Co-reporter:Aaron A. Koch, Douglas A. Hansen, Vikram V. Shende, Lawrence R. Furan, K. N. Houk, Gonzalo Jiménez-Osés, and David H. Sherman
Journal of the American Chemical Society September 27, 2017 Volume 139(Issue 38) pp:13456-13456
Publication Date(Web):August 24, 2017
DOI:10.1021/jacs.7b06436
Macrolactonization of natural product analogs presents a significant challenge to both biosynthetic assembly and synthetic chemistry. In the preceding paper, we identified a thioesterase (TE) domain catalytic bottleneck processing unnatural substrates in the pikromycin (Pik) system, preventing the formation of epimerized macrolactones. Here, we perform molecular dynamics simulations showing the epimerized hexaketide was accommodated within the Pik TE active site; however, intrinsic conformational preferences of the substrate resulted in predominately unproductive conformations, in agreement with the observed hydrolysis. Accordingly, we engineered the stereoselective Pik TE to yield a variant (TES148C) with improved reaction kinetics and gain-of-function processing of an unnatural, epimerized hexaketide. Quantum mechanical comparison of model TES148C and TEWT reaction coordinate diagrams revealed a change in mechanism from a stepwise addition–elimination (TEWT) to a lower energy concerted acyl substitution (TES148C), accounting for the gain-of-function and improved reaction kinetics. Finally, we introduced the S148C mutation into a polyketide synthase module (PikAIII-TE) to impart increased substrate flexibility, enabling the production of diastereomeric macrolactones.
Co-reporter:Shuo-Qing Zhang, Buck L. H. Taylor, Chong-Lei Ji, Yuan Gao, Michael R. Harris, Luke E. Hanna, Elizabeth R. Jarvo, K. N. Houk, and Xin Hong
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:12994-12994
Publication Date(Web):August 25, 2017
DOI:10.1021/jacs.7b04973
Nickel catalysts have shown unique ligand control of stereoselectivity in the Suzuki–Miyaura cross-coupling of boronates with benzylic pivalates and derivatives involving C(sp3)–O cleavage. The SIMes ligand (1,3-dimesityl-4,5-dihydroimidazol-2-ylidene) produces the stereochemically inverted C–C coupling product, while the tricyclohexylphosphine (PCy3) ligand delivers the retained stereochemistry. We have explored the mechanism and origins of the ligand-controlled stereoselectivity with density functional theory (DFT) calculations. The oxidative addition determines the stereoselectivity with two competing transition states, an SN2 back-side attack type transition state that inverts the benzylic stereogenic center and a concerted oxidative addition through a cyclic transition state, which provides stereoretention. The key difference between the two transition states is the substrate–nickel–ligand angle distortion; the ligand controls the selectivity by differentiating the ease of this angle distortion. For the PCy3 ligand, the nickel–ligand interaction involves mainly σ-donation, which does not require a significant energy penalty for the angle distortion. The facile angle distortion with PCy3 ligand allows the favorable cyclic oxidative addition transition state, leading to the stereoretention. For the SIMes ligand, the extra d–p back-donation from nickel to the coordinating carbene increases the rigidity of the nickel–ligand bond, and the corresponding angle distortion is more difficult. This makes the concerted cyclic oxidative addition unfavorable with SIMes ligand, and the back-side SN2-type oxidative addition delivers the stereoinversion.
Co-reporter:Shuo-Qing Zhang, Buck L. H. Taylor, Chong-Lei Ji, Yuan Gao, Michael R. Harris, Luke E. Hanna, Elizabeth R. Jarvo, K. N. Houk, and Xin Hong
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:12994-12994
Publication Date(Web):August 25, 2017
DOI:10.1021/jacs.7b04973
Nickel catalysts have shown unique ligand control of stereoselectivity in the Suzuki–Miyaura cross-coupling of boronates with benzylic pivalates and derivatives involving C(sp3)–O cleavage. The SIMes ligand (1,3-dimesityl-4,5-dihydroimidazol-2-ylidene) produces the stereochemically inverted C–C coupling product, while the tricyclohexylphosphine (PCy3) ligand delivers the retained stereochemistry. We have explored the mechanism and origins of the ligand-controlled stereoselectivity with density functional theory (DFT) calculations. The oxidative addition determines the stereoselectivity with two competing transition states, an SN2 back-side attack type transition state that inverts the benzylic stereogenic center and a concerted oxidative addition through a cyclic transition state, which provides stereoretention. The key difference between the two transition states is the substrate–nickel–ligand angle distortion; the ligand controls the selectivity by differentiating the ease of this angle distortion. For the PCy3 ligand, the nickel–ligand interaction involves mainly σ-donation, which does not require a significant energy penalty for the angle distortion. The facile angle distortion with PCy3 ligand allows the favorable cyclic oxidative addition transition state, leading to the stereoretention. For the SIMes ligand, the extra d–p back-donation from nickel to the coordinating carbene increases the rigidity of the nickel–ligand bond, and the corresponding angle distortion is more difficult. This makes the concerted cyclic oxidative addition unfavorable with SIMes ligand, and the back-side SN2-type oxidative addition delivers the stereoinversion.
Co-reporter:Isaac Chogii, Pradipta Das, Jason S. Fell, Kevin A. Scott, Mark N. Crawford, K. N. Houk, and Jon T. Njardarson
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:13141-13141
Publication Date(Web):September 8, 2017
DOI:10.1021/jacs.7b07319
We report useful new lithium-assisted asymmetric anion-accelerated amino-Cope rearrangement cascades. A strategic nitrogen atom chiral auxiliary serves three critical roles, by (1) enabling in situ assembly of the chiral 3-amino-1,5-diene precursor, (2) facilitating the rearrangement via a lithium enolate chelate, and (3) imparting its influence on consecutive inter- or intramolecular C–C or C–X bond-forming events via resulting chiral enamide intermediates or imine products. The mechanism of the amino-Cope rearrangement was explored with density functional theory. A stepwise dissociation–recombination mechanism was found to be favored. The stereochemistry of the chiral auxiliary determines the stereochemistry of the Cope product by influencing the orientation of the lithium dienolate and sulfinylimine fragments in the recombination step. These robust asymmetric anion-accelerated amino-Cope enabled cascades open the door for rapid and predictable assembly of complex chiral acyclic and cyclic nitrogen-containing motifs in one pot.
Co-reporter:Rajat Maji, Pier Alexandre Champagne, K. N. Houk, and Steven E. Wheeler
ACS Catalysis October 6, 2017 Volume 7(Issue 10) pp:7332-7332
Publication Date(Web):September 13, 2017
DOI:10.1021/acscatal.7b02993
Density functional theory methods were used to elucidate the activation mode and origin of stereoselectivity in chiral phosphoric acid-catalyzed intramolecular oxetane desymmetrizations. Computed enantioselectivities are in excellent agreement with experiment. An unexpected, distortion-driven activation mode was observed, instead of the usual “bifunctional activation”. This mode is favored for only some intramolecular oxetane openings, highlighting an exception to known models. Stereoselectivity in these reactions can be explained by the balance of favorable noncovalent interactions of the substrates with both the aryl substituents and phosphoric acid functionality of the catalysts.Keywords: bifunctional activation; density functional theory; enantioselectivity; intramolecular oxetane desymmetrization;
Co-reporter:Jessica M. Grandner, Huiling Shao, Robert H. Grubbs, Peng Liu, and K. N. Houk
The Journal of Organic Chemistry October 6, 2017 Volume 82(Issue 19) pp:10595-10595
Publication Date(Web):August 25, 2017
DOI:10.1021/acs.joc.7b02129
A comprehensive computational study of stereoretentive olefin metathesis with Ru-dithiolate catalysts has been performed. We have determined how the dithiolate ligand enforces a side-bound mechanism and how the side-bound mechanism allows for stereochemical control over the forming olefin. We have used density functional theory (DFT) and ligand steric contour maps to elucidate the origins of stereoretentive metathesis with the goal of understanding how to design a new class of E-selective metathesis catalysts.
Co-reporter:Pier Alexandre Champagne and K. N. Houk
The Journal of Organic Chemistry October 20, 2017 Volume 82(Issue 20) pp:10980-10980
Publication Date(Web):September 6, 2017
DOI:10.1021/acs.joc.7b01928
The geometries, stabilities, and 1,3-dipolar cycloaddition reactivities of 24 mesoionic azomethine ylides and imines were investigated using density functional theory calculations at the M06-2X/6-311+G-(d,p)/M06-2X/6-31G-(d) level. The computed structures highlight how the commonly used "aromatic" resonance form should be replaced by two more accurate resonance structures. Stabilities of the dipoles were assessed by various homodesmotic schemes and are consistent with these compounds being nonaromatic. The activation free energies with ethylene or acetylene range from 11.8 to 36.6 kcal/mol. Within each dipole type, the predicted cycloaddition reactivities correlate with the reaction energies and the resonance stabilization energies provided by the various substituents. Endocyclic (X) heteroatoms increase the reactivity of the 1,3-dipoles in the order of O > NH ≅ S, whereas exocyclic (Y) substituents increase it in the order of CH2 > NH > O > S. Distortion/interaction analysis indicated that the difference in reactivity between differently substituted 1,3-dipoles is driven by distortion, whereas the difference between azomethine ylides and imines is related to lower interaction energies of imines with the dipolarophiles.
Co-reporter:Cyndi Qixin He, Peiyuan Yu, Yu-hong Lam, and K. N. Houk
Organic Letters October 20, 2017 Volume 19(Issue 20) pp:5685-5685
Publication Date(Web):October 4, 2017
DOI:10.1021/acs.orglett.7b02851
The enantioselective coupling of indoles with racemic α-tosyloxy ketones mediated by a chiral amino alcohol catalyst is studied with density functional theory (DFT) calculations. The addition of indole to an oxyallyl cation intrinsically favors the (S,S) and (R,R) stereoisomeric products through electrostatic interactions in the transition state. Our model shows that the enantioselectivity is controlled by the cyclohexane moiety of the catalyst; selectivity diminishes upon removal of the cyclohexane ring. Substitution to enhance the enantioselectivity of this reaction is proposed.
Co-reporter:Byron A. Boon, Aaron G. Green, Peng Liu, K. N. Houk, and Craig A. Merlic
The Journal of Organic Chemistry May 5, 2017 Volume 82(Issue 9) pp:4613-4613
Publication Date(Web):April 14, 2017
DOI:10.1021/acs.joc.7b00203
Syntheses of strained cyclic dienes were accomplished via palladium(II)-catalyzed oxidative cyclizations of terminal bis(vinylboronate esters). The reactions generate strained (E,E)-1,3-dienes that undergo spontaneous 4π-electrocyclizations to form bicyclic cyclobutenes. Formation of the cyclobutenes is driven by the strain in the medium-ring (E,E)-1,3-diene intermediate. Thermal ring openings of the cyclobutenes give (Z,Z)-1,3-diene products, again for thermodynamic reasons. DFT calculations verified the thermodynamic versus kinetic control of the reactions, and kinetic studies are in excellent agreement with the calculated energy changes. An extension of the tandem coupling/4π-electrocyclization pathway was demonstrated by a palladium(II)-catalyzed oxidative homocoupling/8π-electrocyclization cascade.
Co-reporter:Shibdas Banerjee, Yun-Fang Yang, Ian D. Jenkins, Yong Liang, Anton A. Toutov, Wen-Bo Liu, David P. Schuman, Robert H. Grubbs, Brian M. Stoltz, Elizabeth H. Krenske, Kendall N. Houk, and Richard N. Zare
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:6880-6880
Publication Date(Web):May 2, 2017
DOI:10.1021/jacs.6b13032
Exploiting C–H bond activation is difficult, although some success has been achieved using precious metal catalysts. Recently, it was reported that C–H bonds in aromatic heterocycles were converted to C–Si bonds by reaction with hydrosilanes under the catalytic action of potassium tert-butoxide alone. The use of Earth-abundant potassium cation as a catalyst for C–H bond functionalization seems to be without precedent, and no mechanism for the process was established. Using ambient ionization mass spectrometry, we are able to identify crucial ionic intermediates present during the C–H silylation reaction. We propose a plausible catalytic cycle, which involves a pentacoordinate silicon intermediate consisting of silane reagent, substrate, and the tert-butoxide catalyst. Heterolysis of the Si–H bond, deprotonation of the heteroarene, addition of the heteroarene carbanion to the silyl ether, and dissociation of tert-butoxide from silicon lead to the silylated heteroarene product. The steps of the silylation mechanism may follow either an ionic route involving K+ and tBuO– ions or a neutral heterolytic route involving the [KOtBu]4 tetramer. Both mechanisms are consistent with the ionic intermediates detected experimentally. We also present reasons why KOtBu is an active catalyst whereas sodium tert-butoxide and lithium tert-butoxide are not, and we explain the relative reactivities of different (hetero)arenes in the silylation reaction. The unique role of KOtBu is traced, in part, to the stabilization of crucial intermediates through cation−π interactions.
Co-reporter:Wen-Bo Liu, David P. Schuman, Yun-Fang Yang, Anton A. Toutov, Yong Liang, Hendrik F. T. Klare, Nasri Nesnas, Martin Oestreich, Donna G. Blackmond, Scott C. Virgil, Shibdas Banerjee, Richard N. Zare, Robert H. Grubbs, K. N. Houk, and Brian M. Stoltz
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:6867-6867
Publication Date(Web):April 12, 2017
DOI:10.1021/jacs.6b13031
We recently reported a new method for the direct dehydrogenative C–H silylation of heteroaromatics utilizing Earth-abundant potassium tert-butoxide. Herein we report a systematic experimental and computational mechanistic investigation of this transformation. Our experimental results are consistent with a radical chain mechanism. A trialkylsilyl radical may be initially generated by homolytic cleavage of a weakened Si–H bond of a hypercoordinated silicon species as detected by IR, or by traces of oxygen which can generate a reactive peroxide by reaction with [KOt-Bu]4 as indicated by density functional theory (DFT) calculations. Radical clock and kinetic isotope experiments support a mechanism in which the C–Si bond is formed through silyl radical addition to the heterocycle followed by subsequent β-hydrogen scission. DFT calculations reveal a reasonable energy profile for a radical mechanism and support the experimentally observed regioselectivity. The silylation reaction is shown to be reversible, with an equilibrium favoring products due to the generation of H2 gas. In situ NMR experiments with deuterated substrates show that H2 is formed by a cross-dehydrogenative mechanism. The stereochemical course at the silicon center was investigated utilizing a 2H-labeled silolane probe; complete scrambling at the silicon center was observed, consistent with a number of possible radical intermediates or hypercoordinate silicates.
Co-reporter:Ilke Ugur, Sesil Agopcan Cinar, Burcu Dedeoglu, Viktorya Aviyente, M. Frederick Hawthorne, Peng Liu, Fang Liu, K. N. Houk, and Gonzalo Jiménez-Osés
The Journal of Organic Chemistry May 19, 2017 Volume 82(Issue 10) pp:5096-5096
Publication Date(Web):April 17, 2017
DOI:10.1021/acs.joc.7b00282
The reactions between low-valent Rh(I) and Ir(I) metal–carbonyl complexes and arylnitrile oxides possess the electronic and structural features of 1,3-dipolar cycloadditions. Density functional theory (DFT) calculations on these reactions, involving both cyclopentadienyl and carboranyl ligands on the metal carbonyl, explain the ease of the chemical processes and the stabilities of the resulting metallaisoxazolin-5-ones. The metal–carbonyl bond has partial double bond character according to the Wiberg index calculated through NBO analysis, and so the reaction can be considered a normal 1,3-dipolar cycloaddition involving M═C bonds. The rates of formation of the metallacycloadducts are controlled by distortion energy, analogous to their organic counterparts. The superior ability of anionic Ir complexes to share their electron density and accommodate higher oxidation states explains their calculated higher reactivity toward cycloaddition, as compared to Rh analogues.
Co-reporter:K. N. Houk and Fang Liu
Accounts of Chemical Research March 21, 2017 Volume 50(Issue 3) pp:539-539
Publication Date(Web):March 21, 2017
DOI:10.1021/acs.accounts.6b00532
Computational chemistry and biochemistry began with Isaac Newton’s classical mechanics in the 17th century and the establishment of quantum mechanics in the 1920s. Enabled by extraordinary advances in computers, in the last half century, this field has become a robust partner with experiment. The challenges facing computational chemists and biochemists, the Holy Grails of the field, are described. These include the development of a highly accurate density functional, ideally one that has universal chemical accuracy, and accurate polarizable force fields, as well as methods to handle efficiently the massive number of computations that must be performed for molecular dynamics and for the computation of flexible systems such as proteins. We estimate when the breakthroughs that will make computation a powerful engine for chemical discovery and design will be achieved. The Holy Grails of this field involve methods to enable the accurate and efficient prediction of structures and properties of complex biological systems and materials. The principal Holy Grail is a routine computational method for the prediction and design of multicomponent, often heterogeneous, functional systems and devices.
