooctane sulfonamido)ethyl acrylate (FOSA), which exhibit a microphase-separated morphology with physical cross-links formed by the FOSA nanodomains connected by DMA chains. The stress relaxation behavior following a step strain was fit using seven exponentials with relaxation times that spanned 5 orders of magnitude. The deformation and relaxation of the FOSA nanodomains and network chains were independently resolved using two different contrasts with SANS experiments. Stretching of the hydrogel produced anisotropic scattering at both contrasts examined. The DMA network chains relaxed to an isotropic state at a fast rate that corresponded to the shorter stress relaxation time, while the nanodomain structure relaxed slower and did not fully relax after 7 h. These SANS measurements provide correlations between relaxations at the macroscopic (stress) and microscopic (network chains and nanodomains) scales.
Co-reporter:Sarang M. Bhaway, Zhe Qiang, Yanfeng Xia, Xuhui Xia, Byeongdu LeeKevin G. Yager, Lihua Zhang, Kim Kisslinger, Yu-Ming Chen, Kewei Liu, Yu Zhu, Bryan D. Vogt
ACS Nano 2017 Volume 11(Issue 2) pp:
Publication Date(Web):February 1, 2017
DOI:10.1021/acsnano.6b06708
Emergent lithium-ion (Li+) batteries commonly rely on nanostructuring of the active electrode materials to decrease the Li+ ion diffusion path length and to accommodate the strains associated with the insertion and de-insertion of Li+, but in many cases these nanostructures evolve during electrochemical charging–discharging. This change in the nanostructure can adversely impact performance, and challenges remain regarding how to control these changes from the perspective of morphological design. In order to address these questions, operando grazing-incidence small-angle X-ray scattering and X-ray diffraction (GISAXS/GIXD) were used to assess the structural evolution of a family of model ordered mesoporous NiCo2O4 anode films during battery operation. The pore dimensions were systematically varied and appear to impact the stability of the ordered nanostructure during the cycling. For the anodes with small mesopores (≈9 nm), the ordered nanostructure collapses during the first two charge–discharge cycles, as determined from GISAXS. This collapse is accompanied by irreversible Li-ion insertion within the oxide framework, determined from GIXD and irreversible capacity loss. Conversely, anodes with larger ordered mesopores (17–28 nm) mostly maintained their nanostructure through the first two cycles with reversible Li-ion insertion. During the second cycle, there was a small additional deformation of the mesostructure. This preservation of the ordered structure lead to significant improvement in capacity retention during these first two cycles; however, a gradual loss in the ordered nanostructure from continuing deformation of the ordered structure during additional charge–discharge cycles leads to capacity decay in battery performance. These multiscale operando measurements provide insight into how changes at the atomic scale (lithium insertion and de-insertion) are translated to the nanostructure during battery operation. Moreover, small changes in the nanostructure can build up to significant morphological transformations that adversely impact battery performance through multiple charge–discharge cycles.Keywords: cooperative assembly; metal oxide anode; nanoporous;
Co-reporter:Yiming Yang, Chao Wang, Clinton G. Wiener, Jinkun Hao, Sophia Shatas, R.A. Weiss, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 35) pp:22774
Publication Date(Web):August 22, 2016
DOI:10.1021/acsami.6b08255
Nature uses supramolecular interactions and hierarchical structures to produce water-rich materials with combinations of properties that are challenging to obtain in synthetic systems. Here, we demonstrate hierarchical supramolecular hydrogels from electrospun, self-associated copolymers with unprecedented elongation and toughness for high porosity hydrogels. Hydrophobic association of perfluoronated comonomers provides the physical cross-links for these hydrogels based on copolymers of dimethyl acrylamide and 2-(N-ethylperfluorooctane sulfonamido)ethyl methacrylate (FOSM). Intriguingly, the hydrogel fiber mats show an enhancement in toughness in comparison to compression molded bulk hydrogels. This difference is attributed to the size distribution of the hydrophobic aggregates where narrowing the distribution in the electrospun material enhances the toughness of the hydrogel. These hydrogel fiber mats exhibit extensibility more than double that of the bulk hydrogel and a comparable modulus despite the porosity of the fiber mat leading to >25 wt % increase in water content.Keywords: double network; hierarchical hydrogels; orientation induced toughness; supramolecular hydrogels; tough hydrogels
Co-reporter:Sarang M. Bhaway, Yu-Ming Chen, Yuanhao Guo, Pattarasai Tangvijitsakul, Mark D. Soucek, Miko Cakmak, Yu Zhu, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 30) pp:19484
Publication Date(Web):July 11, 2016
DOI:10.1021/acsami.6b05592
A facile method to fabricate hierarchically structured fiber composites is described based on the electrospinning of a dope containing nickel and manganese nitrate salts, citric acid, phenolic resin, and an amphiphilic block copolymer. Carbonization of these fiber mats at 800 °C generates metallic Ni-encapsulated NiO/MnOx/carbon composite fibers with average BET surface area (150 m2/g) almost 3 times higher than those reported for nonporous metal oxide nanofibers. The average diameter (∼900 nm) of these fiber composites is nearly invariant of chemical composition and can be easily tuned by the dope concentration and electrospinning conditions. The metallic Ni nanoparticle encapsulation of NiO/MnOx/C fibers leads to enhanced electrical conductivity of the fibers, while the block copolymers template an internal nanoporous morphology and the carbon in these composite fibers helps to accommodate volumetric changes during charging. These attributes can lead to lithium ion battery anodes with decent rate performance and long-term cycle stability, but performance strongly depends on the composition of the composite fibers. The composite fibers produced from a dope where the metal nitrate is 66% Ni generates the anode that exhibits the highest reversible specific capacity at high rate for any composition, even when including the mass of the nonactive carbon and Ni0 in the calculation of the capacity. On the basis of the active oxides alone, near-theoretical capacity and excellent cycling stability are achieved for this composition. These cooperatively assembled hierarchical composites provide a platform for fundamentally assessing compositional dependencies for electrochemical performance. Moreover, this electrospinning strategy is readily scalable for the fabrication of a wide variety of nanoporous transition metal oxide fibers.Keywords: block copolymer; electrochemical energy storage; electrospinning; nanofibers; ordered mesopores; self-assembly
Co-reporter:Yuanqing Gu, Yubing Ma, Bryan D. Vogt and Nicole S. Zacharia
Soft Matter 2016 vol. 12(Issue 6) pp:1859-1867
Publication Date(Web):15 Dec 2015
DOI:10.1039/C5SM02313K
Weak polyelectrolyte multilayers (PEMs) prepared by the layer-by-layer assembly technique have recently been found to demonstrate a unique contraction upon exposure to organic solvents. This response is dependent upon which organic solvent is employed, and fundamental questions have not been clarified regarding the correlation of the magnitude of the film contraction with solvent type. In this work, we used solubility parameters to analyze the response of branched poly(ethylene imine)/poly(acrylic acid) (BPEI/PAA) multilayers when exposed to a variety of solvents. BPEI/PAA multilayers were immersed in a series of 16 different organic solvents and solvent mixtures. Immersion in organic solvent caused film dehydration and therefore contraction and also induced changes in the mechanical properties of PEMs. The film thickness was the best predictor of how a film swelled in water or contracted in organic solvent when using different batches of commercially available polyelectrolytes, rather than polyelectrolyte assembly pH conditions. The degree of film contraction was correlated with Hansen and Kamlet–Taft solubility parameters as well as solvent dielectric constant. In most cases, the hydrogen bonding ability of solvents is the primary factor to determine the magnitude of film contraction. For these solvents, increasing the temperature which corresponds to decreasing the strength of hydrogen bonding, also decreases the ability to dehydrate the films. For solvents that do not follow these trends with the strength of hydrogen bonding, a stronger correlation was found between contraction and dielectric constant, indicating that both traditional solvent quality arguments and electrostatics are important to understanding the contraction of PEMs in organic solvents.
Co-reporter:Zhe Qiang, Burcu Gurkan, Jianxing Ma, Xiangyu Liu, Yuanhao Guo, Miko Cakmak, Kevin A. Cavicchi, Bryan D. Vogt
Microporous and Mesoporous Materials 2016 Volume 227() pp:57-64
Publication Date(Web):June 2016
DOI:10.1016/j.micromeso.2016.02.015
•Large-scale (multigram-to-kilogram) fabrication is achieved using roll-to-roll processing.•Pore texture is tunable by processing conditions (concentration, TEOS loading, crosslinking temperature).•Dye adsorption capacity significantly enhanced over standard FDU-15.•Rate of dye adsorption is dependent on the molecule size of bulky dyes.Large-scale (multigram-to-kilogram) fabrication of soft-templated ordered mesoporous carbon (OMC) is enabled by roll-to-roll (R2R) processing via evaporation induced self assembly of Pluronic F127, oligomeric phenolic resin (resol), and tetraorthosilicate (TEOS) from ethanolic solution. The solution concentration, TEOS loading (etchable for microporous framework), and crosslinking temperature impact the pore structure. Here we demonstrate that mesoporous carbons with surface areas up to 2455 m2/g can be obtained under the proper processing conditions. Transmission electron microscopy (TEM), small-angle X-ray scattering (SAXS) and nitrogen adsorption–desorption isotherms reveal (i) suppressed framework shrinkage with increasing solution concentration during casting, (ii) improved long range order and higher surface area with increasing TEOS content up to 3:1 TEOS:resol, and (iii) enhanced porosity with crosslinking at 100 °C. These differences can be explained on the basis of block copolymer thermodynamics and mechanical reinforcement by silica. This family of OMCs are effective adsorbents for bulky aqueous organic dyes, such as methylene green (MG) and methyl blue (MB), with high adsorption capacities of 0.436 g MG/g OMC and 0.378 g MB/g OMC obtained. This R2R method provides a facile method to generate significant quantities of OMCs with tunable pore textures.