Co-reporter:Shu-Shan Gao, Marc Garcia-Borràs, Joyann S. Barber, Yang Hai, Abing Duan, Neil K. Garg, K. N. Houk, and Yi Tang
Journal of the American Chemical Society March 15, 2017 Volume 139(Issue 10) pp:3639-3639
Publication Date(Web):February 27, 2017
DOI:10.1021/jacs.7b01089
Hydroalkoxylation is a powerful and efficient method of forming C–O bonds and cyclic ethers in synthetic chemistry. In studying the biosynthesis of the fungal natural product herqueinone, we identified an enzyme that can perform an intramolecular enantioselective hydroalkoxylation reaction. PhnH catalyzes the addition of a phenol to the terminal olefin of a reverse prenyl group to give a dihydrobenzofuran product. The enzyme accelerates the reaction by 3 × 105-fold compared to the uncatalyzed reaction. PhnH belongs to a superfamily of proteins with a domain of unknown function (DUF3237), of which no member has a previously verified function. The discovery of PhnH demonstrates that enzymes can be used to promote the enantioselective hydroalkoxylation reaction and form cyclic ethers.
Co-reporter:Yun-Fang Yang, Gang Chen, Xin Hong, Jin-Quan Yu, and K. N. Houk
Journal of the American Chemical Society June 28, 2017 Volume 139(Issue 25) pp:8514-8514
Publication Date(Web):June 3, 2017
DOI:10.1021/jacs.7b01801
The origin of the unique effectiveness of six-membered chelates on the β-methylene C(sp3)–H activation reactions by Pd(II) catalyst was explained with density functional theory. The Pd(II) catalysts that involve five-membered chelates are inactive in this transformation. Computational studies suggest that the C(sp3)–H bond activation is the rate-limiting step in both cases. The C(sp3)–H bond activation with a five-membered chelate is unfavorable by 7.7 kcal/mol compared to the corresponding six-membered chelate with Pd(II). Two factors cause the difference: (1) the dimeric Pd species with five-membered chelation square-planar structure is more stable than that with six-membered chelation by 2.0 kcal/mol; (2) steric repulsion between the ArF group of the substrate and the quinoline group of the acetyl-protected aminomethyl quinoline ligand destabilizes the five-membered chelate transition structure by 5.7 kcal/mol. The six-membered chelate of Pd(II) with an acetyl-protected aminoethyl quinoline ligand orients the ligand away from the ArF group of the substrate and alleviates the steric repulsion.
Co-reporter:Peiyuan Yu, Tiffany Q. Chen, Zhongyue Yang, Cyndi Qixin He, Ashay Patel, Yu-hong Lam, Ching-Yang Liu, and K. N. Houk
Journal of the American Chemical Society June 21, 2017 Volume 139(Issue 24) pp:8251-8251
Publication Date(Web):May 23, 2017
DOI:10.1021/jacs.7b02966
The mechanisms and selectivities of the cycloadditions of tropone to dimethylfulvene have been investigated with M06-2X and B3LYP-D3 density functional theory (DFT) calculations and quasi-classical direct molecular dynamics simulations. The originally proposed reaction mechanism (Houk) involves a highly peri-, regio-, and stereoselective [6F + 4T] cycloaddition of tropone [4π] to dimethylfulvene [6π], followed by a [1,5] hydrogen shift, and, finally, a second [6 + 4] cycloaddition of tropone [6π] to the cyclopentadiene moiety [4π]. Paddon-Row and Warrener proposed an alternative mechanism: the initial cycloaddition involves a different [6T + 4F] cycloaddition in which fulvene acts as the 4π component, and a subsequent Cope rearrangement produces the formal [6F + 4T] adduct. Computations now demonstrate that the initial cycloaddition proceeds via an ambimodal transition state that can lead to both of the proposed [6 + 4] adducts. These adducts can interconvert through a [3,3] sigmatropic shift (Cope rearrangement). Molecular dynamics simulations reveal the initial distribution of products and provide insights into the time-resolved mechanism of this ambimodal cycloaddition. Competing [4 + 2] cycloadditions and various sigmatropic shifts are also explored.
Co-reporter:Elizabeth L. Noey, Zhongyue Yang, Yanwei Li, Hannah Yu, Rachel N. Richey, Jeremy M. Merritt, Douglas P. Kjell, and K. N. Houk
The Journal of Organic Chemistry June 2, 2017 Volume 82(Issue 11) pp:5904-5904
Publication Date(Web):May 3, 2017
DOI:10.1021/acs.joc.7b00878
The selective androgen receptor modulator, (S)-(7-cyano-4-(pyridin-2-ylmethyl)-1,2,3,4-tetrahydrocyclopenta[b]indol-2-yl)carbamic acid isopropyl ester, LY2452473, is a promising treatment of side effects of prostate cancer therapies. An acid-catalyzed Fischer indolization is a central step in its synthesis. The reaction leads to only one of the two possible indole regioisomers, along with minor decomposition products. Computations show that the formation of the observed indole is most favored energetically, while the potential pathway to the minor isomer leads instead to decomposition products. The disfavored [3,3]-sigmatropic rearrangement, which would produce the unobserved indole product, is destabilized by the electron-withdrawing phthalimide substituent. The most favored [3,3]-sigmatropic rearrangement transition state is bimodal, leading to two reaction intermediates from one transition state, which is confirmed by molecular dynamics simulations. Both intermediates can lead to the observed indole product, albeit through different mechanisms.
Co-reporter:Peiyuan Yu, Wei Li, and K. N. Houk
The Journal of Organic Chemistry June 16, 2017 Volume 82(Issue 12) pp:6398-6398
Publication Date(Web):May 16, 2017
DOI:10.1021/acs.joc.7b01132
The mechanisms of recently reported Lewis acid-catalyzed Diels–Alder reactions of arylallenes and acrylates were studied using density functional theory calculations. A stepwise mechanism involving short-lived zwitterion intermediates is established. The reaction is endo-selective in the presence of Lewis acid catalyst. The [2 + 2] cycloaddition is not observed because of the greater charge separation in the first step of the [2 + 2] cycloaddition. The origins of chirality transfer in the Diels–Alder reaction using chiral arylallenes are uncovered, and the absolute stereochemistry of the product is predicted.
Co-reporter:Yingzi Li, Chunhui Shan, Yun-Fang Yang, Fuqiang Shi, Xiaotian Qi, K. N. Houk, and Yu Lan
The Journal of Physical Chemistry A June 15, 2017 Volume 121(Issue 23) pp:4496-4496
Publication Date(Web):May 10, 2017
DOI:10.1021/acs.jpca.7b01020
Nitrones have been used for rhodium-catalyzed cyclization C–H bond activation and O atom transfer of arylnitrones with alkynes by Chang et al. ( J. Am. Chem. Soc. 2015, 137, 4908−4911). Density functional theory method has been used to study the mechanism, regio-, and diastereoselectivity of type reactions. The results elucidated that the reaction pathway for Rh(III)-catalyzed cyclization of N-arylnitrones with alkyne contains a C–H bond activation, an alkyne insertion into Rh–C bond, a reductive elimination to form a Rh(I) complex, an oxidative addition leading to N–O cleavage, an imine insertion into the Rh–C bond, and the final protonolysis to regenerate the products and the active catalyst. The regioselectivity of this reaction with asymmetric alkyne is controlled by the electronic effect in alkyne insertion type instead of steric effects. The distortion–interaction analysis is also used to explain the regioselectivity. The diastereoselectivity is controlled by the imine insertion step. In this step, the sterically less hindered transition state is favored, leading to stereoselective product formation.
Co-reporter:Matthew N. Grayson, Zhongyue Yang, and K. N. Houk
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7717-7717
Publication Date(Web):May 30, 2017
DOI:10.1021/jacs.7b03847
CH···O hydrogen bonds involving formyl groups have been invoked as a crucial factor controlling many asymmetric transformations. We conducted quasi-classical direct molecular dynamics simulations on the phosphoric acid-catalyzed allylboration of benzaldehyde to understand the synergy between the phosphoric acid OH···O hydrogen bond and the secondary CH···O formyl hydrogen bond as the reaction occurs. In the gas phase, both the CH···O and OH···O hydrogen bonds are enhanced from reactants to transition states. In toluene, the trend of H-bond enhancement is observed with a smaller magnitude because of solvent caging. The strength of the formyl hydrogen bond in the TS, a second CH···O interaction between the P═O oxygen and ortho-hydrogen of the phenyl ring and the OH···O hydrogen bond were determined using quantum mechanical calculations (4.6, 1.0, and 14.5 kcal mol–1, respectively).
Co-reporter:Paul H.-Y. Cheong, Robert S. Paton, Sarah M. Bronner, G-Yoon J. Im, Neil K. Garg and K. N. Houk
Journal of the American Chemical Society February 3, 2010 Volume 132(Issue 4) pp:1267-1269
Publication Date(Web):January 8, 2010
DOI:10.1021/ja9098643
Density functional theory computations reproduce the surprisingly high regioselectivities in nucleophilic additions and cycloadditions to 4,5-indolynes and the low regioselectivities in the reactions of 5,6-indolynes. Transition-state distortion energies control the regioselectivities, activating the 5 and 6 positions over the 4 and 7 positions, leading to high preferences for 5- and 6-substituted products from 4,5- and 6,7-indolynes, respectively. Orbital and electrostatic interactions have only minor effects, producing low regioselectivities in the reactions of 5,6-indolynes. The distortion model predicts high regioselectivities with 6,7-indolynes; these have been verified experimentally. The regioselectivities found with other arynes are explained on the basis of distortion energies that are reflected in reactant geometries.
Co-reporter:Abing Duan, Peiyuan Yu, Fang Liu, Huang Qiu, Feng Long Gu, Michael P. Doyle, and K. N. Houk
Journal of the American Chemical Society February 22, 2017 Volume 139(Issue 7) pp:2766-2766
Publication Date(Web):January 17, 2017
DOI:10.1021/jacs.6b12371
The first experimental examples of Diels–Alder (DA) reactions of diazo compounds as heterodienophiles with dienes have been studied with density functional theory (DFT) using the M06-2X functional. For comparison, the reactivities of diazo esters as dienophiles or 1,3-dipoles with 1,3-dienes in intermolecular model systems have been analyzed by the distortion/interaction model. The 1,3-dipolar cycloaddition is strongly favored for the intermolecular system. The intramolecular example is unique because the tether strongly favors the (4 + 2) cycloaddition.
Co-reporter:Michael M. Gilbert, Matthew D. DeMars II, Song Yang, Jessica M. Grandner, Shoulei Wang, Hengbin Wang, Alison R. H. Narayan, David H. Sherman, K. N. Houk, and John Montgomery
ACS Central Science December 27, 2017 Volume 3(Issue 12) pp:1304-1304
Publication Date(Web):November 15, 2017
DOI:10.1021/acscentsci.7b00450
The diversification of late stage synthetic intermediates provides significant advantages in efficiency in comparison to conventional linear approaches. Despite these advantages, accessing varying ring scaffolds and functional group patterns from a common intermediate poses considerable challenges using existing methods. The combination of regiodivergent nickel-catalyzed C–C couplings and site-selective biocatalytic C–H oxidations using the cytochrome P450 enzyme PikC addresses this problem by enabling a single late-stage linear intermediate to be converted to macrolactones of differing ring size and with diverse patterns of oxidation. The approach is made possible by a novel strategy for site-selective biocatalytic oxidation using a single biocatalyst, with site selectivity being governed by a temporarily installed directing group. Site selectivities of C–H oxidation by this directed approach can overcome positional bias due to C–H bond strength, acidity, inductive influences, steric accessibility, or immediate proximity to the directing group, thus providing complementarity to existing approaches.
Co-reporter:Adam Simon, Yu-hong Lam, and K. N. Houk
The Journal of Organic Chemistry August 4, 2017 Volume 82(Issue 15) pp:8186-8186
Publication Date(Web):July 5, 2017
DOI:10.1021/acs.joc.7b01606
The mechanism and sources of asymmetric induction in Nazarov reactions reported by Tius and co-workers have been determined with quantum chemical calculations. A chiral vicinal diamine forms an enamine–iminium adduct with α-ketoenones, and this undergoes a cationic conrotatory electrocyclization. The chiral diamine imparts stereocontrol in the enamine–iminium complex by forming a six-membered ring that favors one helicity of the electrocyclization transition state.
Co-reporter:Amy E. Fraley, Marc Garcia-Borràs, Ashootosh Tripathi, Dheeraj Khare, Eduardo V. Mercado-Marin, Hong Tran, Qingyun Dan, Gabrielle P. Webb, Katharine R. Watts, Phillip Crews, Richmond Sarpong, Robert M. Williams, Janet L. Smith, K. N. Houk, and David H. Sherman
Journal of the American Chemical Society August 30, 2017 Volume 139(Issue 34) pp:12060-12060
Publication Date(Web):August 4, 2017
DOI:10.1021/jacs.7b06773
Malbrancheamide is a dichlorinated fungal indole alkaloid isolated from both Malbranchea aurantiaca and Malbranchea graminicola that belongs to a family of natural products containing a characteristic bicyclo[2.2.2]diazaoctane core. The introduction of chlorine atoms on the indole ring of malbrancheamide differentiates it from other members of this family and contributes significantly to its biological activity. In this study, we characterized the two flavin-dependent halogenases involved in the late-stage halogenation of malbrancheamide in two different fungal strains. MalA and MalA′ catalyze the iterative dichlorination and monobromination of the free substrate premalbrancheamide as the final steps in the malbrancheamide biosynthetic pathway. Two unnatural bromo-chloro-malbrancheamide analogues were generated through MalA-mediated chemoenzymatic synthesis. Structural analysis and computational studies of MalA′ in complex with three substrates revealed that the enzyme represents a new class of zinc-binding flavin-dependent halogenases and provides new insights into a potentially unique reaction mechanism.
Co-reporter:Janice B. Lin, Tejas K. Shah, Adam E. Goetz, Neil K. Garg, and K. N. Houk
Journal of the American Chemical Society August 2, 2017 Volume 139(Issue 30) pp:10447-10447
Publication Date(Web):July 4, 2017
DOI:10.1021/jacs.7b05317
We report the design and synthesis of a new class of indole-based conjugated trimers. The targeted compounds are accessed from in situ generated, highly reactive indolyne intermediates using Pd-catalyzed cyclotrimerization reactions. By harnessing three indolyne isomers, six isomeric indole trimers are accessible, none of which have been previously synthesized. Using computational analysis, we describe the structural and photophysical properties of these unique compounds. This study showcases the use of indolynes in transition metal-catalyzed reactions, while providing access to a new class of conjugated trimers, including highly bent heteroaromatic compounds. Computations indicate that, despite differences in planarity between the molecules, the photophysical properties of each trimer are derived from the N-methylindole building block. Excited state behavior follows predicable patterns.
Co-reporter:Chen-Chen Zhou, M. Frederick Hawthorne, K. N. Houk, and Gonzalo Jiménez-Osés
The Journal of Organic Chemistry August 18, 2017 Volume 82(Issue 16) pp:8438-8438
Publication Date(Web):July 13, 2017
DOI:10.1021/acs.joc.7b01169
The thermal decompositions of metallaisoxazolin-5-ones containing Ir, Rh, or Co are investigated using density functional theory. The experimentally observed decarboxylations of these molecules are found to proceed through retro-(3+2)-cycloaddition reactions, generating the experimentally reported η2 side-bonded nitrile complexes. These intermediates can isomerize in situ to yield a η1 nitrile complex. A competitive alternative pathway is also found where the decarboxylation happens concertedly with an aryl migration process, producing a η1 isonitrile complex. Despite their comparable stability, these η1 bonded species were not detected experimentally. The experimentally detected η2 side bound species are likely involved in the subsequent C–H activation reactions with hydrocarbon solvents reported for some of these metallaisoxazolin-5-ones.
Co-reporter:Cyndi Qixin He, Adam Simon, Yu-hong Lam, Andrew P. J. Brunskill, Nobuyoshi Yasuda, Jiajing Tan, Alan M. Hyde, Edward C. Sherer, and K. N. Houk
The Journal of Organic Chemistry August 18, 2017 Volume 82(Issue 16) pp:8645-8645
Publication Date(Web):July 21, 2017
DOI:10.1021/acs.joc.7b01577
A model for the stereoselectivity of intramolecular alkylations by N,N′-disubstituted cinchona alkaloids reported by Xiang et al. was established using density functional theory (DFT) calculations. The stereocontrol is based on the minimal distortion of the transition state (TS) and catalyst required to achieve favorable electrostatic interactions in the favored TS. Counterions must be included in computational modeling of ion-paired catalysis in order to reproduce experimental enantioselectivity.
Co-reporter:Brian J. Levandowski, Trevor A. Hamlin, F. Matthias Bickelhaupt, and K. N. Houk
The Journal of Organic Chemistry August 18, 2017 Volume 82(Issue 16) pp:8668-8668
Publication Date(Web):July 16, 2017
DOI:10.1021/acs.joc.7b01673
The Diels–Alder reactivities of a series of cycloalkenes, from the highly strained cyclopropene to the unstrained cyclohexene, have been studied with density functional theory using the M06-2X functional. The normal electron-demand Diels–Alder reactions with cyclopentadiene and the inverse electron-demand Diels–Alder reactions with 3,6-bis(trifluoromethyl)tetrazine were analyzed using the distortion/interaction-activation strain model. Previous studies showed that activation strain computed from the distorted reactants in the transition structures are larger for unstrained than strained cycloalkenes, and that most of the activation energy differences are accounted for by this difference. We have now analyzed the strain and interaction energy curves for the series of cycloalkenes along the reaction coordinate. Our analyses reveal that the strain curves associated with the distortion of the reactants in the Diels–Alder reactions are nearly identical and that the reactivity differences originate from differences in interaction energies. Analysis of the diene-dienophile interactions reveal that the reactivity trends result from differences in the strength of the primary and secondary orbital interactions.
Co-reporter:Yao Li, Guo-Hui Yang, Cyndi Qixin He, Xin Li, K. N. Houk, and Jin-Pei Cheng
Organic Letters August 18, 2017 Volume 19(Issue 16) pp:
Publication Date(Web):August 4, 2017
DOI:10.1021/acs.orglett.7b01743
N-tert-Butyl sulfinyl squaramides were used for chiral discrimination of α-hydroxyphosphonates using 31P NMR. A free energy relationship study indicates that both steric and the electronic effects influence the chiral recognition of the donors.
Co-reporter:Yao Li, Cyndi Qixin He, Fei-Xiang Gao, Zhen Li, Xiao-Song Xue, Xin Li, K. N. Houk, and Jin-Pei Cheng
Organic Letters April 7, 2017 Volume 19(Issue 7) pp:
Publication Date(Web):March 30, 2017
DOI:10.1021/acs.orglett.7b00727
A new chiral HBD system, N-tert-butyl sulfinyl squaramide, was designed and synthesized. The core N-tert-butyl sulfinyl squaramide with an 1-aminoindan-2-ol skeleton was found to be an efficient catalyst in the enantioselective Friedel–Crafts alkylation of indoles and acyl phosphonates.
Co-reporter:Xu Han;Jiyong Park;Wei Wu;Andres Malagon;Lingyu Wang;Edgar Vargas;Athula Wikramanayake;K. N. Houk;Roger M. Leblanc
Chemical Science (2010-Present) 2017 vol. 8(Issue 3) pp:2003-2009
Publication Date(Web):2017/02/28
DOI:10.1039/C6SC04854D
Amyloid-β peptides (Aβ) fibrillation is the hallmark of Alzheimer's disease (AD). However, it has been challenging to discover potent agents in order to inhibit Aβ fibrillation. Herein, we demonstrated the effect of resorcinarene on inhibiting Aβ fibrillation in vitro via experimental and computational methods. Aβ were incubated with different concentrations of resorcinarene so as to monitor the kinetics by using thioflavin T binding assay. The results, which were further confirmed by far-UV CD spectroscopy and atomic force microscopy, strongly indicated that the higher concentration of resorcinarene, the more effective the inhibition of Aβ fibrillation. A cytotoxicity study showed that when sea urchin embryos were exposed to the resorcinarene, the majority survived due to the resorcinarene low toxicity. In addition, when the resorcinarene was added, the formation of toxic Aβ 42 species was delayed. Computational studies of Aβ fibrillation, including docking simulations and MD simulations, illustrated that the interaction between inhibitor resorcinarene and Aβ is driven by the non-polar interactions. These studies display a novel strategy for the exploration of promising antiamyloiddogenic agents for AD treatments.
Co-reporter:Joel L. Mackey, Zhongyue Yang, K.N. Houk
Chemical Physics Letters 2017 Volume 683(Volume 683) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.cplett.2017.03.011
•Reaction dynamics of the Cope rearrangement of 1,5-hexadiene was conducted.•For reactive trajectories, the time to traverse the transition zone was 35 ± 16 fs.•Dynamically concerted (94%) and stepwise trajectories (6%) were found.•Dynamically concerted trajectory travels across the TS dividing surface.•Dynamically stepwise trajectory “surfs” the TS dividing surface.Molecular dynamics of the [3,3]-sigmatropic (Cope) rearrangement of 1,5-hexadiene were performed with the B3LYP/6-31G(d) density functional theory method. We found that the forming or breaking bond lengths of sampled transition state geometries are 1.97 Å ± 0.15 Å, which is defined as the transition zone. Two hundred and thirty trajectories were propagated. Ninety-five percent of the trajectories connect reactant to product. Five percent of the trajectories involved recrossing. For the reactive trajectories, the time to traverse the transition zone was 35 ± 16 fs. Ninety-four percent of these trajectories are dynamically concerted, while the remaining six percent are dynamically stepwise.Download high-res image (96KB)Download full-size image
Co-reporter:Ilhan YavuzK. N. Houk
The Journal of Physical Chemistry C 2017 Volume 121(Issue 2) pp:
Publication Date(Web):December 15, 2016
DOI:10.1021/acs.jpcc.6b08624
The mesoscale ordering and charge-transport of crystalline spiro-OMeTAD, a hole-transporting material extensively used in perosvkite and dye-sensitized solar cell applications, were explored using molecular dynamics and hole mobility calculations. Morphologies were evaluated through conformational changes, nematic order and paracrystallinity at various temperatures. Charge transport is predicted with electronic structure methods employing a hopping mechanism. Our calculations show that along with strong fluorene backbone packing, phenylenes in the methoxyphenyl–amine substituents of spiro-OMeTAD are an integral part of the material performance. Backbone and substituent paracrystallinity predictions showed highly ordered crystalline phase. The methoxyphenyl substituents have multiple conformations in the unit-cell scale, but interphenylene electronic-coupling remain nearly constant. A thermal increase in positional disorder results in a systematic increase in energetic disorder and a decrease in hole mobility. The predicted crystalline hole mobility is approximately two-orders of magnitude higher than the experimental thin-film measurements, indicating that the performance of spiro-OMeTADs can be improved significantly by exploiting crystallinity.
Co-reporter:F. Matthias Bickelhaupt
Angewandte Chemie 2017 Volume 129(Issue 34) pp:10134-10134
Publication Date(Web):2017/08/14
DOI:10.1002/ange.201707253
Ein Modell zur Analyse und Vorhersage von Reaktionsgeschwindigkeiten wird von F. M. Bickelhaupt und K. N. Houk in ihrem Aufsatz auf S. 10204 vorgestellt. Dieses Modell beruht auf einer Zerlegung von Reaktionsprofilen und Energiebarrieren anhand der Eigenschaften der Reaktanten, insbesondere der Spannungsenergie bei ihrer Deformation und der Wechselwirkung zwischen den deformierten Reaktanten. Beispiele aus organischer und anorganischer Chemie illustrieren die Anwendung des Modells.
Co-reporter:F. Matthias Bickelhaupt
Angewandte Chemie 2017 Volume 129(Issue 34) pp:10204-10221
Publication Date(Web):2017/08/14
DOI:10.1002/ange.201701486
AbstractDas Activation-Strain- oder Distortion/Interaction-Modell ist ein Instrument zur Analyse von Aktivierungsbarrieren und damit von Reaktionsgeschwindigkeiten. Für eine bimolekulare Reaktion ergibt sich die Aktivierungsenergie aus der Summe der Energien, die für die Deformation der Grundzustandsgeometrie der Reaktanten benötigt wird, um die Geometrie des Übergangszustands anzunehmen, plus der Wechselwirkungsenergie zwischen diesen deformierten Reaktanten. Die mit der Deformation assoziierte Energie wird “activation strain” (Aktivierungsspannung) oder “distortion energy” (Deformationsenergie) genannt. Diese Energie bildet den Hauptbeitrag zur Aktivierungsbarriere. Der Übergangszustand wird erreicht, wenn die stabilisierende Wechselwirkung die Aktivierungsspannung überwindet. Die Verlaufsanalyse dieser Energien gibt Einblicke in die reaktivitätsbestimmenden physikalischen Faktoren. Anwendungsbeispiele für das Modell sind die Analyse von Substitutions- und Eliminierungsreaktionen sowie Cycloadditionen.
Co-reporter: Dr. F. Matthias Bickelhaupt; Dr. Dr. Kendall N. Houk
Angewandte Chemie International Edition 2017 Volume 56(Issue 34) pp:10070-10086
Publication Date(Web):2017/08/14
DOI:10.1002/anie.201701486
AbstractThe activation strain or distortion/interaction model is a tool to analyze activation barriers that determine reaction rates. For bimolecular reactions, the activation energies are the sum of the energies to distort the reactants into geometries they have in transition states plus the interaction energies between the two distorted molecules. The energy required to distort the molecules is called the activation strain or distortion energy. This energy is the principal contributor to the activation barrier. The transition state occurs when this activation strain is overcome by the stabilizing interaction energy. Following the changes in these energies along the reaction coordinate gives insights into the factors controlling reactivity. This model has been applied to reactions of all types in both organic and inorganic chemistry, including substitutions and eliminations, cycloadditions, and several types of organometallic reactions.
Co-reporter: Dr. F. Matthias Bickelhaupt; Dr. Dr. Kendall N. Houk
Angewandte Chemie International Edition 2017 Volume 56(Issue 34) pp:10002-10002
Publication Date(Web):2017/08/14
DOI:10.1002/anie.201707253
A model for analyzing and predicting reaction rates is presented by F. M. Bickelhaupt and K. N. Houk in their Review on page 10070 ff. This model is based on dissecting reaction profiles and energy barrier heights in terms of reactant properties, in particular, the strain associated with distorting the reactants and the interaction between the distorted reactants. The model is illustrated with examples from organic and inorganic chemistry.
Co-reporter:Jason S. FellBlanton N. Martin, K. N. Houk
The Journal of Organic Chemistry 2017 Volume 82(Issue 4) pp:
Publication Date(Web):February 2, 2017
DOI:10.1021/acs.joc.6b02524
The reactivities of butadiene, cyclopentadiene, furan, thiophene, pyrrole, and their 1-aza- and 2-aza-derivatives in Diels–Alder reactions with ethylene and fumaronitrile were investigated with density functional theory (M06-2X/6-311G(d,p)). The activation free energies for the Diels–Alder reactions of cyclic 1-azadienes are 10–14 kcal mol–1 higher than those of cyclic 2-azadienes, and the reaction free energies are 17–20 kcal mol–1 more endergonic. The distortion/interaction model shows that the increased activation energies of cyclic 1-azadienes originate from increased transition state distortion energies and unfavorable interaction energies, arising from addition to the nitrogen terminus of the C═N bond.
Co-reporter:Yu-hong Lam, Matthew N. Grayson, Mareike C. Holland, Adam Simon, and K. N. Houk
Accounts of Chemical Research 2016 Volume 49(Issue 4) pp:750
Publication Date(Web):March 11, 2016
DOI:10.1021/acs.accounts.6b00006
Modern density functional theory and powerful contemporary computers have made it possible to explore complex reactions of value in organic synthesis. We describe recent explorations of mechanisms and origins of stereoselectivities with density functional theory calculations. The specific functionals and basis sets that are routinely used in computational studies of stereoselectivities of organic and organometallic reactions in our group are described, followed by our recent studies that uncovered the origins of stereocontrol in reactions catalyzed by (1) vicinal diamines, including cinchona alkaloid-derived primary amines, (2) vicinal amidophosphines, and (3) organo-transition-metal complexes. Two common cyclic models account for the stereoselectivity of aldol reactions of metal enolates (Zimmerman–Traxler) or those catalyzed by the organocatalyst proline (Houk–List). Three other models were derived from computational studies described in this Account.Cinchona alkaloid-derived primary amines and other vicinal diamines are venerable asymmetric organocatalysts. For α-fluorinations and a variety of aldol reactions, vicinal diamines form enamines at one terminal amine and activate electrophilically with NH+ or NF+ at the other. We found that the stereocontrolling transition states are cyclic and that their conformational preferences are responsible for the observed stereoselectivity. In fluorinations, the chair seven-membered cyclic transition states is highly favored, just as the Zimmerman–Traxler chair six-membered aldol transition state controls stereoselectivity. In aldol reactions with vicinal diamine catalysts, the crown transition states are favored, both in the prototype and in an experimental example, shown in the graphic. We found that low-energy conformations of cyclic transition states occur and control stereoselectivities in these reactions. Another class of bifunctional organocatalysts, the vicinal amidophosphines, catalyzes the (3 + 2) annulation reaction of allenes with activated olefins. Stereocontrol here is due to an intermolecular hydrogen bond that activates the electrophilic partner in this reaction. We have also studied complex organometallic catalysts. Krische’s ruthenium-catalyzed asymmetric hydrohydroxyalkylation of butadiene involves two chiral ligands at Ru, a chiral diphosphine and a chiral phosphate. The size of this combination strains the limits of modern computations with over 160 atoms, multiple significant steps, and a variety of ligand coordinations and conformations possible. We found that carbon–carbon bond formation occurs via a chair Zimmerman–Traxler-type transition structure and that a formyl CH···O hydrogen bond from aldehyde CH to phosphate oxygen, as well as steric interactions of the two chiral ligands, control the stereoselectivity.
Co-reporter:Hsiao-Ching Lin; Travis C. McMahon; Ashay Patel; Michael Corsello; Adam Simon; Wei Xu; Muxun Zhao; K. N. Houk; Neil K. Garg;Yi Tang
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:4002-4005
Publication Date(Web):March 10, 2016
DOI:10.1021/jacs.6b01413
Dimeric indole alkaloids are structurally diverse natural products that have attracted significant attention from the synthetic and biosynthetic communities. Here, we describe the characterization of a P450 monooxygenase CnsC from Penicillium that catalyzes the heterodimeric coupling between two different indole moieties, tryptamine and aurantioclavine, to construct vicinal quaternary stereocenters and yield the heptacyclic communesin scaffold. We show, via biochemical characterization, substrate analogues, and computational methods that CnsC catalyzes the C3-C3′ carbon-carbon bond formation and controls the regioselectivities of the pair of subsequent aminal bond formations to yield the communesin core. Use of ω-N-methyltryptamine and tryptophol in place of tryptamine led to the enzymatic synthesis of isocommunesin compounds, which have not been isolated to date.
Co-reporter:Xing Jiang; Zachary J. O’Brien; Song Yang; Lan Huong Lai; Jeffrey Buenaflor; Colleen Tan; Saeed Khan; K. N. Houk;Miguel A. Garcia-Garibay
Journal of the American Chemical Society 2016 Volume 138(Issue 13) pp:4650-4656
Publication Date(Web):March 14, 2016
DOI:10.1021/jacs.6b01398
Low packing densities are key structural features of amphidynamic crystals built with static and mobile components. Here we report a loosely packed crystal of dendrimeric rotor 2 and the fast dynamics of all its aromatic groups, both resulting from the hyperbranched structure of the molecule. Compound 2 was synthesized with a convergent strategy to construct a central phenylene core with stators consisting of two layers of triarylmethyl groups. Single crystal X-ray diffraction analysis confirmed a low-density packing structure consisting of one molecule of 2 and approximately eight solvent molecules per unit cell. Three isotopologues of 2 were synthesized to study the motion of each segment of the molecule in the solid state using variable temperature quadrupolar echo 2H NMR spectroscopy. Line shape analysis of the spectra reveals that the central phenylene, the six branch phenylenes, and the 18 periphery phenyls all display megahertz rotational dynamics in the crystals at ambient temperature. Arrhenius analysis of the data gives similar activation energies and pre-exponential factors for different parts of the structure. The observed pre-exponential factors are 4–6 orders of magnitude greater than those of elementary site-exchange processes, indicating that the dynamics are not dictated by static energetic potentials. Instead, the activation energies for rotations in the crystals of 2 are controlled by temperature dependent local structural fluctuations and crystal fluidity.
Co-reporter:Zhongyue Yang; Peiyuan Yu;K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:4237-4242
Publication Date(Web):March 10, 2016
DOI:10.1021/jacs.6b01028
We report molecular dynamics simulations of the reaction of dimethyldioxirane (DMDO) with isobutane. The reaction involves hydrogen atom abstraction in the transition state, and trajectories branch to the oxygen rebound pathway, which gives tert-butanol and acetone, or a separated radical pair. In the gas phase, only 10% of the reactive trajectories undergo the oxygen rebound pathway, but this increases to 90% in simulations in an implicit acetone solvent (SMD) because the oxygen rebound becomes barrierless in solution. Short-lived diradical species were observed in the oxygen rebound trajectories. The time gap between C–H bond-breaking and C–O bond formation ranges from 30 to 150 fs, close to the <200 fs lifetime of radical pairs from DMDO hydroxylation of trans-1-phenyl-2-ethylcyclopropane measured by Newcomb.
Co-reporter:Shu-Shan Gao; Abing Duan; Wei Xu; Peiyuan Yu; Leibniz Hang; K. N. Houk;Yi Tang
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:4249-4259
Publication Date(Web):March 15, 2016
DOI:10.1021/jacs.6b01528
Phenalenones are polyketide natural products that display diverse structures and biological activities. The core of phenalenones is a peri-fused tricyclic ring system cyclized from a linear polyketide precursor via an unresolved mechanism. Toward understanding the unusual cyclization steps, the phn biosynthetic gene cluster responsible for herqueinone biosynthesis was identified from the genome of Penicillium herquei. A nonreducing polyketide synthase (NR-PKS) PhnA was shown to synthesize the heptaketide backbone and cyclize it into the angular, hemiketal-containing naphtho-γ-pyrone prephenalenone. The product template (PT) domain of PhnA catalyzes only the C4–C9 aldol condensation, which is unprecedented among known PT domains. The transformation of prephenalenone to phenalenone requires an FAD-dependent monooxygenase (FMO) PhnB, which catalyzes the C2 aromatic hydroxylation of prephenalenone and ring opening of the γ-pyrone ring simultaneously. Density functional theory calculations provide insights into why the hydroxylated intermediate undergoes an aldol-like phenoxide–ketone cyclization to yield the phenalenone core. This study therefore unveiled new routes and biocatalysts for polyketide cyclization.