Co-reporter:Siyang Wang, Pattarasai Tangvijitsakul, Zhe Qiang, Sarang M. Bhaway, Kehua Lin, Kevin A. Cavicchi, Mark D. Soucek, and Bryan D. Vogt
Langmuir 2016 Volume 32(Issue 16) pp:4077-4085
Publication Date(Web):April 3, 2016
DOI:10.1021/acs.langmuir.6b01026
Block copolymer templating is a versatile approach for the generation of well-defined porosity in a wide variety of framework chemistries. Here, we systematically investigate how the composition of a poly(methoxy poly[ethylene glycol] methacrylate)-block-poly(butyl acrylate) (PMPEG-PBA) template impacts the pore characteristics of mesoporous cobalt oxide films. Three templates with a constant PMPEG segment length and different hydrophilic block volume fractions of 17%, 51%, and 68% for the PMPEG-PBA are cooperatively assembled with cobalt nitrate hexahydrate and citric acid. Irrespective of template composition, a spherical nanostructure is templated and elliptical mesostructures are obtained on calcination due to uniaxial contraction of the film. The average pore size increases from 11.4 ± 2.8 to 48.5 ± 4.3 nm as the length of the PBA segment increases as determined from AFM. For all three templates examined, a maximum in porosity (∼35% in all cases) and surface area is obtained when the precursor solids contain 35–45 wt % PMPEG-PBA. This invariance suggests that the total polymer content drives the structure through interfacial assembly. The composition for maximizing porosity and surface area with the micelle-templating approach results from a general decrease in porosity with increasing cobalt nitrate hexahydrate content and the increasing mechanical integrity of the framework to resist collapse during template removal/crystallization as the cobalt nitrate hexahydrate content increases. Unlike typical evaporation induced self-assembly with sol–gel chemistry, the hydrophilic/hydrophobic composition of the block copolymer template is not a critical component to the mesostructure developed with micelle-templating using metal nitrate–citric acid as the precursor.
Co-reporter:Zhe Qiang;Changhuai Ye;Kehua Lin;Matthew L. Becker;Kevin A. Cavicchi
Journal of Polymer Science Part B: Polymer Physics 2016 Volume 54( Issue 15) pp:1499-1506
Publication Date(Web):
DOI:10.1002/polb.24043
ABSTRACT
Microwave annealing enables rapid (60 s) ordering and orientation of block copolymer films. The developed morphology in polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) thin films depends on details of the heating rate that is controlled by microwave output energy as well as the sample location in the microwave. Over a wide heating rate (1.1–2.7 °C/s), perpendicular orientation of the cylindrical mesostructure at the surface is >50% after 60 s, but goes through a maximum at 1.8 °C/s leading to approximately 97% perpendicular cylinders at the surface. The propagation of this perpendicular surface morphology through the film thickness is also dependent upon the microwave annealing conditions. The surface structure evolves with the microwave annealing time from imperfect ordering to perpendicular cylinders to parallel cylinders as the annealing time increases. This work demonstrates the importance of controlling the heating rate during microwave annealing, which will be critical for optimizing microwave conditions for directed self-assembly. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2016, 54, 1499–1506
Co-reporter:Kar Tean Tan;Christopher C. White;Donald Hunston;Justin M. Gorham;Michael J. Imburgia;Aaron M. Forster
Polymer Engineering & Science 2016 Volume 56( Issue 1) pp:18-26
Publication Date(Web):
DOI:10.1002/pen.24186
Joints held by polymeric adhesives are commonplace in many engineered products, but normal service can require exposure to environmental conditions that present a significant challenge for maintaining the structural integrity of the interface. In particular, aqueous environments can wreak havoc on the joint strength. Here, a mechanistic approach is used to understand the difference in the debonding behavior of an epoxy/aluminum (oxide) interface when exposed to deionized (DI) water and aqueous sodium chloride by correlating macroscopic failure with the sorption of salt and water into the adhesive and its nanoscale distribution. For the epoxy-aluminum system examined here, the presence of sodium chloride increases the resistance to crack growth in comparison to DI water. The debonding appears to be controlled by water near the buried interface. Salt water decreases the solubility of water in the epoxy and decreases the concentration of water near the buried interface, but the concentration of salt that enters the epoxy is below the detection limit. Thus, even if ions cannot penetrate or sorb into the adhesive, the presence of salt can significantly alter the water distribution within the adhesive and ultimately the strength of the joint. POLYM. ENG. SCI., 56:18–26, 2016. © 2015 Society of Plastics Engineers
Co-reporter:Clinton G. Wiener, Madhusudan Tyagi, Yun Liu, R. A. Weiss, and Bryan D. Vogt
The Journal of Physical Chemistry B 2016 Volume 120(Issue 24) pp:5543-5552
Publication Date(Web):May 26, 2016
DOI:10.1021/acs.jpcb.6b02863
Prevention of ice crystallization is a challenging problem with implications in diverse applications, as well as examining the fundamental low temperature physics of water. Here, we demonstrate a simple route, inspired by water confinement in antifreeze proteins, to inhibit crystallization and provide high water mobility of highly supercooled water using supramolecular hydrogels of copolymers of dimethylacrylamide (DMA) and 2-(N-ethylperfluorooctane sulfonamido)ethyl acrylate (FOSA). These hydrogels can suppress or inhibit freezing of their water, depending on the copolymer composition. Dynamic and static neutron scattering indicate that hydrogels using the copolymer with 22 mol % FOSA partially inhibit ice formation. This behavior is attributed to confinement (<2 nm) of water between the hydrophobic FOSA nanodomains that prevents 45% of the water within the hydrogel from freezing even at 205 K. Very fast dynamics of the amorphous water are observed at 220 K with an effective local diffusivity decreased by only a factor of 2 from that observed at 295 K within the hydrogel using the copolymer with 22 mol % FOSA. The spacing between the hydrophobic nanodomains, tuned through the copolymer composition, appears to modulate the water that can crystallize. These fully hydrated hydrogels (at equilibrium with liquid water at 295 K) can enable a significant fraction of highly supercooled water to be stable down to at least 205 K.
Co-reporter:Chao Wang, Clinton G. Wiener, Ziwei Cheng, Bryan D. Vogt, and R. A. Weiss
Macromolecules 2016 Volume 49(Issue 23) pp:9228-9238
Publication Date(Web):November 30, 2016
DOI:10.1021/acs.macromol.6b01813
A combination of rheology and small-angle neutron scattering (SANS) experiments revealed the mechanism by which a surfactant can stiffen or soften physically cross-linked hydrogels. Here, the structure and rheological properties of a supramolecular hydrogel based on a random copolymer of N,N-dimethylacrylamide (DMA) and 2-(N-ethylperfluorooctane-sulfonamido)ethyl methacrylate (FOSM) were modified by the addition of sodium dodecyl sulfate (SDS). The effect of SDS concentration on the microstructure and properties of the hydrogel was determined by two types of experiments: (1) adding SDS by time-dependent, radial diffusion and (2) using samples where uniform loadings, i.e., no concentration gradient, of the SDS were achieved. Nanodomains consisting of FOSM aggregates were responsible for the physical cross-links in the hydrogel, but the formation of an equilibrium supramolecular network of the hydrogel was limited by conformational pinning of the water-swollen polymer segments by the relatively immobile FOSM groups within the nanodomains. The addition of low concentrations of SDS increased the equilibrium swelling of the hydrogel by as much as 3 times, but also increased the network cross-link density and the elastic modulus of the hydrogel. At sufficiently high SDS concentration, however, the surfactant effectively solvated the supramolecular bonds such that the nanodomain structure was partially destroyed, and the sample broke up into smaller pieces that eventually dissolved. The changes in the mechanical properties with addition of SDS corresponded to changes in the nanoscale morphology of the hydrogel measured by SANS.
Co-reporter:Kehua Lin, Yuanqing Gu, Huan Zhang, Zhe Qiang, Bryan D. Vogt, and Nicole S. Zacharia
Langmuir 2016 Volume 32(Issue 36) pp:9118-9125
Publication Date(Web):August 22, 2016
DOI:10.1021/acs.langmuir.6b02051
Chemical cross-linking of layer-by-layer assembled films promotes mechanical stability and robustness in a wide variety of environments, which can be a challenge for polyelectrolyte multilayers in saline environments or for multilayers made from weak polyelectrolytes in environments with extreme pHs. Heating branched poly(ethylenimine)/poly(acrylic acid) (BPEI/PAA) multilayers at sufficiently high temperatures drives amidization and dehydration to covalently cross-link the film, but this reaction is rather slow, typically requiring heating for hours for appreciable cross-linking to occur. Here, a more than one order of magnitude increase in the amidization kinetics is realized through microwave heating of BPEI/PAA multilayers on indium tin oxide (ITO)/glass substrates. The cross-linking reaction is tracked using infrared spectroscopic ellipsometry to monitor the development of the cross-linking products. For thick films (∼1500 nm), gradients in cross-link density can be readily identified by infrared ellipsometry. Such gradients in cross-link density are driven by the temperature gradient developed by the localized heating of ITO by microwaves. This significant acceleration of reactions using microwaves to generate a well-defined cross-link network as well as being a simple method for developing graded materials should open new applications for these polymer films and coatings.
Co-reporter:Sarang M. Bhaway, Pattarasai Tangvijitsakul, Jeongwoo Lee, Mark D. Soucek and Bryan D. Vogt
Journal of Materials Chemistry A 2015 vol. 3(Issue 42) pp:21060-21069
Publication Date(Web):16 Sep 2015
DOI:10.1039/C5TA04520G
Micelle-templated ordered mesoporous nickel–cobalt carbonates and oxides are fabricated using a metal nitrate–citric acid strategy, which avoids the hydrolysis and aging requirements associated with sol–gel chemistry. A series of mesoporous NixCo(3−x)(CO3)y and NixCo(3−x)O4 films with varying Ni–Co compositions and 14 ± 4 nm mesopores are fabricated with the same block copolymer template. AFM and GISAXS analysis indicates that the mesostructure is maintained through the formation of the carbonate and oxide, while GIXD profiles confirm formation of pure spinel phases of semi-crystalline NixCo(3−x)O4. The micelle templated mesopores are interconnected and provide transport paths for the electrolyte to minimize the solid-state diffusion requirements associated with battery electrodes. These materials exhibit good performance as sodium ion battery anodes even at high current densities of 4 A g−1. Amongst the mixed-metal oxides, Ni2CoO4 exhibits the highest specific capacity of 239 mA h g−1 after galvanostatic cycling at a current density of 1 A g−1 for 10 cycles. We attribute the superior performance of Ni2CoO4 at high rates to the high surface area and short ion-diffusion paths of the nanoporous anode architecture, while the higher nickel content in the mixed metal oxide provides enhanced stability during oxide formation along with enhanced electronic conductivity, leading to improved cycling stability of the anode. This micelle template metal nitrate–citric acid method enables new possibilities for fabricating variety of ordered mesoporous mixed-metal carbonates and oxides that could be used in a wide range of applications.
Co-reporter:Zhe Qiang, Yuanhao Guo, Hao Liu, Stephen Z.D. Cheng, Miko Cakmak, Kevin A. Cavicchi, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 7) pp:4306
Publication Date(Web):January 30, 2015
DOI:10.1021/am5086275
Roll-to-roll (R2R) processing enables the rapid fabrication of large-area sheets of cooperatively assembled materials for production of mesoporous materials. Evaporation induced self-assembly of a nonionic surfactant (Pluronic F127) with sol–gel precursors and phenolic resin oligomers (resol) produce highly ordered mesostructures for a variety of chemistries including silica, titania, and tin oxide. The cast thick (>200 μm) film can be easily delaminated from the carrier substrate (polyethylene terephthalate, PET) after cross-linking the resol to produce meter-long self-assembled sheets. The surface areas of these mesoporous materials range from 240 m2/g to >1650 m2/g with these areas for each material comparing favorably with prior reports in the literature. These R2R methods provide a facile route to the scalable production of kilograms of a wide variety of ordered mesoporous materials that have shown potential for a wide variety of applications with small-batch syntheses.Keywords: continuous production; FDU-15; nanocomposite; SBA-15; tin oxide; titania
Co-reporter:Changhuai Ye, Tamami Takigawa, Oleksandr (Sasha) Burtovvy, Leah Langsdorf, Dane Jablonski, Andrew Bell, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 22) pp:11765
Publication Date(Web):May 18, 2015
DOI:10.1021/acsami.5b02692
The structure and mechanical properties of a novel block copolymer (BCP) system with Tg’s for both segments exceeding 300 °C, poly(butylnorbornene)-block-poly(hydroxyhexafluoroisopropyl norbornene) (BuNB-b-HFANB), are investigated as a function of processing conditions used for solvent vapor annealing (SVA). Solvent selection impacts long-range order markedly, but unexpectedly vertical orientation of cylinders are preferred over a wide range of solubility parameters, as determined by atomic force microscopy and grazing incidence small-angle X-ray scattering. The mechanical properties (elastic modulus, fracture strength, and onset fracture strain) are dependent upon the long-range order induced during SVA and determined using the combination of surface wrinkling and cracking. The modulus and fracture strength of the films increase from 1.44 GPa and 12.1 MPa to 1.77 GPa and 17.5 MPa, respectively, whereas the onset fracture strain decreases from 1.6% to approximately 0.6% as the ordering is improved. The polarity difference in the segments of the BCP is attractive for membrane separations, especially butanol–water. For biobutanol recovery, the titers are typically <3 wt % butanol; exposure of the BCP membrane to aqueous 1 wt % butanol decreases the elastic modulus to approximately 0.90 GPa, irrespective of the morphology, despite the high Tg of both segments and limited swelling (5.0 wt %). Correspondingly, the onset fracture strain of these swollen films is estimated to increase significantly to 6–7%. These results indicate that operating conditions impact the mechanical performance of BCP membranes more than their morphology despite the high Tg of the neat copolymer. Wrinkling and cracking provide a facile route to test the mechanical properties of membranes under simulated operando conditions.Keywords: ABE separations; cracking; QCM-D; wrinkling; Young’s modulus;
Co-reporter:Changhuai Ye, Lei Zhang, Guopeng Fu, Alamgir Karim, Thein Kyu, Alejandro L. Briseno, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 41) pp:23008
Publication Date(Web):September 28, 2015
DOI:10.1021/acsami.5b06344
We demonstrate a simple route to directionally grow crystals of oligothiophenes, based on 2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene with degrees of polymerization of 2 (BTTT-2) and 4 (BTTT-4) via zone annealing (ZA) of preseeded films. ZA of spun-cast films of BTTT-2 does not yield highly aligned crystals. However, if the film is oven-annealed briefly prior to ZA, highly aligned crystals that are millimeters in length can be grown, whose length depends on the velocity of the ZA front. The precrystallized region provides existing nuclei that promote crystal growth and limit nucleation of new crystals in the melted region. Aligned crystals of BTTT-2 can be obtained even when the moving velocity for ZA is an order of magnitude greater than the crystal growth rate. The relative nucleation rate to the crystallization rate for BTTT-4 is greater than that for BTTT-2, which decreases the length over which BTTT-4 can be aligned to ∼500 μm for the conditions examined. The temperature gradient and moving velocity of ZA enable control of the length of the aligned crystalline structure at the macroscale.Keywords: directed crystallization; organic electronics; P3HT; pentacene; thermal gradient
Co-reporter:Yuanzhong Zhang, Sarang M. Bhaway, Yi Wang, Kevin A. Cavicchi, Matthew L. Becker and Bryan D. Vogt
Chemical Communications 2015 vol. 51(Issue 24) pp:4997-5000
Publication Date(Web):16 Feb 2015
DOI:10.1039/C4CC09808K
Rapid chemical transformation from micelle templated precursors (metal nitrate and citric acid) to ordered mesoporous metal carbonates and oxides is demonstrated using microwave heating for cobalt, copper, manganese and zinc. Without aging requirements, <3 min of microwave processing yields highly ordered mesoporous films.
Co-reporter:Zhe Qiang, Yuanzhong Zhang, Yi Wang, Sarang M. Bhaway, Kevin A. Cavicchi, Bryan D. Vogt
Carbon 2015 Volume 82() pp:51-59
Publication Date(Web):February 2015
DOI:10.1016/j.carbon.2014.10.025
Macroscopic alignment of block copolymer (BCP)-templated mesoporous carbon films is challenging, especially for large pores (>10 nm), due to the slow dynamics of the polymer segments that impede re-orientation of the ordered domains. Here, we demonstrate a facile method, solvent vapor annealing with soft shear (SVA–SS), to fabricate unidirectionally aligned, ordered mesoporous carbon films using two different BCP templates, poly(ethylene oxide)-block-poly(n-butyl acrylate) and polystyrene-block-poly(N,N-dimethyl-n-octadecylammonium p-styrenesulfonate), and we illustrate the efficacy of this technique for both cylindrical and spherical morphologies with relatively large accessible pores (≈15 nm). This alignment is preserved through the thermopolymerization of resol and carbonization. The alignment of the mesopores impacts several key properties of these carbon films, especially for the unidirectional cylindrical mesostructures. The highly aligned mesoporous carbon films exhibit a more narrow pore size distribution than the analogous unaligned ordered mesoporous carbon as determined by ellipsometric porosimetry. Moreover, the electrical conductivity becomes anisotropic with nearly 40% difference in conductivity between parallel and perpendicular directions of the cylindrical mesopores. In the parallel orientation, the electrical conductivity is over 20% greater than the analogous unoriented (random) films. These results illustrate the applicability of SVA–SS to obtain unidirectional aligned mesoporous carbon films over large areas without additional physical or chemical templating.
Co-reporter:Changhuai Ye and Bryan D. Vogt
Soft Matter 2015 vol. 11(Issue 43) pp:8499-8507
Publication Date(Web):09 Sep 2015
DOI:10.1039/C5SM01867F
Nanoporous block copolymer thin films are fabricated by selective solvent swelling of the majority phase and subsequent rapid extraction with a miscible non-solvent (water). Selection of the initial solvent provides a facile route to tune the porosity of the films. Poly(butylnorbornene)-block-poly(hydroxyhexafluoroisopropyl norbornene) (BuHFA) is used to generate these porous thin films due to its high Tg (>300 °C) and the selectivity of primary alcohols towards poly(hydroxyhexafluoroisopropyl norbornene) (pHFANB) to enable a relatively environmentally benign process. As the solvent quality for the HFA increases from ethanol to isopropanol to n-butanol, the porosity of the film developed by water extraction increases up to 69%. Aqueous mixtures of ethanol provide an addition handle to tune the porosity between 10 and 54%. These nanoporous films are robust with the porosity nearly unchanged after extended heating at 160 °C. Their elastic moduli are investigated using surface wrinkling and the modulus, E, scales with the film density, ρ, as E ∼ ρ2.2, which is similar to cellular solids. The nanopores are templated by the self-assembled structure of the block copolymer, so these coatings are transparent despite the high porosity. These thin films act as anti-reflective coatings for glass slides. Spin coating provides a coating on both sides and processing to generate 55% porosity leads to an increase in transmittance from approximately 92% to 99.1% (average for the full range of visible light). A maximum transmittance of 99.8% is found at 523 nm. This methodology is simple and highly tunable; extension to other block copolymer systems is likely possible if sufficient solubility contrast between segments exists.
Co-reporter:Christopher White, Kar Tean Tan, Donald Hunston, Kristen Steffens, Deborah L. Stanley, Sushil K. Satija, Bulent Akgun and Bryan D. Vogt
Soft Matter 2015 vol. 11(Issue 20) pp:3994-4001
Publication Date(Web):10 Apr 2015
DOI:10.1039/C4SM02725F
Moisture attack on adhesive joints is a long-standing scientific and engineering problem. A particularly interesting observation is that when the moisture level in certain systems exceeds a critical concentration, the bonded joint shows a dramatic loss of strength. The joint interface plays a dominant role in this phenomenon; however, why a critical concentration of moisture exists and what role is played by the properties of the bulk adhesive have not been adequately addressed. Moreover if the interface is crucial, the local water content near the interface will help elucidate the mechanisms of criticality more than the more commonly examined bulk water concentration in the adhesive. To gain a detailed picture of this criticality, we have combined a fracture mechanics approach to determine joint strength with neutron reflectivity, which provides the moisture distribution near the interface. A well-defined model system, silica glass substrates bonded to a series of polymers based on poly(n-alkyl methacrylate), was utilized to probe the role of the adhesive in a systematic manner. By altering the alkyl chain length, the molecular structure of the polymer can be systematically changed to vary the chemical and physical properties of the adhesive over a relatively wide range. Our findings suggest that the loss of adhesion is dependent on a combination of the build-up of the local water concentration near the interface, interfacial swelling stresses resulting from water absorption, and water-induced weakening of the interfacial bonds. This complexity explains the source of criticality in environmental adhesion failure and could enable design of adhesives to minimize environmental failure.