Co-reporter:Ashay Patel; Zhuo Chen; Zhongyue Yang; Osvaldo Gutiérrez; Hung-wen Liu; K. N. Houk;Daniel A. Singleton
Journal of the American Chemical Society 2016 Volume 138(Issue 11) pp:3631-3634
Publication Date(Web):February 24, 2016
DOI:10.1021/jacs.6b00017
SpnF, an enzyme involved in the biosynthesis of spinosyn A, catalyzes a transannular Diels–Alder reaction. Quantum mechanical computations and dynamic simulations now show that this cycloaddition is not well described as either a concerted or stepwise process, and dynamical effects influence the identity and timing of bond formation. The transition state for the reaction is ambimodal and leads directly to both the observed Diels–Alder and an unobserved [6+4] cycloadduct. The potential energy surface bifurcates and the cycloadditions occur by dynamically stepwise modes featuring an “entropic intermediate”. A rapid Cope rearrangement converts the [6+4] adduct into the observed [4+2] adduct. Control of nonstatistical dynamical effects may serve as another way by which enzymes control reactions.
Co-reporter:Xia Yu, Fang Liu, Yi Zou, Man-Cheng Tang, Leibniz Hang, K. N. Houk, and Yi Tang
Journal of the American Chemical Society 2016 Volume 138(Issue 41) pp:13529-13532
Publication Date(Web):October 3, 2016
DOI:10.1021/jacs.6b09464
Nature synthesizes many strained natural products that have diverse biological activities. Uncovering these biosynthetic pathways may lead to biomimetic strategies for organic synthesis of such compounds. In this work, we elucidated the concise biosynthetic pathway of herquline A, a highly strained and reduced fungal piperazine alkaloid. The pathway builds on a nonribosomal peptide synthetase derived dityrosine piperazine intermediate. Following enzymatic reduction of the P450-cross-linked dicyclohexadienone, N-methylation of the piperazine serves as a trigger that leads to a cascade of stereoselective and nonenzymatic transformations. Computational analysis of key steps in the pathway rationalizes the observed reactivities.
Co-reporter:Brian J. LevandowskiK. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 51) pp:16731-16736
Publication Date(Web):December 1, 2016
DOI:10.1021/jacs.6b10463
The factors controlling the reactivities and stereoselectivities in the Diels–Alder reactions of substituted cyclopropenes with butadiene were explored with M06-2X density functional theory. Differences in reactivities result from differences in the hyperconjugative aromaticities and antiaromaticities of the cyclopropenes. When the 3-substituent is a σ-donor, the ground state is destabilized, and the reactivity is enhanced. Acceptors have the opposite effect. Electrostatic, secondary orbital, and steric effects are all found to influence stereoselectivities.
Co-reporter:Taoufik Ben Halima, Wanying Zhang, Imane Yalaoui, Xin Hong, Yun-Fang YangKendall N. Houk, Stephen G. Newman
Journal of the American Chemical Society 2016 Volume 139(Issue 3) pp:1311-1318
Publication Date(Web):December 21, 2016
DOI:10.1021/jacs.6b12329
The Suzuki–Miyaura coupling is among the most important C–C bond-forming reactions available due to its reliability, chemoselectivity, and diversity. Aryl halides and pseudohalides such as iodides, bromides, and triflates are traditionally used as the electrophilic coupling partner. The expansion of the reaction scope to nontraditional electrophiles is an ongoing challenge to enable an even greater number of useful products to be made from simple starting materials. Herein, we present how an NHC-based Pd catalyst can enable Suzuki–Miyaura coupling where the C(acyl)–O bond of aryl esters takes on the role of electrophile, allowing the synthesis of various ketone-containing products. This contrasts known reactions of similar esters that provide biaryls via nickel catalysis. The underlying cause of this mechanistic divergence is investigated by DFT calculations, and the robustness of esters compared to more electrophilic acylative coupling partners is analyzed.
Co-reporter:L. E. Rosebrugh; T. S. Ahmed; V. M. Marx; J. Hartung; P. Liu; J. G. López; K. N. Houk;R. H. Grubbs
Journal of the American Chemical Society 2016 Volume 138(Issue 4) pp:1394-1405
Publication Date(Web):January 4, 2016
DOI:10.1021/jacs.5b12277
The microstructures of polymers produced by ring-opening metathesis polymerization (ROMP) with cyclometalated Ru-carbene metathesis catalysts were investigated. A strong bias for a cis,syndiotactic microstructure with minimal head-to-tail bias was observed. In instances where trans errors were introduced, it was determined that these regions were also syndiotactic. Furthermore, hypothetical reaction intermediates and transition structures were analyzed computationally. Combined experimental and computational data support a reaction mechanism in which cis,syndio-selectivity is a result of stereogenic metal control, while microstructural errors are predominantly due to alkylidene isomerization via rotation about the Ru═C double bond.
Co-reporter:Matthew N. Grayson;K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 29) pp:9041-9044
Publication Date(Web):July 9, 2016
DOI:10.1021/jacs.6b05074
The cinchona alkaloid-derived urea-catalyzed asymmetric conjugate addition of aromatic thiols to cycloalkenones was studied using density functional theory (DFT). Deprotonation of the thiol gives a protonated amine that activates the electrophile by Brønsted acid catalysis, while the urea group binds the nucleophilic thiolate by hydrogen bonding. These results demonstrate the generality of the Brønsted acid−hydrogen bonding transition state (TS) model for cinchona alkaloid catalysis that we recently showed to be favored over Wynberg’s widely accepted ion pair−hydrogen bonding model and represent the first detailed mechanistic study of a cinchona urea-catalyzed reaction. The conformation of the catalyst methoxy group has a strong effect on the TS, an effect overlooked in previous mechanistic studies of reactions catalyzed by cinchona alkaloids.
Co-reporter:Shao-Xiong Luo, Jeffrey S. Cannon, Buck L. H. Taylor, Keary M. Engle, K. N. Houk, and Robert H. Grubbs
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:14039-14046
Publication Date(Web):September 30, 2016
DOI:10.1021/jacs.6b08387
Olefin metathesis reactions with 3E-1,3-dienes using Z-selective cyclometalated ruthenium benzylidene catalysts are described. In particular, a procedure for employing 3E-1,3-dienes in Z-selective homodimerization and cross-metathesis with terminal alkenes is detailed. The reaction takes advantage of the pronounced chemoselectivity of a recently reported ruthenium-based catalyst containing a cyclometalated NHC ligand for terminal alkenes in the presence of internal E-alkenes. A wide array of commonly encountered functional groups can be tolerated, and only a small excess (1.5 equiv) of the diene coupling partner is required to achieve high yields of the desired internal E,Z-diene cross-metathesis product. Computational studies have been performed to elucidate the reaction mechanism. The computations are consistent with a diene-first pathway. The reaction can be used to quickly assemble structurally complex targets. The power of this cross-metathesis reaction is demonstrated by the concise syntheses of two insect pheromones.
Co-reporter:Peiyuan Yu; Zhongyue Yang; Yong Liang; Xin Hong; Yanwei Li;K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 26) pp:8247-8252
Publication Date(Web):June 10, 2016
DOI:10.1021/jacs.6b04113
We report density functional theory (M06-2X) studies of a series of dehydro-Diels–Alder (DDA) reactions. For these and the parent reaction, the stepwise mechanisms have similar barriers, whereas the barriers of the concerted mechanisms differ significantly. The reactivity of DDA reactions is controlled by distortion energy. The concerted and stepwise mechanisms of the hexadehydro-Diels–Alder (HDDA) reaction are competitive with activation barriers of ∼36 kcal/mol. This is because a large distortion energy (∼43 kcal/mol) is required to achieve the concerted transition state geometry. MD simulations reveal that productive concerted trajectories display a strong angle bending oscillation (∼25° oscillation amplitude), while the stepwise trajectories show only a chaotic pattern and less pronounced bending vibrations.
Co-reporter:Yun-Fang Yang; K. N. Houk;Yun-Dong Wu
Journal of the American Chemical Society 2016 Volume 138(Issue 21) pp:6861-6868
Publication Date(Web):May 13, 2016
DOI:10.1021/jacs.6b03424
The selective rhodium-catalyzed functionalization of arenes is greatly facilitated by oxidizing directing groups that act both as directing groups and internal oxidants. We report density functional theory (B3LYP and M06) investigations on the mechanism of rhodium(III)-catalyzed redox coupling reaction of N-phenoxyacetamides with alkynes. The results elucidated the role of the internal oxidizing directing group, and the role of RhIII/RhI and RhIII/RhV catalysis of C–H functionalizations. A novel RhIII–RhV–RhIII cycle successfully rationalizes recent experimental observations by Liu and Lu et al. (Liu, G. Angew. Chem. Int. Ed. 2013, 52, 6033) on the reactions of N-phenoxyacetamides with alkynes in different solvents. Natural Bond Orbital (NBO) analysis confirms the identity of RhV intermediate in the catalytic cycle.
Co-reporter:Pier Alexandre Champagne and K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:12356-12359
Publication Date(Web):September 14, 2016
DOI:10.1021/jacs.6b08276
The origins of the high enantioselectivity of chiral phosphoric acid-catalyzed oxetane desymmetrizations were investigated by density functional theory (DFT) calculations. Distortion of the catalyst structure, caused by steric crowding in the catalyst pocket of one enantiomeric transition state, is the main cause for stereochemical preference. A general model was developed to assist in the rational design of new catalysts for related transformations.
Co-reporter:Adam Simon; Yu-hong Lam;K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 2) pp:503-506
Publication Date(Web):January 4, 2016
DOI:10.1021/jacs.5b12097
The transition states of aldol reactions catalyzed by vicinal diamines are characterized with density functional calculations. It was found that a cyclic transition state involving a nine-membered hydrogen-bonded ring is preferred. The crown (chair–chair) conformations of the transition state account for the observed stereoselectivity of these reactions.
Co-reporter:Yun-Fang Yang; Yong Liang; Fang Liu;K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 5) pp:1660-1667
Publication Date(Web):January 23, 2016
DOI:10.1021/jacs.5b12054
The cycloadditions of benzene and ten different azabenzenes (pyridine, three diazines, three triazines, and three tetrazines) with the ethylene dienophile have been explored with density functional theory (M06-2X) and analyzed with the distortion/interaction model. Activation barriers correlate closely with both distortion energies and interaction energies over an activation energy range of 45 kcal/mol. The replacement of CH with N increases Diels–Alder reactivity due not only to the more favorable orbital interaction, but also to a decrease in distortion energy. The rates of reactions are greatly influenced by the nature of the bonds being formed: two C—C bonds > one C—C bond, and one C—N bond > two C—N bonds. The activation energy of Diels–Alder reactions correlates very well with reaction energies and with the NICS(0) values of the aromatic dienes. The distortion energy of the Diels–Alder reaction transition states mostly arises from the diene out-of-plane distortion energy.
Co-reporter:Dionicio Martinez-Solorio; Bruno Melillo; Luis Sanchez; Yong Liang; Erwin Lam; K. N. Houk;Amos B. SmithIII
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:1836-1839
Publication Date(Web):February 2, 2016
DOI:10.1021/jacs.5b13260
A reusable silicon-based transfer agent (1) has been designed, synthesized, and validated for effective room-temperature palladium-catalyzed cross-coupling reactions (CCRs) of aryl and heteroaryl chlorides with readily accessible aryl lithium reagents. The crystalline, bench-stable siloxane transfer agent (1) is easily prepared via a one-step protocol. Importantly, this “green” CCR protocol circumvents prefunctionalization, isolation of organometallic cross-coupling partners, and/or stoichiometric waste aside from LiCl. DFT calculations support a σ-bond metathesis mechanism during transmetalation and lead to insights on the importance of the CF3 groups.
Co-reporter:Joyann S. Barber; Evan D. Styduhar; Hung V. Pham; Travis C. McMahon; K. N. Houk;Neil K. Garg
Journal of the American Chemical Society 2016 Volume 138(Issue 8) pp:2512-2515
Publication Date(Web):February 8, 2016
DOI:10.1021/jacs.5b13304
We report the first 1,3-dipolar cycloadditions of 1,2-cyclohexadiene, a rarely exploited strained allene. 1,2-Cyclohexadiene is generated in situ under mild conditions and trapped with nitrones to give isoxazolidine products in synthetically useful yields. The reactions occur regioselectively and exhibit a notable endo preference, thus resulting in the controlled formation of two new bonds and two stereogenic centers. DFT calculations of stepwise and concerted reaction pathways are used to rationalize the observed selectivities. Moreover, the strategic manipulation of nitrone cycloadducts demonstrates the utility of this methodology for the assembly of compounds bearing multiple heterocyclic units. These studies showcase the exploitation of a traditionally avoided reactive intermediate in chemical synthesis.
Co-reporter:Matthew N. Grayson;K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 4) pp:1170-1173
Publication Date(Web):January 19, 2016
DOI:10.1021/jacs.5b13275
Wynberg’s report from 1977 that natural cinchona alkaloids catalyze the asymmetric conjugate addition of aromatic thiols to cycloalkenones is a landmark discovery in hydrogen bonding organocatalysis. Wynberg proposed that this reaction proceeded via the formation of a thiolate-alkylammonium tight ion pair and activation of the enone electrophile by a hydrogen bond from the catalyst’s hydroxyl group. This reaction model provided the mechanistic basis for understanding Wynberg’s reaction and many other asymmetric transformations since. Our quantum mechanical calculations reveal a different model should be used to explain the results: the alkylammonium ion activates the enone by Brønsted acid catalysis, and the catalyst’s hydroxyl group orients the thiolate nucleophile. The new model rationalizes the stereoselective outcome of Wynberg’s reaction and provides a new, general model for asymmetric cinchona organocatalysis.
Co-reporter:Maruthi Kumar Narayanam, Yong Liang, K. N. Houk and Jennifer M. Murphy
Chemical Science 2016 vol. 7(Issue 2) pp:1257-1261
Publication Date(Web):2015/11/11
DOI:10.1039/C5SC03259H
Density functional theory (DFT) calculations and experiments in tandem led to discoveries of new reactivities and selectivities involving bioorthogonal sydnone cycloadditions. Dibenzocyclooctyne derivatives (DIBAC and BARAC) were identified to be especially reactive dipolarophiles, which undergo the (3 + 2) cycloadditions with N-phenyl sydnone with the rate constant of up to 1.46 M−1 s−1. Most significantly, the sydnone-dibenzocyclooctyne and norbornene-tetrazine cycloadditions were predicted to be mutually orthogonal. This was validated experimentally and used for highly selective fluorescence labeling of two proteins simultaneously.
Co-reporter:Jessica M. Grandner, Ralph A. Cacho, Yi Tang, and K. N. Houk
ACS Catalysis 2016 Volume 6(Issue 7) pp:4506
Publication Date(Web):May 31, 2016
DOI:10.1021/acscatal.6b01068
Griseofulvin is an antifungal agent that has recently been determined to have potential antiviral and anticancer applications. The role of specific enzymes involved in the biosynthesis of this natural product has previously been determined, but the mechanism by which a p450 (GsfF), catalyzes the key oxidative cyclization of griseophenone B remains unknown. Using density functional theory (DFT), we have determined the mechanism of this oxidation that forms the oxa-spiro core of griseofulvin. Computations show GsfF preferentially performs phenolic O–H abstraction over epoxidation to catalyze the oxidation.Keywords: computations; DFT; griseofulvin; mechanism; O−H abstraction; p450
Co-reporter:Elsa Rodríguez, Matthew N. Grayson, Amparo Asensio, Pablo Barrio, K. N. Houk, and Santos Fustero
ACS Catalysis 2016 Volume 6(Issue 4) pp:2506
Publication Date(Web):March 7, 2016
DOI:10.1021/acscatal.6b00209
Chiral Brønsted acid-catalyzed allyl(propargyl)boration of ortho-alkynyl benzaldehydes gives rise to ω-alkynyl homoallylic(homopropargylic)alcohols that can be further transformed to complex molecular scaffolds via subsequent hydroalkoxylation, ring-closing enyne metathesis (RCEYM), or intramolecular Pauson–Khand reaction (PKR). Optimizations of each two-step transformation is reported. A strong dependence between enantioselectivities and the nature of the substitution at the alkynyl moiety is observed, showcasing that the triple bond is not merely a spectator in this transformation. Density functional theory (DFT) calculations (M06-2X/6-311+G(d,p)–IEFPCM//B3LYP/6-31G(d)) show that this dependence is the result of the steric and electronic properties of the alkyne substituent.Keywords: asymmetric allylboration; chiral Brønsted acids; DFT calculations; diversity-oriented synthesis; organocatalysis
Co-reporter:Michael E. Jung, Courtney A. Roberts, Felix Perez, Hung V. Pham, Lufeng Zou, and K. N. Houk
Organic Letters 2016 Volume 18(Issue 1) pp:32-35
Publication Date(Web):December 15, 2015
DOI:10.1021/acs.orglett.5b03112
The rearrangements of 4-substituted bicyclo[2.2.2]oct-5-en-2-yl radicals, generated from the corresponding Diels–Alder adducts with phenylseleno acrylates by radical-induced reductive deselenocarbonylations, give the 2-substituted bicyclo[3.2.1]oct-6-en-2-yl radicals with some substituents, e.g., alkoxy and phenyl, but not for silyloxymethyl or benzyl substituents. Theoretical calculations with DFT give the thermodynamics of these reactions and the origins of these processes.
Co-reporter:Michael E. Jung, Daniel L. Sun, Timothy A. Dwight, Peiyuan Yu, Wei Li, and K. N. Houk
Organic Letters 2016 Volume 18(Issue 19) pp:5138-5141
Publication Date(Web):September 12, 2016
DOI:10.1021/acs.orglett.6b02588
The efficient synthesis of trans-2-ethenylcyclopropyl aryl ketones via an intramolecular SN2-like displacement of an allylic ester is reported. A novel 1,5-acyl shift process is also observed that contributes to the product mixture. Theoretical calculations provide a rationale for the observed product ratio.
Co-reporter:Jiyong Park, Joseph J. McDonald, Russell C. Petter, and K. N. Houk
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 4) pp:2066-2078
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.jctc.5b01221
Epidermal growth factor receptor (EGFR) inhibitors interrupt EGFR-dependent cellular signaling pathways that lead to accelerated tumor growth and proliferation. Mutation of a threonine in the inhibitor binding pocket, known as the “gatekeeper”, to methionine (T790M) confers acquired resistance to several EGFR-selective inhibitors. We studied interactions between EGFR inhibitors and the gatekeeper residues of the target protein. Thermodynamic integration (TI) with Amber14 indicates that the binding energies of gefitinib and AEE788 to the active state of the T790M mutant EGFR is 3 kcal/mol higher than to the wild type (WT), whereas ATP binding energy to the mutant is similar to the WT. Using metadynamics MD simulations with NAMD v2.9, the conformational equilibrium between the inactive resting state and the catalytically competent activate state was determined for the WT EGFR. When combined with the results obtained by Sutto and Gervasio, our simulations showed that the T790M point mutation lowers the free energy of the active state by 5 kcal/mol relative to the inactive state of the enzyme. Relative to the WT, the T790M mutant binds gefitinib more strongly. The T790M mutation is nevertheless resistant due to its increased binding of ATP. By contrast, the binding of AEE788 to the active state causes a conformational change in the αC-helix adjacent to the inhibitor binding pocket, that results in a 2 kcal/mol energy penalty. The energy penalty explains why the binding of AEE788 to the T790M mutant is unfavorable relative to binding to WT EGFR. These results establish the role of the gatekeeper mutation on inhibitor selectivity. Additional molecular dynamics (MD) simulations, TI, and metadynamics MD simulations reveal the origins of the changes in binding energy of WT and mutants.
Co-reporter:Bernice Lin, Peiyuan Yu, Cyndi Qixin He, K.N. Houk
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 20) pp:4787-4790
Publication Date(Web):15 October 2016
DOI:10.1016/j.bmc.2016.07.032
Density functional theory (M06-2X) studies of the regioselectivity of 1,3-dipolar cycloaddition reactions of benzo and mesitonitrile oxides with alkynyl pinacol and MIDA boronates are reported. Calculated relative free energies of activation reproduce the experimentally observed product ratios. The electronic energies of activation are found to be mainly controlled by distortion energies required to achieve the transition states. Both electronic and steric effects influence regioselectivities.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Zhe Li, Thomas Bally, Kendall N. Houk, and Weston Thatcher Borden
The Journal of Organic Chemistry 2016 Volume 81(Issue 20) pp:9576-9584
Publication Date(Web):October 3, 2016
DOI:10.1021/acs.joc.6b01530
High accuracy quantum chemical calculations show that the barriers to rotation of a CH2 group in the allyl cation, radical, and anion are 33, 14, and 21 kcal/mol, respectively. The benzyl cation, radical, and anion have barriers of 45, 11, and 24 kcal/mol, respectively. These barrier heights are related to the magnitude of the delocalization stabilization of each fully conjugated system. This paper addresses the question of why these rotational barriers, which at the Hückel level of theory are independent of the number of nonbonding electrons in allyl and benzyl, are in fact calculated to be factors that are of 2.4 and 4.1 higher in the cations and 1.5 and 1.9 higher in the anions than in the radicals. We also investigate why the barrier to rotation is higher for benzyl than for allyl in the cations and in the anions. Only in the radicals is the barrier for benzyl lower than that for allyl, as Hückel theory predicts should be the case. These fundamental questions in electronic structure theory, which have not been addressed previously, are related to differences in electron–electron repulsions in the conjugated and nonconjugated systems, which depend on the number of nonbonding electrons.
Co-reporter:Nima Zargari, Pierre Winter, Yong Liang, Joo Ho Lee, Andrew Cooksy, K. N. Houk, and Kyung Woon Jung
The Journal of Organic Chemistry 2016 Volume 81(Issue 20) pp:9820-9825
Publication Date(Web):September 29, 2016
DOI:10.1021/acs.joc.6b01903
Thorough mechanistic studies and DFT calculations revealed a background radical pathway latent in metal-catalyzed oxidation reactions of methane at low temperatures. Use of hydrogen peroxide with TFAA generated a trifluoromethyl radical (•CF3), which in turn reacted with methane gas to selectively yield acetic acid. It was found that the methyl carbon of the product was derived from methane, while the carbonyl carbon was derived from TFAA. Computational studies also support these findings, revealing the reaction cycle to be energetically favorable.
Co-reporter:Elizabeth H. Krenske, Russell C. Petter, and K. N. Houk
The Journal of Organic Chemistry 2016 Volume 81(Issue 23) pp:11726-11733
Publication Date(Web):November 8, 2016
DOI:10.1021/acs.joc.6b02188
Additions of cysteine thiols to Michael acceptors underpin the mechanism of action of several covalent drugs (e.g., afatinib, osimertinib, ibrutinib, neratinib, and CC-292). Reversible Michael acceptors have been reported in which an additional electron-withdrawing group was added at the α-carbon of a Michael acceptor. We have performed density functional theory calculations to determine why thiol additions to these Michael acceptors are reversible. The α-EWG group stabilizes the anionic transition state and intermediate of the Michael addition, but less intuitively, it destabilizes the neutral adduct. This makes the reverse reaction (elimination) both faster and more thermodynamically favorable. For thiol addition to be reversible, the Michael acceptor must also contain a suitable substituent on the β-carbon, such as an aryl or branched alkyl group. Computations explain how these structural elements contribute to reversibility and the ability to tune the binding affinities and the residence times of covalent inhibitors.
Co-reporter:Nico Santschi, Christian Thiehoff, Mareike C. Holland, Constantin G. Daniliuc, K. N. Houk, and Ryan Gilmour
Organometallics 2016 Volume 35(Issue 17) pp:3040-3044
Publication Date(Web):August 25, 2016
DOI:10.1021/acs.organomet.6b00564
Controlling the rotation about unhindered C(sp3)–C(sp3) bonds by simple structural changes has obvious benefits in molecular design. While the avoidance of nonbonding interactions remains one of the cornerstones of acyclic conformational control, stabilizing stereoelectronic effects have the added benefit that conformer populations can be fine-tuned by augmenting or diminishing the central interaction. Strategies may include adjusting the oxidation state of a substituent or reversible formation of a complex to modulate MO levels. In the case of the sulfur–fluorine gauche effect, the propensity of the S–C–C–F motif to adopt a synclinal arrangement (ΦFCCS = 60°), the conformer population distribution of the three dominant rotamers partitioned by 120° can be biased by oxidation of the S atom. Motivated by the importance of sulfur-based ligands in main structural chemistry, the sulfur–fluorine gauche effect was translated to an organometallic paradigm as a potential tool to achieve structural preorganization. This would allow the influence of coinage-metal complexation on conformer population to be initially assessed. The synthesis and characterization of a model gold(I) and silver(I) metal complex featuring a ligand system containing a freely rotatable SCCF motif is disclosed. In both complexes, the title stereoelectronic effect manifests itself in the expected conformation, with the synclinal-endo conformer being preferred. This was corroborated by X-ray crystallography and DFT analysis, and the molar fraction of rotamers was extrapolated from a detailed solution-phase NMR spectroscopic analysis. Complexation was found to reinforce the sulfur–fluorine gauche effect.
Co-reporter:Dr. Mareike C. Holl;Dr. Ryan Gilmour;Dr. K. N. Houk
Angewandte Chemie 2016 Volume 128( Issue 6) pp:2062-2067
Publication Date(Web):
DOI:10.1002/ange.201508980
Abstract
The origin of stereoselectivity in the (3+2) annulation of allenes and enones catalyzed by an amino acid derived phosphine catalyst has been investigated by the use of dispersion-corrected density functional theory. An intermolecular hydrogen bond between the intermediate zwitterion and the enone was found to be the key interaction in the two enantiomeric transition states. Additional stabilization is provided by intermolecular hydrogen-bonding interactions between acidic positions on the catalyst backbone and the substrate. Enantioselectivity occurs because the intermolecular hydrogen bond in the transition state leading to the minor enantiomer is only possible at the expense of reactant distortion.
Co-reporter:Liana Hie;Noah F. FineNathel;Xin Hong;Yun-Fang Yang; Kendall N. Houk; Neil K. Garg
Angewandte Chemie 2016 Volume 128( Issue 8) pp:2860-2864
Publication Date(Web):
DOI:10.1002/ange.201511486
Abstract
We report the first catalytic method for activating the acyl C−O bonds of methyl esters through an oxidative-addition process. The oxidative-addition adducts, formed using nickel catalysis, undergo in situ trapping to provide anilide products. DFT calculations are used to support the proposed reaction mechanism, to understand why decarbonylation does not occur competitively, and to elucidate the beneficial role of the substrate structure and the Al(OtBu)3 additive on the kinetics and thermodynamics of the reaction.
Co-reporter:Liana Hie;Noah F. FineNathel;Xin Hong;Yun-Fang Yang; Kendall N. Houk; Neil K. Garg
Angewandte Chemie International Edition 2016 Volume 55( Issue 8) pp:2810-2814
Publication Date(Web):
DOI:10.1002/anie.201511486
Abstract
We report the first catalytic method for activating the acyl C−O bonds of methyl esters through an oxidative-addition process. The oxidative-addition adducts, formed using nickel catalysis, undergo in situ trapping to provide anilide products. DFT calculations are used to support the proposed reaction mechanism, to understand why decarbonylation does not occur competitively, and to elucidate the beneficial role of the substrate structure and the Al(OtBu)3 additive on the kinetics and thermodynamics of the reaction.
Co-reporter:Yingzi Li;Dr. Sílvia Osuna;Dr. Marc Garcia-Borràs;Xiaotian Qi;Song Liu;Dr. Kendall N. Houk;Dr. Yu Lan
Chemistry - A European Journal 2016 Volume 22( Issue 36) pp:12819-12824
Publication Date(Web):
DOI:10.1002/chem.201601799
Abstract
Diels–Alder cycloaddition is one of the most powerful tools for the functionalization of single-walled carbon nanotubes (SWCNTs). Density functional theory at the B3-LYP level of theory has been used to investigate the reactivity of different-diameter SWCNTs (4–9,5) in Diels–Alder reactions with 1,3-butadiene; the reactivity was found to decrease with increasing SWCNT diameter. Distortion/interaction analysis along the whole reaction pathway was found to be a better way to explore the reactivity of this type of reaction. The difference in interaction energy along the reaction pathway is larger than that of the corresponding distortion energy. However, the distortion energy plots for these reactions show the same trend. Therefore, the formation of the transition state can be determined from the interaction energy. A lower interaction energy leads to an earlier transition state, which indicates a lower activation energy. The computational results also indicate that the original distortion of the SWCNTs leads to an increase in the reactivity of the SWCNTs.
Co-reporter:Adam SimonAlexander J. Yeh, Yu-hong Lam, K. N. Houk
The Journal of Organic Chemistry 2016 Volume 81(Issue 24) pp:12408-12415
Publication Date(Web):November 14, 2016
DOI:10.1021/acs.joc.6b02542
The sources of asymmetric induction in aldol reactions catalyzed by cinchona alkaloid-derived amines, and chiral vicinal diamines in general, have been determined by density functional theory calculations. Four vicinal diamine-catalyzed aldol reactions were examined. The cyclic transition states of these reactions involve nine-membered hydrogen-bonded rings in distinct conformations. Using nomenclature from eight-membered cycloalkanes, the heavy atoms of the low-energy transition states are in crown (chair–chair) and chair-boat conformations. The factors that control which of these are favored have been identified.
Co-reporter:Fengjiao Liu, Zhongyue Yang, Ye Mei, and K. N. Houk
The Journal of Physical Chemistry B 2016 Volume 120(Issue 26) pp:6250-6254
Publication Date(Web):April 19, 2016
DOI:10.1021/acs.jpcb.6b02336
A QM/QM′ direct molecular dynamics study of a water-accelerated Diels–Alder reaction in aqueous solution is reported. Cyclopentadiene and methyl vinyl ketone are known to react faster in water than in nonpolar solvents. We have explored how polarization of water molecules afforded by PM3 influences the nature of the transition state, and the reaction dynamics. We compare the results with previous studies on QM/MM and QM/MM+3QM water simulations from our laboratory. Transition state sampling in vacuum PM3 water boxes indicates that the asynchronicity is 0.54 Å in QM/QM′, as compared to 0.48 Å in QM/MM, and 0.54 Å in QM/MM+3QM water. The mean time gap between the formation of two C–C bonds is 19 fs for QM/QM′, compared to 20 fs for QM/MM, and 25 fs for QM/MM+3QM water. The samplings and time gaps are qualitatively consistent, indicating that water polarization is not significant in sampling and dynamics of bonding changes. The dynamics of hydrogen bonding between reacting molecules and water molecules was also analyzed. From reactants to transition states, H-bond shortening is 0.4 Å by QM/QM′, while only 0.15 Å for QM/MM and QM/MM+3QM water. From reactants to transition states, the mean value of the H-bond angle increases by 19° in QM/QM′, but only 4° in QM/MM, and 10° in QM/MM+3QM water. These suggest that water polarization is essential for the correct representation of dynamical formation of hydrogen bonds in the transition state by water reorientation. QM/QM′ overestimates the hydrogen bonding enhancement because of its underestimation of neutral hydrogen bonding within the reactants, a general deficiency of PM3.
Co-reporter:Dr. Mareike C. Holl;Dr. Ryan Gilmour;Dr. K. N. Houk
Angewandte Chemie International Edition 2016 Volume 55( Issue 6) pp:2022-2027
Publication Date(Web):
DOI:10.1002/anie.201508980
Abstract
The origin of stereoselectivity in the (3+2) annulation of allenes and enones catalyzed by an amino acid derived phosphine catalyst has been investigated by the use of dispersion-corrected density functional theory. An intermolecular hydrogen bond between the intermediate zwitterion and the enone was found to be the key interaction in the two enantiomeric transition states. Additional stabilization is provided by intermolecular hydrogen-bonding interactions between acidic positions on the catalyst backbone and the substrate. Enantioselectivity occurs because the intermolecular hydrogen bond in the transition state leading to the minor enantiomer is only possible at the expense of reactant distortion.
Co-reporter:Elizabeth L. Noey, Gonzalo Jiménez-Osés, Derrick J. L. Clive, and K. N. Houk
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4290-4294
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.joc.6b00691
Intramolecular conjugate displacement (ICD) reactions, developed by the Clive group, form carbocycles and polycyclic amines by intramolecular nucleophilic attack on a Michael acceptor with an allylic leaving group. Quantum mechanical investigations with density functional theory show that ICDs involve a stepwise addition, forming an intermediate stabilized carbanion, followed by elimination. The electron-withdrawing nature of the allylic leaving group facilitates the addition by negative hyperconjugation; the twist-boat conformation of the addition and intermediate is stabilized by this interaction. In the absence of an activating electron-withdrawing group as part of the Michael acceptor, a high energy concerted SN2′ reaction occurs. The reactions of carbon nucleophiles have lower activation energies than those of amines.