Co-reporter:Jeongwoo Lee, Yu-Ming Chen, Yu Zhu and Bryan D. Vogt
RSC Advances 2015 vol. 5(Issue 120) pp:99329-99338
Publication Date(Web):10 Nov 2015
DOI:10.1039/C5RA14907J
The morphology of composite materials used in battery electrodes is critical to provide the requisite transport paths for ions and electrons to enable high performance. In this work, we describe a simple and scalable method to fine tune the morphology of carbon/TiO2 composite through polymerization-induced phase separation of a mixture containing commercial TiO2 nanoparticles, poly(hydroxyethyl methacrylate) (PHEMA), and photoacid generator (PAG) dissolved in furfuryl alcohol (FA, monomer). UV exposure converts the PAG to a strong acid that catalyzes the FA polymerization to quickly initiate the polymerization. The morphology is modulated by the molecular weight of PHEMA and FA concentration that impact the miscibility and mobility during phase separation. The polymerized composite is carbonized to yield porous carbon/TiO2 electrodes. The cycling performance is dictated by the morphology that develops during phase separation. Electrochemical impedance spectroscopy (EIS) analysis illustrates that subtle changes in synthetic conditions can dramatically impact the electrical or ion conductance, primarily through modulation in the solid electrolyte interphase (SEI). A careful investigation of the SEI layer on the porous carbon/TiO2 composites demonstrates a clear correlation between the SEI and the surface area of the porous anode as determined by transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). With selection of synthetic conditions to yield a modest surface area composite, sustainable anodes with stable capacity can be fabricated for use in Na ion batteries.
Co-reporter:Michael C. Burroughs
The Journal of Physical Chemistry C 2015 Volume 119(Issue 22) pp:12138-12148
Publication Date(Web):May 5, 2015
DOI:10.1021/acs.jpcc.5b02177
The conversion of cooperatively assembled metal nitrate, citric acid, and an amphiphilic block copolymer, poly(methoxypoly[ethylene glycol methacrylate])-block-poly(butyl acrylate), films to their associated carbonate is investigated using Fourier transform infrared spectroscopy (FTIR) and spectroscopic ellipsometry for both cobalt and copper. The processing conditions associated with the formation of the carbonate significantly impact the mesostructure generated. Ex situ FTIR measurements tracked the carbonate formation and consumption of citric acid to elucidate the kinetics of the reactions and were compared to the evolution in the film thickness and refractive index by in situ spectroscopic ellipsometry. From ellipsometry, the initial rate of thickness change appears to follow an Arrhenius temperature dependence with the apparent activation energy for Co (43 kJ/mol) approximately double that for Cu (23 kJ/mol). These data elucidating the reaction kinetics enable optimization of the temperature and reaction time for improved properties and decreased fabrication time. The temperature utilized to form the carbonate impacts the mesostructure that develops and the porosity in the resultant oxide film. The optimum temperature to maximize the porosity of the oxide films is an intermediate carbonate formation temperature where the rate of conversion is not too fast to disrupt the nanostructure, but the final conversion is sufficiently high to provide thermal resilience to the framework through calcination. This knowledge enables fabrication of ordered mesoporous oxides with porosities in excess of 60%.
Co-reporter:Jessica M. Torres, Nathan Bakken, Jian Li, and Bryan D. Vogt
The Journal of Physical Chemistry B 2015 Volume 119(Issue 35) pp:11928-11934
Publication Date(Web):July 31, 2015
DOI:10.1021/acs.jpcb.5b05814
Ultrastable glasses are generated by vapor deposition on substrates heated near the glass transition temperature (Tg), but it is unclear if the remarkable properties of such glasses are present in ultrathin (<100 nm) films. Here, we demonstrate that the moduli of 50 nm thick N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPD) film can be increased from 1.5 to 2.5 GPa by simply increasing the temperature of the substrate during deposition with a maximum in modulus found at T/Tg = 0.94. This maximum in modulus is the same modulus obtained for very thin (<15 nm) NPD films deposited at 295 K (T/Tg = 0.80). However, the modulus of films deposited at this lower temperature abruptly decreases to approximately 1.5 GPa for thicker films; the modulus from deposition at T/Tg = 0.94 is thickness independent. In addition to the thin film modulus, the substrate temperature significantly impacts the water uptake in NPD films. From QCM, the volume fraction of water at equilibrium with nearly saturated water vapor decreases from nearly 4% to less than 1% as the substrate temperature increases from T/Tg = 0.82 to T/Tg = 0.93. The substrate temperature provides a simple route to control mechanical properties and water uptake into vapor-deposited NPD, and these concepts are likely extendable to other organic electronic materials; the increased moduli and decreased water uptake could enable improved performance and lifetime of small molecule glasses for a variety of organic electronic applications.
Co-reporter:Changhuai Ye, Yan Sun, Alamgir Karim, and Bryan D. Vogt
Macromolecules 2015 Volume 48(Issue 20) pp:7567-7573
Publication Date(Web):October 13, 2015
DOI:10.1021/acs.macromol.5b02128
Scalable and low-cost methods to align and orient block copolymer (BCP) films and membranes are critical for their adaptation for nonlithographic applications. Cold zone annealing (CZA) can align BCP microdomains and is scalable via roll-to-roll (R2R) manufacturing. However, the efficacy of orientation by CZA is strongly dependent on the thermal zone velocity (Vcza). Optimization of this rate can be time-consuming and tedious. To address this shortcoming, we report rotational or radial CZA (RCZA) that provides a combinatorial approach to efficiently determine how linear Vcza rate impacts microdomain orientation. RCZA rapidly identifies the optimal CZA velocities for perpendicular orientation of cylinders in polystyrene-block-poly(methyl methacrylate) films that previously required tens of measurements [ Macromolecules 2012, 45, 7107], demonstrated here with much finer velocity resolution using three overlapping radial regimes. Notably, the efficacy of CZA for perpendicular alignment rapidly decays for Vcza > 10 μm/s. To overcome this limitation, the addition of 2 wt % 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide sufficiently alters the surface tension and segmental relaxations via reduced viscosity to increase the processing window for perpendicular cylinders, approximately 75% at Vcza ≈ 330 μm/s, approaching R2R speeds. Further increasing ionic liquid content to 5 wt % leads to mostly parallel orientation due to surface wetting. Ionic liquids can dramatically increase BCP processing speeds for applications, such as membranes, and RCZA can efficiently map out the optimal processing parameters.
Co-reporter:Zhe Qiang;Maurice L. Wadley;Kevin A. Cavicchi
Journal of Polymer Science Part B: Polymer Physics 2015 Volume 53( Issue 15) pp:1058-1064
Publication Date(Web):
DOI:10.1002/polb.23740
ABSTRACT
Thin films (monolayer and bilayer) of cylinder forming polystyrene-block-polydimethylsiloxane (PS-b-PDMS) were shear aligned by the swelling and deswelling of a crosslinked PDMS pad that was physically adhered to the film during solvent vapor annealing. The nanostructures formed by self-assembly were exposed to ultraviolet-ozone to partially oxidize the PDMS, followed by calcination in air at 500 °C. In this process, the PS segments were fully decomposed, while the PDMS yielded silica nanostructures. The highly aligned PDMS cylinders were thus deposited as silica nanolines on the silicon substrate. Using a bilayer film, the center-to-center distance of these features were effectively halved from 38 to 19 nm. Similarly, by sequential shear-alignment of two distinct layers, a rhombic array of silica nanolines was fabricated. This methodology provides a facile route to fabricating complex topographically patterned nanostructures. © 2015 Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2015, 53, 1058–1064
Co-reporter:Sarang M. Bhaway, Kim Kisslinger, Lihua Zhang, Kevin G. Yager, Andrew L. Schmitt, Mahesh K. Mahanthappa, Alamgir Karim, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 21) pp:19288
Publication Date(Web):October 15, 2014
DOI:10.1021/am505307t
Unlike other crystalline metal oxides amenable to templating by the combined assemblies of soft and hard chemistries (CASH) method, vanadium oxide nanostructures templated by poly(ethylene oxide-b-1,4-butadiene-b-ethylene oxide) (OBO) triblock copolymers are not preserved upon high temperature calcination in argon. Triconstituent cooperative assembly of a phenolic resin oligomer (resol) and an OBO triblock in a VOCl3 precursor solution enhances the carbon yield and can prevent breakout crystallization of the vanadia during calcination. However, the calcination environment significantly influences the observed mesoporous morphology in these composite thin films. Use of an argon atmosphere in this processing protocol leads to nearly complete loss of carbon–vanadium oxide thin film mesostructure, due to carbothermal reduction of vanadium oxide. This reduction mechanism also explains why the CASH method is not more generally successful for the fabrication of ordered mesoporous vanadia. Carbonization under a nitrogen atmosphere at temperatures up to 800 °C instead enables formation of a block copolymer-templated mesoporous structure, which apparently stems from the formation of a minor fraction of a stabilizing vanadium oxynitride. Thus, judicious selection of the inert gas for template removal is critical for the synthesis of well-defined, mesoporous vanadia–carbon composite films. This resol-assisted assembly method may generally apply to the fabrication of other mesoporous materials, wherein inorganic framework crystallization is problematic due to kinetically competitive carbothermal reduction processes.Keywords: block copolymer; FDU-16; nanopores; self-assembly; templated synthesis; vanadia
Co-reporter:Jeongwoo Lee, Yu-Ming Chen, Yu Zhu, and Bryan D. Vogt
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 23) pp:21011
Publication Date(Web):November 14, 2014
DOI:10.1021/am5058037
Polymerization-induced phase separation of nanoparticle-filled solution is demonstrated as a simple approach to control the structure of porous composites. These composites are subsequently demonstrated as the active component for sodium ion battery anode. To synthesize the composites, we dissolved/dispersed titanium oxide (anatase) nanoparticles (for sodium insertion) and poly(hydroxybutyl methacrylate) (PHBMA, porogen) in furfuryl alcohol (carbon precursor) containing a photoacid generator (PAG). UV exposure converts the PAG to a strong acid that catalyzes the furfuryl alcohol polymerization. This polymerization simultaneously decreases the miscibility of the PHBMA and reduces the mobility in the mixture to kinetically trap the phase separation. Carbonization of this polymer composite yields a porous nanocomposite. This nanocomposite exhibits nearly 3-fold greater gravimetric capacity in Na-ion batteries than the same titanium oxide nanoparticles that have been coated with carbon. This improved performance is attributed to the morphology as the carbon content in the composite is five times that of the coated nanoparticles. The porous composite materials exhibit stable cyclic performance. Moreover, the battery performance using materials from this polymerization-induced phase separation method is reproducible (capacity within 10% batch-to-batch). This simple fabrication methodology may be extendable to other systems and provides a facile route to generate reproducible hierarchical porous morphology that can be beneficial in energy storage applications.Keywords: furfuryl alcohol; Na-ion battery; porous carbon; TiO2 nanoparticles
Co-reporter:Guodong Deng, Yuanzhong Zhang, Changhuai Ye, Zhe Qiang, Gila E. Stein, Kevin A. Cavicchi and Bryan D. Vogt
Chemical Communications 2014 vol. 50(Issue 84) pp:12684-12687
Publication Date(Web):03 Sep 2014
DOI:10.1039/C4CC02471K
Bicontinuous mesoporous carbon films are fabricated by cooperative self-assembly of phenolic resin and amphiphilic triblock copolymer via an order–order transition from cylinders to gyroid. The film morphology is strongly influenced by the details of processing, including age of the resol, resol:template ratio, and the solvent vapor annealing process.
Co-reporter:Zhe Qiang, Yuanzhong Zhang, Jesse A. Groff, Kevin A. Cavicchi and Bryan D. Vogt
Soft Matter 2014 vol. 10(Issue 32) pp:6068-6076
Publication Date(Web):20 Jun 2014
DOI:10.1039/C4SM00875H
One of the key issues associated with the utilization of block copolymer (BCP) thin films in nanoscience and nanotechnology is control of their alignment and orientation over macroscopic dimensions. We have recently reported a method, solvent vapor annealing with soft shear (SVA-SS), for fabricating unidirectional alignment of cylindrical nanostructures. This method is a simple extension of the common SVA process by adhering a flat, crosslinked poly(dimethylsiloxane) (PDMS) pad to the BCP thin film. The impact of processing parameters, including annealing time, solvent removal rate and the physical properties of the PDMS pad, on the quality of alignment quantified by the Herman's orientational factor (S) is systematically examined for a model system of polystyrene-block-polyisoprene-block-polystyrene (SIS). As annealing time increases, the SIS morphology transitions from isotropic rods to highly aligned cylinders. Decreasing the rate of solvent removal, which impacts the shear rate imposed by the contraction of the PDMS, improves the orientation factor of the cylindrical domains; this suggests the nanostructure alignment is primarily induced by contraction of PDMS during solvent removal. Moreover, the physical properties of the PDMS controlled by the crosslink density impact the orientation factor by tuning its swelling extent during SVA-SS and elastic modulus. Decreasing the PDMS crosslink density increases S; this effect appears to be primarily driven by the changes in the solubility of the SVA-SS solvent in the PDMS. With this understanding of the critical processing parameters, SVA-SS has been successfully applied to align a wide variety of BCPs including polystyrene-block-polybutadiene-block-polystyrene (SBS), polystyrene-block-poly(N,N-dimethyl-n-octadecylammonium p-styrenesulfonate) (PS-b-PSS-DMODA), polystyrene-block-polydimethylsiloxane (PS-b-PDMS) and polystyrene-block-poly(2-vinlypyridine) (PS-b-P2VP). These results suggest that SVA-SS is a generalizable method for the alignment of BCP thin films.
Co-reporter:Clinton G. Wiener, R. A. Weiss and Bryan D. Vogt
Soft Matter 2014 vol. 10(Issue 35) pp:6705-6712
Publication Date(Web):08 Jul 2014
DOI:10.1039/C4SM00815D
The thin film behavior of poly(N-isopropylacrylamide-stat-2-(N-ethylperfluorooctane sulfonamido)ethyl acrylate) (NIPAAm-stat-FOSA) based hydrogels containing 5 mol% FOSA was elucidated using quartz crystal microbalance with dissipation (QCM-D) in combination with spectroscopic ellipsometry (SE) through examination of the lower critical solution temperature (LCST) and temperature dependent swelling for (dry) thicknesses ranging from 10 nm to 121 nm. For all thin films measured, the LCST was shown to slightly increase (>3 °C) in comparison to that of the bulk sample. However for these films, the increase in LCST was statistically identical, irrespective of thickness. Surprisingly, the volumetric swelling of the hydrogel in thin films, even at temperatures less than the LCST, was similar (within 20%) to the volumetric swelling of the bulk hydrogel, despite the expected significant decrease associated with the hydrogel being constrained by the substrate as predicted by one dimensional Flory–Rehner theory. We attribute this enhancement in swelling compared to theoretical expectations to the ability of the hydrophobic crosslinks to re-arrange under stress, which provides a mechanism to alleviate the decreased dimensionality imposed by the substrate; this mechanism is consistent with a large hysteresis in the swelling when cycling between 35 °C and 5 °C. Unlike the LCST, the swelling ratio increases with decreasing film thickness. At low temperatures (below the LCST), the volume swelling ratio increased from 3.9 to 4.9, while at temperatures above the LCST the swelling ratio increased from 1.5 to 2.5 when the film thickness decreased from 121 nm to 10 nm. The combination of facile processing through solution casting without the need for additional crosslinking chemistry and limited thickness dependent variation of swelling and LCST behavior in these physically crosslinked hydrogels makes these materials attractive for applications requiring thermoresponsive soft coatings.
Co-reporter:Jiachen Xue, Christopher Henry, Jeongwoo Lee and Bryan D. Vogt
RSC Advances 2014 vol. 4(Issue 8) pp:3675-3683
Publication Date(Web):27 Nov 2013
DOI:10.1039/C3RA44723E
Mesoporous carbon films are generally fabricated on rigid substrates that act to provide mechanical stability. Here, instead of utilizing rigid substrates, a thin sheet of polyimide (Kapton) is utilized as the substrate to enable continuous production of sheets of soft templated mesoporous carbon films. The mesoporous carbon film is well adhered to the carbonized Kapton. Although mesoporous carbon is generally considered brittle, carbonization of the coated polyimide yields a tough flexible sheet that can be gently bent or even cut to the desired shape. Characterization with transmission electron microscopy, small angle X-ray scattering and N2 sorption (B.E.T.) confirms the presence of ordered mesopores that conform to expected structures based upon analogous powders. A simple demonstration of the flexible mesoporous carbons as electrodes for aqueous supercapacitors illustrates that the carbonized Kapton can act as the current collector without significant degradation in performance based on the estimated mass of the active mesoporous carbon film at low rates. The fabrication of ordered mesoporous carbon on Kapton provides a facile route to large area, robust high surface area, conductive materials that could be used for a wide variety of electrochemical applications, especially as sensors.
Co-reporter:Yuanzhong Zhang, Zhe Qiang and Bryan D. Vogt
RSC Advances 2014 vol. 4(Issue 85) pp:44858-44867
Publication Date(Web):10 Sep 2014
DOI:10.1039/C4RA08316D
Although the fabrication of mesoporous carbon films through the cooperative assembly of phenolic resin oligomers (resol) and block copolymers is well-established, these methods generally rely upon simply following protocols developed for analogous bulk powders despite growing evidence of significant differences between thin films and bulk powders. Here, we examine the chemical evolution of resol through spectroscopically tracking the methylol content for films with and without a common templating agent (Pluronic F127). At lower temperature (100 °C and 120 °C), the crosslinking rate is not impacted by the presence of the template, while the addition of the Pluronic template decreases the reaction rate at higher temperatures (140 °C and 160 °C). At all conditions examined, the crosslinking kinetics of resol can be fit using the Jander model. in situ grazing incidence small angle X-ray scattering illustrates that the mesostructural evolution is not highly correlated with the chemical changes during crosslinking. With this knowledge, a shortened processing schedule (3 h total) was devised in place of the standard 24 h thermopolymerization without adversely impacting the ordered structure of the mesoporous film and this protocol significantly increases (almost double) the porosity of the film.
Co-reporter:Guodong Deng, Zhe Qiang, Willis Lecorchick, Kevin A. Cavicchi, and Bryan D. Vogt
Langmuir 2014 Volume 30(Issue 9) pp:2530-2540
Publication Date(Web):2017-2-22
DOI:10.1021/la404964c
Cooperative self-assembly of block copolymers with (in)organic precursors effectively generates ordered nanoporous films, but the porosity is typically limited by the need for a continuous (in)organic phase. Here, a network of homogeneous fibrous nanostructures (≈20 nm diameter cylinders) having high porosity (≈ 60%) is fabricated by cooperative self-assembly of a phenolic resin oligomer (resol) with a novel, nonfrustrated, ABC amphiphilic triblock copolymer template, poly(ethylene oxide)-block-poly(ethyl acrylate)-block-polystyrene (PEO-b-PEA-b-PS), via a thermally induced self-assembly process. Due to the high glass transition temperature (Tg) of the PS segments, the self-assembly behavior is kinetically hindered as a result of competing effects associated with the ordering of the self-assembled system and the cross-linking of resol that suppresses segmental mobility. The balance in these competing processes reproducibly yields a disordered fibril network with a uniform fibril diameter. This nonequilibrium morphology is dependent on the PEO-b-PEA-b-PS to resol ratio with an evolution from a relatively open fibrous structure to an apparent poorly ordered mixed lamellae-cylinder morphology. Pyrolysis of these former films at elevated temperatures yields a highly porous carbon film with the fibril morphology preserved through the carbonization process. These results illustrate a simple method to fabricate thin films and coatings with a well-defined fiber network that could be promising materials for energy and separation applications.