Co-reporter:Sílvia Osuna, Gonzalo Jiménez-Osés, Elizabeth L. Noey, and K. N. Houk
Accounts of Chemical Research 2015 Volume 48(Issue 4) pp:1080
Publication Date(Web):March 4, 2015
DOI:10.1021/ar500452q
This Account describes the use of molecular dynamics (MD) simulations to reveal how mutations alter the structure and organization of enzyme active sites. As proposed by Pauling about 70 years ago and elaborated by many others since then, biocatalysis is efficient when functional groups in the active site of an enzyme are in optimal positions for transition state stabilization. Changes in mechanism and covalent interactions are often critical parts of enzyme catalysis. We describe our explorations of the dynamical preorganization of active sites using MD, studying the fluctuations between active and inactive conformations normally concealed to static crystallography. MD shows how the various arrangements of active site residues influence the free energy of the transition state and relates the populations of the catalytic conformational ensemble to the enzyme activity. This Account is organized around three case studies from our laboratory. We first describe the importance of dynamics in evaluating a series of computationally designed and experimentally evolved enzymes for the Kemp elimination, a popular subject in the enzyme design field. We find that the dynamics of the active site is influenced not only by the original sequence design and subsequent mutations but also by the nature of the ligand present in the active site. In the second example, we show how microsecond MD has been used to uncover the role of remote mutations in the active site dynamics and catalysis of a transesterase, LovD. This enzyme was evolved by Tang at UCLA and Codexis, Inc., and is a useful commercial catalyst for the production of the drug simvastatin. X-ray analysis of inactive and active mutants did not reveal differences in the active sites, but relatively long time scale MD in solution showed that the active site of the wild-type enzyme preorganizes only upon binding of the acyl carrier protein (ACP) that delivers the natural acyl group to the active site. In the absence of bound ACP, a noncatalytic arrangement of the catalytic triad is dominant. Unnatural truncated substrates are inactive because of the lack of protein–protein interactions provided by the ACP. Directed evolution is able to gradually restore the catalytic organization of the active site by motion of the protein backbone that alters the active site geometry. In the third case, we demonstrate the key role of MD in combination with crystallography to identify the origins of substrate-dependent stereoselectivities in a number of Codexis-engineered ketoreductases, one of which is used commercially for the production of the antibiotic sulopenem. Here, mutations alter the shape of the active site as well as the accessibility of water to different regions of it. Each of these examples reveals something different about how mutations can influence enzyme activity and shows that directed evolution, like natural evolution, can increase catalytic activity in a variety of remarkable and often subtle ways.
Co-reporter:Peiyuan Yu; Ashay Patel;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 42) pp:13518-13523
Publication Date(Web):October 5, 2015
DOI:10.1021/jacs.5b06656
The transannular [6 + 4] cycloaddition proposed as a step in the biosynthesis of heronamide A has been modeled using density functional theory. The proposed cycloaddition is highly stereoselective, affording a single product. The reaction proceeds through an ambimodal transition state that directly leads to a [4 + 2] adduct in addition to the observed [6 + 4] adduct. Interconversion of these adducts is possible via a facile Cope rearrangement. The [6 + 4] adduct is thermodynamically more stable than the [4 + 2] adduct by 5.2 kcal mol–1 due to a combination of the ring and steric strain in the [4 + 2] product. The results strongly support the plausibility of the proposed transannular [6 + 4] cycloaddition in the biogenesis of heronamide A and may provide insights to designing substrates that selectively undergo [6 + 4] cycloaddition to form unbridged 10-membered rings.
Co-reporter:Austin H. Asari; Yu-hong Lam; Marcus A. Tius;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 40) pp:13191-13199
Publication Date(Web):October 1, 2015
DOI:10.1021/jacs.5b08969
The origins of stereoselectivity of the Nazarov reactions of α-hydroxydivinylketones catalyzed by a vicinal thiourea–primary amine first reported by Tius have been explored with density functional theory. The electrocyclization transition structures in which the thiourea group of the catalyst donates two hydrogen bonds to the keto carbonyl group of the Nazarov reactant and the primary amine accepts a hydrogen bond from the hydroxyl group of the reactant have been modeled. The enantiomeric Nazarov transition structures, which are conventionally described by the absolute sense of conrotation of the dienone termini (“clockwise” or “counterclockwise”) in the literature, are nonplanar and adopt helically chiral conformations. The interactions of these helical electrocyclization transition structures with the chiral catalyst are studied in detail. The organocatalyst is found to employ a combination of hydrogen bonding and steric effects to achieve helical recognition of the Nazarov transition state.
Co-reporter:Sharon R. Neufeldt; Gonzalo Jiménez-Osés; John R. Huckins; Oliver R. Thiel;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 31) pp:9843-9854
Publication Date(Web):July 21, 2015
DOI:10.1021/jacs.5b03535
The origin of the high reactivity and site selectivity of pyridine N-oxide substrates in O-pivaloyl hydroxamic acid-directed Rh(III)-catalyzed (4+2) annulation reactions with alkynes was investigated computationally. The reactions of the analogous pyridine derivatives were previously reported to be slower and to display poor site selectivity for functionalization of the C(2)–H vs the C(4)–H bonds of the pyridine ring. The N-oxide substrates are found to be more reactive overall because the directing group interacts more strongly with Rh. For N-oxide substrates, alkyne insertion is rate-limiting and selectivity-determining in the reaction with a dialkyl alkyne, but C–H activation can be selectivity-determining with other coupling partners such as terminal alkynes. The rates of reaction with a dialkyl alkyne at the two sites of a pyridine substrate are limited by two different steps: C–H activation is limiting for C(2)-functionalization, while alkyne insertion is limiting for C(4)-functionalization. Consistent with the observed poor site selectivity in the reaction of a pyridine substrate, the overall energy barriers for functionalization of the two positions are nearly identical. High C(2)-selectivity in the C–H activation step of the reaction of the N-oxide is due to a cooperative effect of the C–H Brønsted acidity, the strength of the forming C–Rh bond, and intramolecular electrostatic interactions between the [Rh]Cp* and the heteroaryl moieties. On the other hand, some of these forces are in opposition in the case of the pyridine substrate, and C(4)–H activation is moderately favored overall. The alkyne insertion step is favored at C(2) over C(4) for both substrates, and this preference is largely influenced by electrostatic interactions between the alkyne and the heteroarene. Experimental results that support these calculations, including kinetic isotope effect studies, H/D exchange studies, and results using a substituted pyridine, are also described.
Co-reporter:Matthew N. Grayson; Michael J. Krische;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 27) pp:8838-8850
Publication Date(Web):June 24, 2015
DOI:10.1021/jacs.5b04844
The catalyst generated in situ from RuH2(CO)(PPh3)3, (S)-SEGPHOS, and a chiral phosphoric acid promotes asymmetric hydrohydroxyalkylation of butadiene and affords enantioenriched α-methyl homoallylic alcohols. The observed diastereo- and enantioselectivities are determined by both the chiral phosphine and chiral phosphate ligands. Density functional theory calculations (M06/SDD-6-311G(d,p)–IEFPCM(acetone)//B3LYP/SDD-6-31G(d)) predict that the product distribution is controlled by the kinetics of carbon–carbon bond formation, and this process occurs via a closed-chair Zimmerman–Traxler-type transition structure (TS). Chiral-phosphate-dependent stereoselectivity arising from this TS is enabled through a hydrogen bond between the phosphoryl oxygen and the aldehyde formyl proton present in TADDOL-derived catalysts. This interaction is absent in the corresponding BINOL-derived systems, and the opposite diastereo- and enantioselectivity is observed. Additional factors influencing the stereochemical control are determined.
Co-reporter:Xin Hong; Daniel A. Bercovici; Zhongyue Yang; Nezar Al-Bataineh; Ramya Srinivasan; Ram C. Dhakal; K. N. Houk;Matthias Brewer
Journal of the American Chemical Society 2015 Volume 137(Issue 28) pp:9100-9107
Publication Date(Web):July 7, 2015
DOI:10.1021/jacs.5b04474
The 1-aza-2-azoniaallene salts, generated from α-chloroazo compounds by treatment with halophilic Lewis acids, undergo intramolecular C–H amination reactions to form pyrazolines in good to excellent yields. This intramolecular amination occurs readily at both benzylic and tertiary aliphatic positions and proceeds at an enantioenriched chiral center with retention of stereochemistry. Competition experiments show that insertion occurs more readily at an electron-rich benzylic position than it does at an electron-deficient one. The C–H amination reaction occurs only with certain tethers connecting the heteroallene cation and the pendant aryl groups. With a longer tether or when the reaction is intermolecular, electrophilic aromatic substitution occurs instead of C–H amination. The mechanism and origins of stereospecificity and chemoselectivity were explored with density functional theory (B3LYP and M06-2X). The 1-aza-2-azoniaallene cation undergoes C–H amination through a hydride transfer transition state to form the N–H bond, and the subsequent C–N bond formation occurs spontaneously to generate the heterocyclic product. This concerted two-stage mechanism was shown by IRC and quasi-classical molecular dynamics trajectory studies.
Co-reporter:Hung V. Pham; Alexander S. Karns; Christopher D. Vanderwal;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6956-6964
Publication Date(Web):May 11, 2015
DOI:10.1021/jacs.5b03718
The fascinating intramolecular arene/allene cycloaddition discovered by Himbert affords dearomatized, polycyclic intermediates with sufficient strain energy to drive rearrangement processes of the newly formed ring system. We disclose a detailed examination of a thermally induced stepwise dyotropic skeletal rearrangement of these cycloadducts, a reaction also first described by Himbert. We offer computational evidence for a two-stage mechanism for this formal dyotropic rearrangement and provide rationalizations for the significant substitution-dependent rate differences observed in experiments. These investigations led to the development of a Lewis-acid-catalyzed rearrangement of precursors that were unreactive under simple thermal instigation. The isolation of the product of an “interrupted” rearrangement under Lewis acidic conditions provides further support for the proposed stepwise mechanism. Computational results also matched experiments in terms of regiochemical preferences in unsymmetrical rearrangement precursors and explained why lactam O-, S-, and C-heterologues do not easily undergo this rearrangement.
Co-reporter:Tao Yang; Shigeru Nagase; Takeshi Akasaka; Josep M. Poblet; K. N. Houk; Masahiro Ehara;Xiang Zhao
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6820-6828
Publication Date(Web):May 13, 2015
DOI:10.1021/jacs.5b01444
The reaction mechanism and origin of regioselectivity of (2 + 2) cycloadditions of benzyne to endohedral metallofullerenes M3N@C80 (M = Sc, Y) were investigated with density functional calculations. The reaction was demonstrated to follow a diradical mechanism rather than a carbene mechanism, in which the formation of the diradical intermediate is the rate-determining step. Through rotation of benzyne moiety on the fullerene surface, the diradical intermediate on 566 site could isomerize to two new diradical intermediates which give rise to two distinct [5,6] and [6,6] benzoadducts, respectively. However, the diradical intermediate on 666 site only produces the [6,6] benzoadduct. The nature of the endohedral cluster not only influences the regioselectivity, but also determines the cycloadduct geometry. For Sc3N@C80, the [5,6] benzoadduct is preferred kinetically and thermodynamically, whereas in the case of Y3N@C80, both [5,6] and [6,6] benzoadducts are favorable. In contrast to closed-cage benzoadducts of Sc3N@C80, Y3N@C80 affords open-cage benzoadducts, making it the first example that the endohedral cluster could alter cycloadducts from the closed cage to open cage. With further analysis, it is revealed that the origin of regioselectivity results from the local strain energy of the fullerene cage.
Co-reporter:Chendan Zhu; Yong Liang; Xin Hong; Heqing Sun; Wei-Yin Sun; K. N. Houk;Zhuangzhi Shi
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7564-7567
Publication Date(Web):June 2, 2015
DOI:10.1021/jacs.5b03488
A new strategy is reported for intramolecular sp3 C–H amination under mild reaction conditions using iodoarene as catalyst and m-CPBA as oxidant. This C–H functionalization involving iodine(III) reagents generated in situ occurs readily at sterically hindered tertiary C–H bonds. DFT (M06-2X) calculations show that the preferred pathway involves an iodonium cation intermediate and proceeds via an energetically concerted transition state, through hydride transfer followed by the spontaneous C–N bond formation. This leads to the experimentally observed amination at a chiral center without loss of stereochemical information.
Co-reporter:Salvador Pérez-Estrada; Braulio Rodrı́guez-Molina; Leilei Xiao; Rosa Santillan; Gonzalo Jiménez-Osés; K. N. Houk;Miguel A. Garcia-Garibay
Journal of the American Chemical Society 2015 Volume 137(Issue 6) pp:2175-2178
Publication Date(Web):January 30, 2015
DOI:10.1021/ja512053t
A molecular rotor built with a stator formed by two rigid 9β-mestranol units having a 90° bent angle linked to a central phenylene rotator has an ideal structure to examine aromatic CH/π interactions. Energies and populations of the multiple solution conformations from quantum-mechanical calculations and molecular dynamics simulations were combined with variable-temperature (VT) 1H NMR data to establish the enthalpy of this interaction and the entropy associated with rotation about a single bond. Rotational dynamics in the solid state were determined via VT cross-polarization magic-angle spinning 13C NMR spectroscopy.
Co-reporter:Keary M. Engle; Gang Lu; Shao-Xiong Luo; Lawrence M. Henling; Michael K. Takase; Peng Liu; K. N. Houk;Robert H. Grubbs
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5782-5792
Publication Date(Web):April 21, 2015
DOI:10.1021/jacs.5b01144
A series of second-generation ruthenium olefin metathesis catalysts was investigated using a combination of reaction kinetics, X-ray crystallography, NMR spectroscopy, and DFT calculations in order to determine the relationship between the structure of the chelating o-alkoxybenzylidene and the observed initiation rate. Included in this series were previously reported catalysts containing a variety of benzylidene modifications as well as four new catalysts containing cyclopropoxy, neopentyloxy, 1-adamantyloxy, and 2-adamantyloxy groups. The initiation rates of this series of catalysts were determined using a UV/vis assay. All four new catalysts were observed to be faster-initiating than the corresponding isopropoxy control, and the 2-adamantyloxy catalyst was found to be among the fastest-initiating Hoveyda-type catalysts reported to date. Analysis of the X-ray crystal structures and computed energy-minimized structures of these catalysts revealed no correlation between the Ru–O bond length and Ru–O bond strength. On the other hand, the initiation rate was found to correlate strongly with the computed Ru–O bond strength. This latter finding enables both the rationalization and prediction of catalyst initiation through the calculation of a single thermodynamic parameter in which no assumptions about the mechanism of the initiation step are made.
Co-reporter:Xiao-Na Wang; Elizabeth H. Krenske; Ryne C. Johnston; K. N. Houk;Richard P. Hsung
Journal of the American Chemical Society 2015 Volume 137(Issue 16) pp:5596-5601
Publication Date(Web):April 20, 2015
DOI:10.1021/jacs.5b02561
We report the first experimental evidence for the generation of highly strained cis,trans-cycloheptadienones by electrocyclic ring opening of 4,5-fused cyclobutenamides. In the presence of AlCl3, the cyclobutenamides rearrange to [2.2.1]-bicyclic ketones; DFT calculations provide evidence for a mechanism involving torquoselective 4π-electrocyclic ring opening to a cis,trans-cycloheptadienone followed by a Nazarov-like recyclization and a 1,2-alkyl shift. Similarly, 4,6-fused cyclobutenamides undergo AlCl3-catalyzed rearrangements to [3.2.1]-bicyclic ketones through cis,trans-cyclooctadienone intermediates. The products can be further elaborated via facile cascade reactions to give complex tri- and tetracyclic molecules.
Co-reporter:Travis C. McMahon; Jose M. Medina; Yun-Fang Yang; Bryan J. Simmons; K. N. Houk;Neil K. Garg
Journal of the American Chemical Society 2015 Volume 137(Issue 12) pp:4082-4085
Publication Date(Web):March 13, 2015
DOI:10.1021/jacs.5b01589
We report the generation of the first 3,4-piperidyne and its use as a building block for the synthesis of annulated piperidines. Experimental and computational studies of this intermediate are disclosed, along with comparisons to the well-known 3,4-pyridyne. The distortion/interaction model is used to explain the observed regioselectivities.
Co-reporter:Lisa Törk; Gonzalo Jiménez-Osés; Charles Doubleday; Fang Liu;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4749-4758
Publication Date(Web):March 2, 2015
DOI:10.1021/jacs.5b00014
The cycloadditions of tetrazines with cyclopropenes and other strained alkenes have become among the most valuable bioorthogonal reactions. These reactions lead to bicyclic Diels–Alder adducts that spontaneously lose N2. We report quantum mechanical (QM) and quasiclassical trajectory simulations on a number of these reactions, with special attention to stereoelectronic and dynamic effects on spontaneous N2 loss from these adducts. QM calculations show that the barrier to N2 loss is low, and molecular dynamics calculations show that the intermediate is frequently bypassed dynamically. There is a large preference for N2 loss anti to the cyclopropane moiety rather than syn from adducts formed from reactions with cyclopropenes. This is explained by the interactions of the Walsh orbitals of the cyclopropane group with the breaking C–N bonds in N2 loss. Dynamical effects opposing the QM preferences have also been discovered involving the coupling of vibrations associated with the formation of the new C–C bonds in the cycloaddition step, and those of the breaking C–N bonds during subsequent N2 loss. This dynamic matching leads to pronounced nonstatistical effects on the lifetimes of Diels–Alder intermediates. An unusual oscillatory behavior of the intermediate decay rate has been identified and attributed to specific vibrational coupling.