Co-reporter:Jeongwoo Lee, Murthy V. S. N. Maddipatla, Abraham Joy, and Bryan D. Vogt
Macromolecules 2014 Volume 47(Issue 9) pp:2891-2898
Publication Date(Web):April 22, 2014
DOI:10.1021/ma500328r
Photoresponsive thin films are commonly encountered as high performance coatings as well as critical component, photoresists, for microelectronics manufacture. Despite intensive investigations into the dynamics of thin glassy polymer films, studies involving reactions of thin films have typically been limited by difficulties in decoupling segregation of reacting components or catalysts due to the interfaces. Here, thin films of coumarin polyesters overcome this limitation where the polyester undergoes predominately cross-linking upon irradiation at 350 nm, while chain scission occurs with exposure to 254 nm light. Spectroscopic ellipsometry is utilized to track these reactions as a function of exposure time to elucidate the associated reaction kinetics for films as thin as 15 nm. The cross-linking appears to follow a second order kinetic rate law, while oxidation of the coumarin that accompanies the chain scission and enables this reaction to be tracked spectroscopically appears to be a first order reaction in coumarin concentration. Because of the asymmetry in the coumarin diol monomer and the associated differences in local structure that result during formation of the polyester, two populations of coumarin are required to fit the reaction kinetics; 10–20% of the coumarin is significantly more reactive, but these groups appear to undergo chain scission/oxidation at both wavelengths. These reaction rate constants are nearly independent (within 1 order of magnitude) of film thickness down to 15 nm. There is maximum rate at a finite thickness for the 254 nm exposure, which we attribute to constructive interference of the UV radiation within the polymer film, rather than typical confinement effects; no thickness dependence in reaction rates is observed for the 350 nm exposure. The utilization of a single polymer with two distinct reactions enables unambiguous investigation of thickness effects on reactions.
Co-reporter:Zhe Qiang, Longhe Zhang, Gila E. Stein, Kevin A. Cavicchi, and Bryan D. Vogt
Macromolecules 2014 Volume 47(Issue 3) pp:1109-1116
Publication Date(Web):January 28, 2014
DOI:10.1021/ma402131j
One challenge associated with the utilization of block copolymers in nanotechnology is the difficulties associated with alignment and orientation of the self-assembled nanostructure on macroscopic length scales. Here we demonstrate a simple method to generate unidirectional alignment of the cylindrical domains of polystyrene-block-polyisoprene-block-polystyrene, SIS, based on a modification of the commonly utilized solvent vapor annealing (SVA) process. In this modification, cross-linked poly(dimethylsiloxane) (PDMS) is physically adhered to the SIS film during SVA; differential swelling of the PDMS and SIS produces a shear force to align the ordered domains of SIS in the areas covered by PDMS. This method is termed solvent vapor annealing with soft shear (SVA-SS). The alignment direction can be readily controlled by the shape and placement of the PDMS with the alignment angle equal to the diagonal across the rectangular PDMS pad due to a propagating deswelling front from directional drying of the PDMS by a dry air stream. Herman’s (second order) orientational parameter, S, can quantify the quality of the alignment over large areas with S > 0.94 obtainable using SVA-SS.
Co-reporter:Jiachen Xue, Gurpreet Singh, Zhe Qiang, Kevin G. Yager, Alamgir Karim and Bryan D. Vogt
Nanoscale 2013 vol. 5(Issue 24) pp:12440-12447
Publication Date(Web):04 Oct 2013
DOI:10.1039/C3NR03591C
Ordered mesoporous carbons exhibit appealing properties for many applications, but their function and performance can depend critically on their structure. The in-plane orientation of 2D cylinders from the cooperative assembly of Pluronic P123 and resol has been controlled by application of cold zone annealing (CZA). By varying the moving rate, the preferential in-plane orientation of the self-assembled cylinders can be tuned through the entire 180° range possible from ϕ = 50° to ϕ = −130° (relative to the moving direction). At a moving rate of 2 μm s−1, this simple and easy CZA process leads to cylinders that are well aligned parallel to the moving direction with a high orientational factor of S = 0.98. Moreover, the in-plane oriented cylinders can be nearly perfectly aligned transverse to the moving direction (S = 0.95) by simply decreasing the moving velocity to 0.5 μm s−1. We attribute the parallel alignment to the flow that develops from the motion of the thermal gradients, while the transverse alignment is related to flow cessation (inertial effect). The preferential orientation is retained through the carbonization process, but there is some degradation in orientation due to insufficient crosslinking of the resol during CZA; this effect is most prominent for the higher moving rates (less time for crosslinking), but can be overcome by post-CZA annealing at uniform elevated temperatures to further crosslink the resol. CZA is a simple and powerful method for fabricating well-aligned and self-assembled mesoporous carbon films over large areas.
Co-reporter:Jiachen Xue, Gurpreet Singh, Zhe Qiang, Alamgir Karim and Bryan D. Vogt
Nanoscale 2013 vol. 5(Issue 17) pp:7928-7935
Publication Date(Web):05 Jul 2013
DOI:10.1039/C3NR02821F
Surfactant or block copolymer-templated mesoporous films have been extensively explored, but achieving mesostructure coherence and unidirectional orientation over macroscopic dimensions has remained quite challenging for these self-assembled systems. Here, we extend the concepts associated with zone refinement of crystalline materials to soft templated mesoporous carbon films based on the cooperative assembly of commercial non-ionic surfactants (block copolymers) and phenolic resin oligomers (resol) to provide macroscopic alignment of both cubic (FDU-16) and hexagonal (FDU-15) mesostructures. The average orientation of these mesophases is determined from rotation grazing incidence small angle X-ray scattering (GISAXS) measurements. For FDU-15 templated by Pluronic P123, the orientation factor for the zone-annealed film is 0.98 based on the average of the second Legendre polynomial, but this orientation deteriorates significantly during carbonization. Notably, a thermal stabilization step following zone annealing preserves the orientation of the mesostructure during carbonization. The orientation factor for an isotropic cubic structure (FDU-16 templated by Pluronic F127) is only 0.48 (based on the 111 reflection with incident angle 0.15°) for the same zone annealing protocol, but this illustrates the versatility of zone annealing to different mesostructures. Unexpectedly, zone annealing of FDU-15 templated by Pluronic F127 leads to stabilization of the mesostructure through carbonization, whereas this structure collapses fully during carbonization even after extended oven annealing; despite no clear macroscopic orientation of the cylindrical mesostructure from zone annealing. Thermal zone annealing provides a simple methodology to produce highly ordered and macroscopically oriented stable mesoporous carbon films, but the efficacy is strongly tied to the mobility of the template during the zone annealing.
Co-reporter:Mingzhi Dai, Brittney Haselwood, Bryan D. Vogt, Jeffrey T. La Belle
Analytica Chimica Acta 2013 Volume 788() pp:32-38
Publication Date(Web):25 July 2013
DOI:10.1016/j.aca.2013.06.019
•Screen printed norepinephrine sensors are prepared using high surface area carbon inks.•The impact of surface area and pore size of the carbons to the sensitivity of the sensors has been investigated.•Increase in surface area and pore size improves the sensitivity of the sensor.•The detection limit of norepinephrine in whole rabbit blood is as low as 100 pg mL−1.Norepinephrine (NE) is detected amperometrically using the enzyme Phenylethanolamine N-methyl transferase and cofactor S-(5′-Adenosyl)-l-methionine chloride dihydrochloride with disposable screen printed mesoporous carbon electrodes. The role of internal surface area and pore size of the mesoporous carbon is systematically examined using soft-templated, mesoporous silica–carbon powders with highly microporous walls obtained from etching of the silica to produce powders with surface areas ranging from 671–2339 m2 g−1. As the surface area increases, the sensitivity of the biosensor at very low NE concentrations (0–500 pg mL−1) in phosphate buffered saline (PBS) increases just as the current signal increases with respect to the NE concentration of 81–1581 μA mL ng−1 cm−2 for the mesoporous carbons. The best performing electrode provides similar sensitivity in whole rabbit blood in comparison to PBS despite no membrane layer to filter the non-desired reactants; the small (<5 nm) pore size and large internal surface area acts to minimize non-specific events that decrease sensitivity.
Co-reporter:Todd D. Prichard
Polymer Engineering & Science 2013 Volume 53( Issue 1) pp:69-77
Publication Date(Web):
DOI:10.1002/pen.23237
Abstract
Single-wall carbon nanotubes (SWNTs) are promising filler materials for advanced polymer composites, but the impressive properties that have been predicted theoretically have not been realized experimentally. This gap is generally attributed to aggregation and nonideal dispersion of the SWNTs. Here, nonionic surfactants based on poly(ethylene oxide) are used to disperse SWNTs in either water or ethanol using sonication. The dispersed aqueous SWNTs are stable, while the analogous ethanol system yields loosely flocculated SWNTs. After drying these dispersions, the electrical conductivity of the flocculated system is at least an order of magnitude greater than the dispersed system at the same SWNT loading with conductivity greater than 20 S/cm obtained for the flocculated systems containing unsorted, commercial SWNTs. These flocculated systems can be readily sprayed to create conductive coatings. Despite their high electrical conductivity, these coatings provide only modest electromagnetic interference shielding (<20 dB) when testing large areas (30.5 × 30.5 cm2), which suggests significant heterogeneity or defects in these coatings that are not readily visible by eye or scanning electron microscopy. This defect mechanism is consistent with a decrease in shield efficacy at high SWNT loadings, despite no statistical change in the electrical conductivity of the coating. POLYM. ENG. SCI., 2013. © 2012 Society of Plastics Engineers
Co-reporter:Todd D. Prichard, Sudhanshu S. Singh, Nikhilesh Chawla, Bryan D. Vogt
Polymer 2013 Volume 54(Issue 3) pp:1130-1135
Publication Date(Web):5 February 2013
DOI:10.1016/j.polymer.2012.12.061
Co-reporter:Zhe Qiang, Jiachen Xue, Kevin A. Cavicchi, and Bryan D. Vogt
Langmuir 2013 Volume 29(Issue 10) pp:3428-3438
Publication Date(Web):February 8, 2013
DOI:10.1021/la304915j
Ordered mesoporous (2–50 nm) carbon films were fabricated using cooperative self-assembly of a phenolic resin oligomer with a novel block copolymer template (poly(styrene-block-N,N-dimethyl-n-octadecylamine p-styrenesulfonate), (PS-b-PSS-DMODA)) synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization. Due to the high Tg of the PS segment and the strong interactions between the phenolic resin and the PSS-DMODA, the segmental rearrangement is kinetically hindered relative to the cross-linking rate of the phenolic resin, which inhibits long-range ordering and yields a poorly ordered mesoporous carbon with a broad pore size distribution. However, relatively short exposure (2 h) to controlled vapor pressures of methyl ethyl ketone (MEK) yields significant improvements in the long-range ordering and narrows the pore size distribution. The average pore size increases as the solvent vapor pressure during annealing increases, but an upper limit of p/p0 = 0.85 exists above which the films dewet rapidly during solvent vapor annealing. This approach can be extended using mesityl oxide, which has similar solvent qualities to MEK, but is not easily removed by ambient air drying after solvent annealing. This residual solvent can impact the morphology that develops during cross-linking of the films. These results illustrate the ability to fine-tune the mesostructure of ordered mesoporous carbon films through simple changes in the processing without any compositional changes in the initial cast film.