Co-reporter:Yu-hong Lam;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 5) pp:2116-2127
Publication Date(Web):January 28, 2015
DOI:10.1021/ja513096x
The intramolecular aldol condensation of 4-substituted heptane-2,6-diones leads to chiral cyclohexenones. The origins of the enantioselectivities of this reaction, disclosed by List et al. using a cinchona alkaloid-derived primary amine (cinchona amine) organocatalyst, have been determined with dispersion-corrected density functional theory (DFT). The stereocontrol hinges on the chair preference of the substrate–enamine intermediate and the conformational preferences of a hydrogen-bonded nine-membered aldol transition state containing eight heavy atoms. The conformations of the hydrogen-bonded ring in the various stereoisomeric transition structures have been analyzed in detail and shown to closely resemble the conformers of cyclooctane. A model of stereoselectivity is proposed for the cinchona amine catalysis of this reaction. The inclusion of Grimme’s dispersion corrections in the DFT calculations (B3LYP-D3(BJ)) substantially improves the agreement of the computed energetics and experiment, attesting to the importance of dispersion effects in stereoselectivity.
Co-reporter:Xin Hong, Jinglin Wang, Yun-Fang Yang, Lisi He, Chun-Yu Ho, and K. N. Houk
ACS Catalysis 2015 Volume 5(Issue 9) pp:5545
Publication Date(Web):August 13, 2015
DOI:10.1021/acscatal.5b01075
The [LNiH]+-catalyzed hydroalkenylation between styrene and α-olefins gives distinctive chemo- and regioselectivities with N-heterocyclic carbene (L = NHC) ligands: (a) the reaction with NHC ligands produces the branched tail-to-tail products, whereas the reaction with phosphine ligands (L = PR3) favors the tail-to-head regio-isomers; (b) the reaction stops at heterodimerization with no further oligomerization even with excess α-olefin substrates; (c) typical side reactions with α-olefins, such as isomerization to internal olefins or polymerization, are either significantly diminished or eliminated. To understand the operating mechanism and origins of selectivities, density functional theory (DFT) calculations were performed, and several additional experiments were conducted. The olefin insertion step is found to determine both the regioselectivity and chemoselectivity, leading to the tail-to-tail heterohydroalkenylation product. With a small NHC ligand (1,3-dimethylimidazol-2-ylidene), the intrinsic electronic effects of ligand favor the tail-to-head regioisomer by about 1 kcal/mol in the olefin insertion step. With bulky NHC ligands (1,3-bis(2,6-dimethylphenyl)imidazol-2-ylidene or SIPr), the steric repulsions between the ligand and the substituent of the inserting alkene override the intrinsic electronic preference, making the tail-to-tail regioisomer favored (about 3 kcal/mol with both ligands). In the competition between homo- and heterodimerization, the insertion of the secondary styrene breaks its π-conjugation, making the insertion of styrene about 2 kcal/mol less favorable than that of alkyl-substituted alkenes. In addition, the interaction between nickel and phenyl group of styrene stabilizes the resting state and inhibits the side reactions with α-olefins, suggesting that styrene, or similar aryl olefins, is not only a substrate, but also an inhibitor for side reactions. This unique effect of styrene is verified by control experiments.Keywords: density functional theory; hydrovinylation; NHC ligand; regioselectivity; β-hydride transfer
Co-reporter:C. Thiehoff, M. C. Holland, C. Daniliuc, K. N. Houk and R. Gilmour
Chemical Science 2015 vol. 6(Issue 6) pp:3565-3571
Publication Date(Web):17 Apr 2015
DOI:10.1039/C5SC00871A
The gauche conformation of the 1,2-difluoroethane motif is known to involve stabilising hyperconjugative interactions between donor (bonding, σC–H) and acceptor (antibonding, σ*C–F) orbitals. This model rationalises the generic conformational preference of F–Cβ–Cα–X systems (ϕFCCX ≈ 60°), where X is an electron deficient substituent containing a Period 2 atom. Little is known about the corresponding Period 3 systems, such as sulfur and phosphorus, where multiple oxidation states are possible. Conformational analyses of β-fluorosulfides, -sulfoxides and -sulfones are disclosed here, thus extending the scope of the fluorine gauche effect to the 3rd Period (F–C–C–S(O)n; ϕFCCS ≈ 60°). Synergy between experiment and computation has revealed that the gauche effect is only pronounced in structures bearing an electropositive vicinal sulfur atom (S+–O−, SO2).
Co-reporter:Mary C. Walton, Yun-Fang Yang, Xin Hong, K. N. Houk, and Larry E. Overman
Organic Letters 2015 Volume 17(Issue 24) pp:6166-6169
Publication Date(Web):November 25, 2015
DOI:10.1021/acs.orglett.5b03171
Copper-catalyzed reactions of glycine ester arylimines and methacrylonitrile provide selective access to either the endo or exo pyrrolidine cycloadducts. DFT calculations have elucidated the origins of ligand-controlled diastereoselectivity.
Co-reporter:Zhi-Xiong Ma, Ashay Patel, K. N. Houk, and Richard P. Hsung
Organic Letters 2015 Volume 17(Issue 9) pp:2138-2141
Publication Date(Web):April 10, 2015
DOI:10.1021/acs.orglett.5b00727
Highly torquoselective electrocyclizations of chiral 1-azatrienes are described. These 1-azatrienes contain an allylic stereocenter that is substituted with a silyl group and are derived in situ from condensation of γ-silyl-substituted enals with vinylogous amides. The ensuing stereoselective ring closures are part of a tandem sequence that constitutes an aza-[3 + 3] annulation method for constructing 1,2-dihydropyridines. Several mechanisms for the formal 1,7-hydrogen shift of these 1-azatrienes were evaluated computationally.
Co-reporter:Tim S. Chung, Steven A. Lopez, K. N. Houk, and Miguel A. Garcia-Garibay
Organic Letters 2015 Volume 17(Issue 18) pp:4568-4571
Publication Date(Web):September 4, 2015
DOI:10.1021/acs.orglett.5b02290
Crystalline cis- or trans-Δ2-1,2,3-triazolines prepared by highly stereospecific and regioselective hydrogen bonding-catalyzed dipolar cycloaddition of activated cis- or trans-alkenes with aryl azides undergo a highly stereospecific photodenitrogenation to form the corresponding cis- or trans- azidirines in high chemical yields. While examples involving disubstituted and trisubstituted triazolines highlight steric challenges encountered in the dipolar cycloaddition reaction, the stereochemical control exerted by the crystalline lattice is enhanced by bulky substituents in the triazoline precursors to generate aziridines photochemically.
Co-reporter:Zhongyue Yang, Charles Doubleday, and K. N. Houk
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 12) pp:5606-5612
Publication Date(Web):November 4, 2015
DOI:10.1021/acs.jctc.5b01029
We describe a solvent-perturbed transition state (SPTS) sampling scheme for simulating chemical reaction dynamics in condensed phase. The method, adapted from Truhlar and Gao’s ensemble-averaged variational transition state theory, includes the effect of instantaneous solvent configuration on the potential energy surface of the reacting system (RS) and allows initial conditions for the RS to be sampled quasiclassically by TS normal mode sampling. We use a QM/MM model with direct dynamics, in which QM forces of the RS are computed at each trajectory point. The SPTS scheme is applied to the acceleration of the Diels–Alder reaction of cyclopentadiene (CP) + methyl vinyl ketone (MVK) in water. We explored the effect of the number of SPTS and of solvent box size on the distribution of bond lengths in the TS. Statistical sampling of the sampling was achieved when distribution of forming bond lengths converged. We describe the region enclosing the partial bond lengths as the transition zone. Transition zones in the gas phase, SMD implicit solvent, QM/MM, and QM/MM+QM (3 water molecules treated by QM) vary according to the ability of the medium to stabilize zwitterionic structures. Mean time gaps between formation of C–C bonds vary from 11 fs for gas phase to 25 fs for QM/MM+QM. Mean H-bond lengths to O(carbonyl) in QM/MM+QM are 0.14 Å smaller at the TS than in MVK reactant, and the mean O(carbonyl)–H(water)–O(water) angle of H-bonds at the TS is 10° larger than in MVK reactant.
Co-reporter:Edward Richmond, Kenneth B. Ling, Nicolas Duguet, Lois B. Manton, Nihan Çelebi-Ölçüm, Yu-Hong Lam, Sezen Alsancak, Alexandra M. Z. Slawin, K. N. Houk and Andrew D. Smith
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 6) pp:1807-1817
Publication Date(Web):12 Dec 2014
DOI:10.1039/C4OB02526A
The reaction of L-serine derived N-arylnitrones with alkylarylketenes generates asymmetric 3-alkyl-3-aryloxindoles in good to excellent yields (up to 93%) and excellent enantioselectivity (up to 98% ee) via a pericyclic cascade process. The optimization, scope and applications of this transformation are reported, alongside further synthetic and computational investigations. The preparation of the enantiomer of a Roche anti-cancer agent (RO4999200) 1 (96% ee) in three steps demonstrates the potential utility of this methodology.
Co-reporter:Myles B. Herbert, Benjamin A. Suslick, Peng Liu, Lufeng Zou, Peter K. Dornan, K. N. Houk, and Robert H. Grubbs
Organometallics 2015 Volume 34(Issue 12) pp:2858-2869
Publication Date(Web):June 2, 2015
DOI:10.1021/acs.organomet.5b00185
In order to design improved ruthenium catalysts for Z-selective olefin metathesis reactions, four cyclometalated catalysts with new chelated architectures were synthesized, structurally characterized, and tested in metathesis assays. The mechanism of formation of each was explored using DFT calculations. Of note, two complexes are derived from activation of a tertiary C–H bond, and one features a four-membered chelating architecture. In addition, two dipivalate complexes that did not undergo further C–H activation were isolated and studied to elucidate information about the factors affecting cyclometalation.
Co-reporter:Jiyong Park, Hung V. Pham, Kristian Mogensen, Theis Ivan Solling, Martin Vad Bennetzen, and K. N. Houk
The Journal of Organic Chemistry 2015 Volume 80(Issue 2) pp:997-1005
Publication Date(Web):December 19, 2014
DOI:10.1021/jo502488e
In order to identify potential de novo enzyme templates for the cleavage of C–C single bonds in long-chain hydrocarbons, we analyzed protein structures that bind substrates containing alkyl and alkenyl functional groups. A survey of ligand-containing protein structures deposited in the Protein Data Bank resulted in 874 entries, consisting of 194 unique ligands that have ≥10 carbons in a linear chain. Fatty acids and phospholipids are the most abundant types of ligands. Hydrophobic amino acids forming α-helical structures frequently line the binding pockets. Occupation of these binding sites was evaluated by calculating both the buried surface area and volume employed by the ligands; these quantities are similar to those computed for drug–protein complexes. Surface complementarity is relatively low due to the nonspecific nature of the interaction between the long-chain hydrocarbons and the hydrophobic amino acids. The selected PDB structures were annotated on the basis of their SCOP and EC identification numbers, which will facilitate design template searches based on structural and functional homologies. Relatively low surface complementarity and ∼55% volume occupancy, also observed in synthetic-host, alkane-guest systems, suggest general principles for the recognition of long-chain linear hydrocarbons.
Co-reporter:Ashay Patel, F. G. West, and K. N. Houk
The Journal of Organic Chemistry 2015 Volume 80(Issue 5) pp:2790-2795
Publication Date(Web):January 29, 2015
DOI:10.1021/acs.joc.5b00070
The 6π electrocyclizations and Nazarov cyclizations of a series of bridged bicyclic substrates were modeled with the M06-2X density functional and the def2-TZVPP basis set, and the factors responsible for the reactivities of these substrates and the stereoselectivities of their ring closures were identified. The ring closures of these bridged bicyclic trienes are up to a million-fold faster (ΔΔG⧧ = 10 kcal mol–1) than that of 1,3,5-hexatriene, despite the absence of any activating functional groups. Three effects, preorganization, predistortion, and a CH π interaction, are responsible for this sizable difference in reactivity. Stereoselectivity is partially controlled by torsional effects, but for highly exo selective electrocyclizations, it is reinforced by a second effect (either a CH π interaction or a steric clash). The absence of this second effect in the ring closures of several divinyl ketones explains the reduced selectivity of these ring closures. In one case, a divinyl ketone (ketone 6) undergoes Nazarov cyclization to yield the endo product preferentially. For this example, through-space interaction of a nonconjugated alkene with the divinyl ketone π system in the endo transition state and a steric effect override the intrinsic exo selectivity.
Co-reporter:Brian J. Levandowski and K. N. Houk
The Journal of Organic Chemistry 2015 Volume 80(Issue 7) pp:3530-3537
Publication Date(Web):March 5, 2015
DOI:10.1021/acs.joc.5b00174
The Diels–Alder reactions of cyclopentadiene, cyclohexadiene, and cycloheptadiene with a series of dienophiles were studied with quantum mechanical calculations (M06-2X density functional theory) and analyzed with the distortion/interaction model. The poor reactivities of cyclohexadiene and cycloheptadiene with dienophiles that give relatively synchronous transition states result from the substantial distortion required to achieve a transition state involving the formation of two bonds of the diene simultaneously. However, highly asynchronous or stepwise reactions result in less distortion of the diene and less differences in reactivities of different dienes. The transition state geometry of cyclopentadiene is less distorted in the asynchronous reaction with 1,1-dicyanoethylene compared to that with cis- and trans-1,2-dicyanoethylenes, which react through synchronous transition states.
Co-reporter:Ryan D. Baxter, Yong Liang, Xin Hong, Timothy A. Brown, Richard N. Zare, K. N. Houk, Phil S. Baran, and Donna G. Blackmond
ACS Central Science 2015 Volume 1(Issue 8) pp:456
Publication Date(Web):November 2, 2015
DOI:10.1021/acscentsci.5b00332
Kinetic, spectroscopic, and computational studies of radical C–H arylations highlight the interplay between chemical and physical rate processes in these multiphase reactions. Anomalous concentration dependences observed here may be reconciled by considering the role of phase transfer processes that mediate concentrations in each phase. In addition, understanding interactions through phase boundaries enables their use in optimization of reaction performance.
Co-reporter:Ilhan Yavuz
The Journal of Physical Chemistry C 2015 Volume 119(Issue 1) pp:158-165
Publication Date(Web):December 15, 2014
DOI:10.1021/jp510567d
Semiconductor poly(3,3″-didodecyl-quaterthiophene) (PQT-12) polymer for which the hole mobility exceeds 0.1 cm2/(V s) exhibits promising charge-transport characteristics as an organic thin-film transistor. A family of its oligomeric analogs, DDQT-n (3,3″-didedocylquaterthiophene-n) has been synthesized (with n = 1–6) and extensively characterized [Zhang, L.; et al. J. Am. Chem. Soc. 2013, 135, 844−854]. Through atomistic molecular dynamics and charge-transport simulations, we have studied the morphologies and electronic properties of crystalline didodecylquaterthiophenes (DDQT-1, DDQT-2, and DDQT-3). The morphologies are characterized by molecular ordering and paracrystallinity, while charge-transport is characterized by electronic-coupling, reorganization energy, energetic disorder, and hole mobility, calculated with VOTCA package. We observed increasing transport efficiency with increasing molecule size, as the morphologies evolve from oligomeric to polymeric packing arrangements. The trend is related to decreasing hole reorganization energy, energetic disorder, and increasing efficacy of transport topology. We also elucidate a direct link between molecular ordering and charge-carrier mobility of different DDQT-n oligomers.
Co-reporter:Cyndi Qixin He, Tiffany Q. Chen, Ashay Patel, Sedef Karabiyikoglu, Craig A. Merlic, and K. N. Houk
The Journal of Organic Chemistry 2015 Volume 80(Issue 21) pp:11039-11047
Publication Date(Web):October 14, 2015
DOI:10.1021/acs.joc.5b02288
Density functional theory calculations were performed on a set of 13 transannular Diels–Alder (TADA) reactions with 10–18-membered rings. The results were compared with those for bimolecular and intramolecular Diels–Alder reactions in order to investigate the controlling factors of the high TADA reactivities. The effects of tether length, heteroatoms, and alkynyl dienophiles on reactivity were analyzed. We found a correlation between tether length and reactivity, specifically with 12-membered macrocycles undergoing cycloaddition most readily. Furthermore, modifying 12-membered macrocycles by heteroatom substitution and utilizing alkynyl dienophiles enhances the reaction rates up to 105-fold.
Co-reporter:Kazuya Honda, Steven A. Lopez, K. N. Houk, and Koichi Mikami
The Journal of Organic Chemistry 2015 Volume 80(Issue 23) pp:11768-11772
Publication Date(Web):August 24, 2015
DOI:10.1021/acs.joc.5b01361
The reactivities and torquoselectivities of electrocyclic ring opening reactions of fluoromethyl-substituted cyclobutenes and oxetenes were studied with M06-2X density functional theory. The torquoselectivities of a series of mono-, di-, and trifluoromethylcyclobutenes and oxetenes result from the interplay of favorable orbital interactions and closed-shell repulsions. When the substituent rotates inward, there can be a favorable interaction between the breaking σCO bond and the σCF* orbital (σCO → σCF*) of the fluoromethyl group in fluoromethyloxetenes. The preference for rotation of a fluoromethyl group is decreased in trifluoromethyloxetenes because closed-shell repulsions between the breaking σCO bond and trifluoromethyl substituent orbitals compete with the σCO → σCF* interaction.