Co-reporter:Changhuai Ye, Gurpreet Singh, Maurice L. Wadley, Alamgir Karim, Kevin A. Cavicchi, and Bryan D. Vogt
Macromolecules 2013 Volume 46(Issue 21) pp:8608-8615
Publication Date(Web):October 23, 2013
DOI:10.1021/ma401780r
Mechanical properties of multicomponent polymers such as block copolymers (BCP) are critical to their utilization in thin film applications, yet the effect of block alignment of even the simplest BCPs on thin film modulus has not been thoroughly explored. Polystyrene-block-polydimethylsiloxane (PS-b-PDMS) thin films with cylindrical PDMS domains (fPDMS = 0.23) are aligned by cold zone annealing–soft shear mode (CZA-SS). Increasing the maximum temperature of the thermal zone and/or decreasing the CZA-SS velocity improves the alignment of the PS-b-PDMS domains as determined by atomic force microscopy (AFM) and small-angle neutron scattering (SANS). The maximum Hermans orientation factor (S) for this system using the CZA-SS conditions examined is S ≈ 0.85, yielding anisotropic mechanical properties as determined by surface wrinkling. While the in-plane modulus perpendicular to PDMS cylinder axis is nearly invariant of S, the modulus parallel to the cylinder axis is increased by a maximum of 31% for S ≈ 0.85, compared to its unaligned state (S ≈ 0). These results demonstrate the highly anisotropic response of the glassy matrix strength to alignment of internal rubbery cylindrical channels. This anisotropic structure–property coupling is consistent with expectations associated with highly aligned composites, but this CZA-SS methodology enables facile examination of the influence of extent of alignment on physical properties that has not been quantitatively investigated in the past.
Co-reporter:Zhe Qiang, Jiachen Xue, Gila E. Stein, Kevin A. Cavicchi, and Bryan D. Vogt
Langmuir 2013 Volume 29(Issue 27) pp:8703-8712
Publication Date(Web):June 5, 2013
DOI:10.1021/la401505h
The structure of ordered mesoporous carbons fabricated using poly(styrene-block-N,N,-dimethyl-n-octadecylamine p-styrenesulfonate) (PS-b-PSS-DMODA) as the template and phenolic resin (resol) as the carbon source can be easily manipulated by inclusion of low concentrations of low volatility selective solvents in the casting solution. Casting from neat methyl ethyl ketone yields a disordered structure even upon thermal annealing. However, addition of both dioctyl phthalate (DOP, PS selective) and dimethyl sulfoxide (DMSO, resol and PSS-DMODA selective) at modest concentrations to this casting solution provides sufficient mobility to produce highly ordered films with cylindrical mesopores. The DOP acts to swell the hydrophobic domain and can more than double the mesopore size, while the DMSO acts to swell the resol phase. Moreover, the surface area of the mesoporous carbons increases significantly as the meosopore size increases. This is a result of the decrease in wall thickness, which can be ascertained by the constant d-spacing of the mesostructure as the pore size increases. This behavior is counter to the typical effect of pore swelling agents that increase the pore size and decrease the surface area. Moreover, with only 4 wt % DOP/DMSO in the solution (20 wt % relative to solids), the scattering profiles exhibit many orders of diffraction, even upon carbonization, which is not typically observed for soft templated films. Variation in the concentration of DOP and DMSO during casting enables facile tuning of the structure of mesoporous carbon films.
Co-reporter:James G. Gaillard, Chelsea Hendrus, and Bryan D. Vogt
Langmuir 2013 Volume 29(Issue 48) pp:15083-15089
Publication Date(Web):2017-2-22
DOI:10.1021/la4038167
Addition of a small fraction of hydrophobic photoacid generator (PAG) to furfuryl alcohol provides a facile route to generate wrinkle topology by acid-catalyzed polymerization that is induced by ultraviolet (UV) light. Here, we describe how the primary characteristic parameters, wavelength and amplitude, of these periodic wrinkles can be tuned through control of the thickness of this furfuryl alcohol–PAG solution prior to UV exposure and the environmental humidity. As the initial coating thickness is increased, the wavelength remains unchanged at fixed temperature and PAG concentration, but the amplitude of the wrinkles increases exponentially with increased coating thickness. A wrinkle to crease transition is observed in some cases as the thickness of the solution coating is increased; this behavior is dependent on the PAG selection. Conversely, variation in relative humidity does not significantly impact the amplitude of the wrinkles, but there is a step change in the wavelength of the wrinkles near approximately 45% relative humidity with a factor of 3 decrease in the wavelength at high humidity. Through this knowledge, we have been able to fabrication wrinkles with an aspect ratio greater than 0.7 in a single step by UV exposure. These simple processing parameters to independently control wavelength and amplitude provide a facile route to systematically examine the role of aspect ratio of wrinkles on physical properties.
Co-reporter:Mingzhi Dai, Stephanie Maxwell, Bryan D. Vogt, Jeffrey T. La Belle
Analytica Chimica Acta 2012 Volume 738() pp:27-34
Publication Date(Web):13 August 2012
DOI:10.1016/j.aca.2012.05.038
Two ordered, soft-templated mesoporous carbon powders with cubic and hexagonal framework structure and four different commercial, low cost methacrylate-based polymer binders with widely varying physical properties are investigated as screen printed electrodes for glucose sensors using glucose oxidase and ferricyanide as the mediator. Both the chemistry and concentration of the binder in the electrode formulation can significantly impact the performance. Poly(hydroxybutyl methacrylate) as the binder provides hydrophilicity to enable transport of species in the aqueous phase to the carbon surface, but yet is sufficiently hydrophobic to provide mechanical robustness to the sensor. The current from the mesoporous carbon electrodes can be more than an order of magnitude greater than for a commercial printed carbon electrode (Zensor) with improved sensitivity for model glucose solutions. Even when applying these sensors to rabbit whole blood, the performance of these glucose sensors compares favorably to a standard commercial glucose meter with the lower detection limit of the mesoporous electrode being approximately 20 mg dL−1 despite the lack of a separation membrane to prevent non-specific events; these results suggest that the small pore sizes and high surface areas associated with ordered mesoporous carbons may effectively decrease some non-specific inferences for electrochemical sensing.Graphical abstract.Highlights► We prepared screen printed glucose sensor with two different mesoporous carbons. ► We investigated low cost, fluorine-free methacrylate-based as polymer binders. ► Cubic structure carbon provided higher signal than hexagonal structure carbon. ► Hydrophilicity of the binder greatly impacted the performance of the sensor. ► The best sensor accurately measured glucose level in rabbit blood.
Co-reporter:Jessica M. Torres, Christopher M. Stafford and Bryan D. Vogt
Soft Matter 2012 vol. 8(Issue 19) pp:5225-5232
Publication Date(Web):27 Mar 2012
DOI:10.1039/C2SM00037G
Periodic wrinkled surfaces have generated significant interest over the past decade as these structures can be easily fabricated over large areas with minimal fabrication cost, but these structures have generally been limited to thin films on soft elastomeric substrates, which limits their general applicability and utility. Here we present a simple methodology to generate wrinkled surfaces on rigid substrates by surface segregation of a photocatalyst. Upon ultraviolet light (UV) induced photopolymerization, increased catalyst concentration yields a cross-linked layer at the free surface that is supported on top of a more liquid-like bulk film due to differences in polymerization rate. Further polymerization of the underlayer provides the requisite mechanical stress (contraction due to polymerization) to create a wrinkled pattern. A system based upon the renewable monomer, furfuryl alcohol, that is cross-linked with a photoacid generator, triphenyl sulfonium triflate, is utilized to illustrate this concept. Moreover, the polymerized furfuryl alcohol can be transformed into amorphous carbon by heating at elevated temperatures in an inert environment. The role of photoacid generator concentration and substrate temperature on the wrinkle formation and morphology is presented. Finally, exposure through a simple mask can generate hierarchical structures with the wrinkled structure conforming to the geometric constraints of the photopatterned area, including curvilinear structures. This photocatalyzed surface segregation-based methodology provides a promising route to the facile fabrication of microstructured surfaces based upon the wrinkling instability.
Co-reporter:Mingzhi Dai, Bryan D. Vogt
Journal of Colloid and Interface Science 2012 Volume 387(Issue 1) pp:127-134
Publication Date(Web):1 December 2012
DOI:10.1016/j.jcis.2012.06.062
Mesoporous carbons containing cobalt nanoparticles are synthesized by tri-or quad-constituent self assembly of Pluronic F127, phenol-formaldehyde oligomer (resol), cobalt acetylacetonate (acac), and optionally tetraethyl orthosilicate (TEOS, optional). Upon pyrolysis in N2 atmosphere, the resol provides sufficient carbon yield to maintain the ordered structure, while decomposition of the Co(acac) yields cobalt nanoparticles. To provide increased surface area, the dispersed silicate from condensation of TEOS can be etched after carbonization to yield micropores, Without silica templated micropores, the surface area decreases as the cobalt content increases, but there is a concurrent increase in the volume-average pore diameter (BHJ) and a dramatic increase in the adsorption capacity of methylene green with the equilibrium adsorption capacity from 2 to 90 mg/g with increasing Co content. Moreover, the surface area and pore size of mesoporous composites can be dramatically increased by addition of TEOS and subsequent etching. These composites exhibit extremely high adsorption capacity up to 1151 mg/g, which also increases with increases in the Co content. Additionally, the inclusion of cobalt nanoparticles provides magnetic separation from aqueous suspension. The in situ synthesis of the Co nanoparticles yields to a carbon shell that can partially protect the Co from leaching in acidic media; after 96 h in 2 M HCl, the powders remain magnetic.Graphical abstractHighlights► High surface mesoporous carbon–Co composites exhibit large adsorption capacity. ► Micropores from co-assembly and etching of silica enhance adsorption capacity. ► Adsorption capacity of the composites increases with Co content. ► High magnetization enables facile separation from aqueous suspension.
Co-reporter:Alpha Labiano, Mingzhi Dai, David Taylor, Wen-Shiue Young, Thomas H. Epps III, Kaushal Rege, Bryan D. Vogt
Microporous and Mesoporous Materials 2012 160() pp: 143-150
Publication Date(Web):
DOI:10.1016/j.micromeso.2012.05.003
Co-reporter:Jessica M. Torres;Christopher M. Stafford;David Uhrig
Journal of Polymer Science Part B: Polymer Physics 2012 Volume 50( Issue 5) pp:370-377
Publication Date(Web):
DOI:10.1002/polb.23014
Abstract
The modulus and glass transition temperature (Tg) of ultrathin films of polystyrene (PS) with different branching architectures are examined via surface wrinkling and the discontinuity in the thermal expansion as determined from spectroscopic ellipsometry, respectively. Branching of the PS is systematically varied using multifunctional monomers to create comb, centipede, and star architectures with similar molecular masses. The bulk-like (thick film) Tg for these polymers is 103 ± 2 °C and independent of branching and all films thinner than 40 nm exhibit reductions in Tg. There are subtle differences between the architectures with reductions in Tg for linear (25 °C), centipede (40 °C), comb (9 °C), and 4 armed star (9 °C) PS for ≈ 5 nm films. Interestingly, the room temperature modulus of the thick films is dependent upon the chain architecture with the star and comb polymers being the most compliant (≈2 GPa) whereas the centipede PS is most rigid (≈4 GPa). The comb PS exhibits no thickness dependence in moduli, whereas all other PS architectures examined show a decrease in modulus as the film thickness is decreased below ∼40 nm. We hypothesize that the chain conformation leads to the apparent susceptibility of the polymer to reductions in moduli in thin films. These results provide insight into potential origins for thickness dependent properties of polymer thin films. © 2011 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys, 2012
Co-reporter:Jessica M. Torres, Chengqing Wang, E. Bryan Coughlin, John P. Bishop, Richard A. Register, Robert A. Riggleman, Christopher M. Stafford, and Bryan D. Vogt
Macromolecules 2011 Volume 44(Issue 22) pp:9040-9045
Publication Date(Web):October 25, 2011
DOI:10.1021/ma201482b
The effect of main chain stiffness and side chain flexibility on the elastic modulus and glass transition temperature (Tg) of thin polymer films is investigated using nontraditional polymers formed from 5-(2-phenylethylnorbornene). Depending on the polymerization route chosen, the resulting polymer backbone is comprised of either bicyclic (norbornyl) units, which leads to a relatively rigid polymer with a high bulk Tg, or monocyclic (cyclopentyl) units, which leads to a more flexible structure with a lower bulk Tg. The modulus and Tg of the rigid bicyclic polymer are thickness independent down to <10 nm, whereas the modulus of the more flexible monocyclic polymer decreases with decreasing thickness. By hydrogenation of the pendant phenyl ring to the cyclohexyl counterpart, we illustrate that minor changes in the relative flexibility of the side chain do not impact the observed thin film behavior. To further support our hypothesis that main chain stiffness (persistence length) plays a central role in confinement-induced changes in physical properties, a series of arylate/phosphonate copolymers, where the addition of phosphonate groups decreases the overall stiffness of the polymer backbone, are also examined. Similar to the polynorbornenes, the poly(arylate-co-phosphonate) polymers exhibit more pronounced thickness-dependent properties as the stiffness of the backbone decreases. We believe based upon our results that examination of chain flexibility in the thin film glass community for future works would be extremely useful in elucidating the physical origins of observed phenomena.
Co-reporter:Zhe Qiang, Yu-Ming Chen, Burcu Gurkan, Yuanhao Guo, Miko Cakmak, Kevin A. Cavicchi, Yu Zhu, Bryan D. Vogt
Carbon (May 2017) Volume 116() pp:
Publication Date(Web):May 2017
DOI:10.1016/j.carbon.2017.01.093
Single pot, multi-component assembly of resol (carbon source), Pluronic F127, tetraethylorthosilicate (TEOS), dicyandiamide (nitrogen source) and iron nitrate (iron oxide source) yields nitrogen-doped ordered mesoporous carbon/iron oxide nanocomposites on carbonization. Etching the silica derived from the TEOS generates micropores in the carbon framework. The incorporation of iron oxide and nitrogen doping of the carbon tends to decrease the surface area; at approximately 3.7 wt% nitrogen and 21 wt% iron oxide (OMC-3.7-21), the surface area decreases from 2162 m2/g for the undoped ordered mesoporous carbon (OMC) to 974 m2/g and the pore volume decreases from 1.62 cm3/g to 0.6 cm3/g due to deformation of the nanostructure by crystallization of γ-Fe2O3 nanoparticles and N-doping of the carbon. Despite the decreased surface area, the reversible capacity when used as anodes in a sodium ion battery is increased from 110 mAh/g (undoped OMC) to 275 mAh/g (OMC-3.7-21) due to high capacity from the γ-Fe2O3 nanoparticles and the improved capacity associated with nitrogen doping of carbon. These nitrogen-doped ordered mesoporous carbon/iron oxide nanocomposites exhibit good cycle stability with almost 99% capacity retention after 350 charge/discharge cycles. The combination of low-cost and excellent electrochemical performance makes these mesoporous nanocomposites promising anode materials for sodium-ion batteries.
Co-reporter:Guodong Deng, Yuanzhong Zhang, Changhuai Ye, Zhe Qiang, Gila E. Stein, Kevin A. Cavicchi and Bryan D. Vogt
Chemical Communications 2014 - vol. 50(Issue 84) pp:NaN12687-12687
Publication Date(Web):2014/09/03
DOI:10.1039/C4CC02471K
Bicontinuous mesoporous carbon films are fabricated by cooperative self-assembly of phenolic resin and amphiphilic triblock copolymer via an order–order transition from cylinders to gyroid. The film morphology is strongly influenced by the details of processing, including age of the resol, resol:template ratio, and the solvent vapor annealing process.
Co-reporter:Yuanzhong Zhang, Sarang M. Bhaway, Yi Wang, Kevin A. Cavicchi, Matthew L. Becker and Bryan D. Vogt
Chemical Communications 2015 - vol. 51(Issue 24) pp:NaN5000-5000
Publication Date(Web):2015/02/16
DOI:10.1039/C4CC09808K
Rapid chemical transformation from micelle templated precursors (metal nitrate and citric acid) to ordered mesoporous metal carbonates and oxides is demonstrated using microwave heating for cobalt, copper, manganese and zinc. Without aging requirements, <3 min of microwave processing yields highly ordered mesoporous films.
Co-reporter:Sarang M. Bhaway, Pattarasai Tangvijitsakul, Jeongwoo Lee, Mark D. Soucek and Bryan D. Vogt
Journal of Materials Chemistry A 2015 - vol. 3(Issue 42) pp:NaN21069-21069
Publication Date(Web):2015/09/16
DOI:10.1039/C5TA04520G
Micelle-templated ordered mesoporous nickel–cobalt carbonates and oxides are fabricated using a metal nitrate–citric acid strategy, which avoids the hydrolysis and aging requirements associated with sol–gel chemistry. A series of mesoporous NixCo(3−x)(CO3)y and NixCo(3−x)O4 films with varying Ni–Co compositions and 14 ± 4 nm mesopores are fabricated with the same block copolymer template. AFM and GISAXS analysis indicates that the mesostructure is maintained through the formation of the carbonate and oxide, while GIXD profiles confirm formation of pure spinel phases of semi-crystalline NixCo(3−x)O4. The micelle templated mesopores are interconnected and provide transport paths for the electrolyte to minimize the solid-state diffusion requirements associated with battery electrodes. These materials exhibit good performance as sodium ion battery anodes even at high current densities of 4 A g−1. Amongst the mixed-metal oxides, Ni2CoO4 exhibits the highest specific capacity of 239 mA h g−1 after galvanostatic cycling at a current density of 1 A g−1 for 10 cycles. We attribute the superior performance of Ni2CoO4 at high rates to the high surface area and short ion-diffusion paths of the nanoporous anode architecture, while the higher nickel content in the mixed metal oxide provides enhanced stability during oxide formation along with enhanced electronic conductivity, leading to improved cycling stability of the anode. This micelle template metal nitrate–citric acid method enables new possibilities for fabricating variety of ordered mesoporous mixed-metal carbonates and oxides that could be used in a wide range of applications.









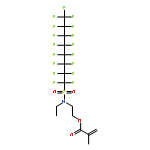
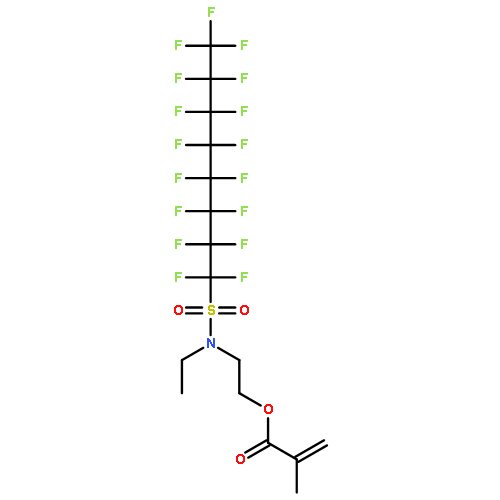






![Bicyclo[2.2.1]hept-5-ene-2-ethanol,a,a-bis(trifluoromethyl)-](http://img.cochemist.com/ccimg/196400/196314-61-1.png)
![Bicyclo[2.2.1]hept-5-ene-2-ethanol,a,a-bis(trifluoromethyl)-](http://img.cochemist.com/ccimg/196400/196314-61-1_b.png)