Co-reporter:Joel F. Hooper, Natalie C. James, Esra Bozkurt, Viktorya Aviyente, Jonathan M. White, Mareike C. Holland, Ryan Gilmour, Andrew B. Holmes, and K. N. Houk
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12058-12075
Publication Date(Web):November 11, 2015
DOI:10.1021/acs.joc.5b02037
The intramolecular Diels–Alder reaction has been used as a powerful method to access the tricyclic core of the eunicellin natural products from a number of 9-membered-ring precursors. The endo/exo selectivity of this reaction can be controlled through a remarkable organocatalytic approach, employing MacMillan’s imidazolidinone catalysts, although the mechanistic origin of this selectivity remains unclear. We present a combined experimental and density functional theory investigation, providing insight into the effects of medium-ring constraints on the organocatalyzed intramolecular Diels–Alder reaction to form the isobenzofuran core of the eunicellins.
Co-reporter:Eric M. Phillips;Tehetena Mesganaw;Ashay Patel;Simon Duttwyler;Bron Q. Mercado;Dr. Kendall N. Houk;Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2015 Volume 54( Issue 41) pp:12044-12048
Publication Date(Web):
DOI:10.1002/anie.201505604
Abstract
The asymmetric synthesis of ent-ketorfanol from simple and commercially available precursors is reported. A RhI-catalyzed intramolecular CH alkenylation/torquoselective 6π electrocyclization cascade provides a fused bicyclic 1,2-dihydropyridine as a key intermediate. Computational studies were performed to understand the high torquoselectivity of the key 6π electrocyclization. The computational results demonstrate that a conformational effect is responsible for the observed selectivity. The ketone functionality and final ring are introduced in a single step by a redox-neutral acid-catalyzed rearrangement of a vicinal diol to give the requisite carbonyl, followed by intramolecular Friedel–Crafts alkylation.
Co-reporter:Eric M. Phillips;Tehetena Mesganaw;Ashay Patel;Simon Duttwyler;Bron Q. Mercado;Dr. Kendall N. Houk;Dr. Jonathan A. Ellman
Angewandte Chemie 2015 Volume 127( Issue 41) pp:12212-12216
Publication Date(Web):
DOI:10.1002/ange.201505604
Abstract
The asymmetric synthesis of ent-ketorfanol from simple and commercially available precursors is reported. A RhI-catalyzed intramolecular CH alkenylation/torquoselective 6π electrocyclization cascade provides a fused bicyclic 1,2-dihydropyridine as a key intermediate. Computational studies were performed to understand the high torquoselectivity of the key 6π electrocyclization. The computational results demonstrate that a conformational effect is responsible for the observed selectivity. The ketone functionality and final ring are introduced in a single step by a redox-neutral acid-catalyzed rearrangement of a vicinal diol to give the requisite carbonyl, followed by intramolecular Friedel–Crafts alkylation.
Co-reporter:Elizabeth L. Noey;Nidhi Tibrewal;Gonzalo Jiménez-Osés;Sílvia Osuna;Jiyong Park;Carly M. Bond;Duilio Cascio;Jack Liang;Xiyun Zhang;Gjalt W. Huisman;Yi Tang
PNAS 2015 112 (51 ) pp:E7065-E7072
Publication Date(Web):2015-12-22
DOI:10.1073/pnas.1507910112
Mutants of Lactobacillus kefir short-chain alcohol dehydrogenase, used here as ketoreductases (KREDs), enantioselectively reduce the pharmaceutically relevant
substrates 3-thiacyclopentanone and 3-oxacyclopentanone. These substrates differ by only the heteroatom (S or O) in the ring,
but the KRED mutants reduce them with different enantioselectivities. Kinetic studies show that these enzymes are more efficient
with 3-thiacyclopentanone than with 3-oxacyclopentanone. X-ray crystal structures of apo- and NADP+-bound selected mutants show that the substrate-binding loop conformational preferences are modified by these mutations. Quantum
mechanical calculations and molecular dynamics (MD) simulations are used to investigate the mechanism of reduction by the
enzyme. We have developed an MD-based method for studying the diastereomeric transition state complexes and rationalize different
enantiomeric ratios. This method, which probes the stability of the catalytic arrangement within the theozyme, shows a correlation
between the relative fractions of catalytically competent poses for the enantiomeric reductions and the experimental enantiomeric
ratio. Some mutations, such as A94F and Y190F, induce conformational changes in the active site that enlarge the small binding
pocket, facilitating accommodation of the larger S atom in this region and enhancing S-selectivity with 3-thiacyclopentanone. In contrast, in the E145S mutant and the final variant evolved for large-scale production
of the intermediate for the antibiotic sulopenem, R-selectivity is promoted by shrinking the small binding pocket, thereby destabilizing the pro-S orientation.
Co-reporter:Jose M. Medina ; Travis C. McMahon ; Gonzalo Jiménez-Osés ; K. N. Houk ;Neil K. Garg
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14706-14709
Publication Date(Web):October 5, 2014
DOI:10.1021/ja508635v
We report the strategic use of cyclohexyne and the more elusive intermediate, cyclopentyne, as a tool for the synthesis of new heterocyclic compounds. Experimental and computational studies of a 3-substituted cyclohexyne are also described. The observed regioselectivities are explained by the distortion/interaction model.
Co-reporter:Xin Hong ; Hatice Başpınar Küçük ; Modhu Sudan Maji ; Yun-Fang Yang ; Magnus Rueping ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13769-13780
Publication Date(Web):September 2, 2014
DOI:10.1021/ja506660c
Brønsted acid catalyzed (3+ + 2) cycloadditions between hydrazones and alkenes provide a general approach to pyrazolidines. The acidity of the Brønsted acid is crucial for the catalytic efficiency: the less acidic phosphoric acids are ineffective, while highly acidic chiral N-triflylphosphoramides are very efficient and can promote highly enantioselective cycloadditions. The mechanism and origins of catalytic efficiencies and selectivities of these reactions have been explored with density functional theory (M06-2X) calculations. Protonation of hydrazones by N-triflylphosphoramide produces hydrazonium–phosphoramide anion complexes. These ion-pair complexes are very reactive in (3+ + 2) cycloadditions with alkenes, producing pyrazolidine products. Alternative 1,3-dipolar (3 + 2) cycloadditions with the analogous azomethine imines are much less favorable due to the endergonic isomerization of hydrazone to azomethine imine. With N-triflylphosphoramide catalyst, only a small distortion of the ion-pair complex is required to achieve its geometry in the (3+ + 2) cycloaddition transition state. In contrast, the weak phosphoric acid does not protonate the hydrazone, and only a hydrogen-bonded complex is formed. Larger distortion energy is required for the hydrogen-bonded complex to achieve the “ion-pair” geometry in the cycloaddition transition state, and a significant barrier is found. On the basis of this mechanism, we have explained the origins of enantioselectivities when a chiral N-triflylphosphoramide catalyst is employed. We also report the experimental studies that extend the substrate scope of alkenes to ethyl vinyl ethers and thioethers.
Co-reporter:Zhongyu Yang ; Gonzalo Jiménez-Osés ; Carlos J. López ; Michael D. Bridges ; K. N. Houk ;Wayne L. Hubbell
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15356-15365
Publication Date(Web):October 7, 2014
DOI:10.1021/ja5083206
Site-directed spin labeling in combination with EPR is a powerful method for providing distances on the nm scale in biological systems. The most popular strategy, double electron–electron resonance (DEER), is carried out at cryogenic temperatures (50–80 K) to increase the short spin–spin relaxation time (T2) upon which the technique relies. A challenge is to measure long-range distances (20–60 Å) in proteins near physiological temperatures. Toward this goal we are investigating an alternative approach based on the distance-dependent enhancement of spin–lattice relaxation rate (T1–1) of a nitroxide spin label by a paramagnetic metal. With a commonly used nitroxide side chain (R1) and Cu2+, it has been found that interspin distances ≤25 Å can be determined in this way (Jun et al. Biochemistry 2006, 45, 11666). Here, the upper limit of the accessible distance is extended to ≈40 Å using spin labels with long T1, a high-affinity 5-residue Cu2+ binding loop inserted into the protein sequence, and pulsed saturation recovery to measure relaxation enhancement. Time-domain Cu2+ electron paramagnetic resonance, quantum mechanical calculations, and molecular dynamics simulations provide information on the structure and geometry of the Cu2+ loop and indicate that the metal ion is well-localized in the protein. An important aspect of these studies is that both Cu2+/nitroxide DEER at cryogenic temperatures and T1 relaxation measurements at room temperature can be carried out on the same sample, allowing both validation of the relaxation method and assessment of the effect of freezing on protein structure.
Co-reporter:Jose M. Medina ; Joel L. Mackey ; Neil K. Garg ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 44) pp:15798-15805
Publication Date(Web):October 10, 2014
DOI:10.1021/ja5099935
The distortion/interaction model has been used to explain and predict reactivity in a variety of reactions where more common explanations, such as steric and electronic factors, do not suffice. This model has also provided new fundamental insight into regioselectivity trends in reactions of unsymmetrical arynes, which in turn has fueled advances in aryne methodology and natural product synthesis. This article describes a systematic experimental and computational study of one particularly important class of arynes, 3-halobenzynes. 3-Halobenzynes are useful synthetic building blocks whose regioselectivities have been explained by several different models over the past few decades. Our efforts show that aryne distortion, rather than steric factors or charge distribution, are responsible for the regioselectivities observed in 3-haloaryne trapping experiments. We also demonstrate the synthetic utility of 3-halobenzynes for the efficient synthesis of functionalized heterocycles, using a tandem aryne-trapping/cross-coupling sequence involving 3-chlorobenzyne.
Co-reporter:Jun Zhu ; Yong Liang ; Lijia Wang ; Zhong-Bo Zheng ; K. N. Houk ;Yong Tang
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:6900-6903
Publication Date(Web):April 25, 2014
DOI:10.1021/ja503117q
Stereocontrol in the synthesis of structurally complex molecules, especially those with all-carbon quaternary stereocenters, remains a challenge. Here, we reported the preparation of a class of tetracyclic cyclopenta-fused spiroindoline skeletons through Cu(II)-catalyzed intramolecular [3 + 2] annulation reactions of donor–acceptor cyclopropanes with indoles. Both cis- and trans-diastereomers of tetracyclic spiroindolines are accessed with high selectivities by altering the remote ester groups of cyclopropanes. The origins of this stereocontrol are identified using DFT calculations: attractive interactions between the ester group and arene favor the generation of the trans isomer, while the formation of the cis isomer is preferred when steric repulsions become predominant.
Co-reporter:Xin Hong ; Matthew C. Stevens ; Peng Liu ; Paul A. Wender ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 49) pp:17273-17283
Publication Date(Web):November 7, 2014
DOI:10.1021/ja5098308
Allenes are important 2π building blocks in organic synthesis and engage as 2-carbon components in many metal-catalyzed reactions. Wender and co-workers discovered that methyl substituents on the terminal allene double bond counterintuitively change the reactivities of allenes in [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions with vinylcyclopropanes (VCPs). More sterically encumbered allenes afford higher cycloadduct yields, and such effects are also observed in other Rh(I)-catalyzed intermolecular cycloadditions. Through density functional theory calculations (B3LYP and M06) and experiment, we explored this enigmatic reactivity and selectivity of allenes in [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions with VCPs. The apparent low reactivity of terminally unsubstituted allenes is associated with a competing allene dimerization that irreversibly sequesters rhodium. With terminally substituted allenes, steric repulsion between the terminal substituents significantly increases the barrier of allene dimerization while the barrier of the (5 + 2) cycloaddition is not affected, and thus the cycloaddition prevails. Computation has also revealed the origin of chemoselectivity in (5 + 2) cycloadditions with allene-ynes. Although simple allene and acetylene have similar reaction barriers, intermolecular (5 + 2) cycloadditions of allene-ynes occur exclusively at the terminal allene double bond. The terminal double bond is more reactive due to the enhanced d−π* backdonation. At the same time, insertion of the internal double bond of an allene-yne has a higher barrier as it would break π conjugation. Substituted alkynes are more difficult to insert compared with acetylene, because of the steric repulsion from the additional substituents. This leads to the greater reactivity of the allene double bond relative to the alkynyl group in allene-ynes.
Co-reporter:M. Taylor Haynes ; II; Peng Liu ; Ryan D. Baxter ; Alex J. Nett ; K. N. Houk ;John Montgomery
Journal of the American Chemical Society 2014 Volume 136(Issue 50) pp:17495-17504
Publication Date(Web):November 17, 2014
DOI:10.1021/ja508909u
The mechanism of nickel(0)-catalyzed reductive coupling of aldehydes and alkynes has been studied. Extensive double-labeling crossover studies have been conducted. While previous studies illustrated that phosphine- and N-heterocyclic carbene-derived catalysts exhibited differing behavior, the origin of these effects has now been evaluated in detail. Many variables, including ligand class, sterics of the ligand and alkyne, temperature, and ring size being formed in intramolecular versions, all influence the extent of crossover observed. A computational evaluation of these effects suggests that dimerization of a key metallacyclic intermediate provides the origin of crossover. Protocols that proceed with crossover are typically less efficient than those without crossover given the thermodynamic stability and low reactivity of the dimeric metallacycles involved in crossover pathways.
Co-reporter:Puneet Kumar ; Ashish Thakur ; Xin Hong ; K. N. Houk ;Janis Louie
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17844-17851
Publication Date(Web):November 21, 2014
DOI:10.1021/ja5105206
A Ni/N-heterocyclic carbene catalyst couples diynes to the C(α)–C(β) double bond of tropone, a type of reaction that is unprecedented for metal-catalyzed cycloadditions with aromatic tropone. Many different diynes were efficiently coupled to afford [5–6–7] fused tricyclic products, while [5–7–6] fused tricyclic compounds were obtained as minor byproducts in a few cases. The reaction has broad substrate scope and tolerates a wide range of functional groups, and excellent regioselectivity is found with unsymmetrical diynes. Theoretical calculations show that the apparent enone cycloaddition occurs through a distinctive 8π insertion of tropone. The initial intramolecular oxidative cyclization of diyne produces the nickelacyclopentadiene intermediate. This intermediate undergoes an 8π insertion of tropone, and subsequent reductive elimination generates the [5–6–7] fused tricyclic product. This initial product undergoes two competing isomerizations, leading to the observed [5–6–7] and [5–7–6] fused tricyclic products.
Co-reporter:Hung V. Pham ; Robert S. Paton ; Audrey G. Ross ; Samuel J. Danishefsky ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 6) pp:2397-2403
Publication Date(Web):January 13, 2014
DOI:10.1021/ja410220w
The intramolecular Diels–Alder reactions of cycloalkenones and terminal dienes occur with high endo stereoselectivity, both thermally and under Lewis-acidic conditions. Through computations, we show that steric repulsion and tether conformation govern the selectivity of the reaction, and incorporation of either BF3 or α-halogenation increases the rate of cycloaddition. With a longer tether, isomerization from a terminal diene to the more stable internal diene results in a more facile cycloaddition.
Co-reporter:Sheng Xie; Steven A. Lopez; Olof Ramström; Mingdi Yan;K. N. Houk
Journal of the American Chemical Society 2014 Volume 137(Issue 8) pp:2958-2966
Publication Date(Web):December 30, 2014
DOI:10.1021/ja511457g
The reactivities of enamines and predistorted (strained) dipolarophiles toward perfluoroaryl azides (PFAAs) were explored experimentally and computationally. Kinetic analyses indicate that PFAAs undergo (3 + 2) cycloadditions with enamines up to 4 orders of magnitude faster than phenyl azide reacts with these dipolarophiles. DFT calculations were used to identify the origin of this rate acceleration. Orbital interactions between the cycloaddends are larger due to the relatively low-lying LUMO of PFAAs. The triazolines resulting from PFAA–enamine cycloadditions rearrange to amidines at room temperature, while (3 + 2) cycloadditions of enamines and phenyl azide yield stable, isolable triazolines. The 1,3-dipolar cycloadditions of norbornene and DIBAC also show increased reactivity toward PFAAs over phenyl azide but are slower than enamine–azide cycloadditions.
Co-reporter:Xin Hong ; Yong Liang ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:2017-2025
Publication Date(Web):January 15, 2014
DOI:10.1021/ja4118413
Many experiments have shown that nickel with monodentate phosphine ligands favors the C(aryl)–O activation over the C(acyl)–O activation for aryl esters. However, Itami and co-workers recently discovered that nickel with bidentate phosphine ligands can selectively activate the C(acyl)–O bond of aryl esters of aromatic carboxylic acids. The chemoselectivity with bidentate phosphine ligands can be switched back to C(aryl)–O activation when aryl pivalates are employed. To understand the mechanisms and origins of this switchable chemoselectivity, density functional theory (DFT) calculations have been conducted. For aryl esters, nickel with bidentate phosphine ligands cleaves C(acyl)–O and C(aryl)–O bonds via three-centered transition states. The C(acyl)–O activation is more favorable due to the lower bond dissociation energy (BDE) of C(acyl)–O bond, which translates into a lower transition-state distortion energy. However, when monodentate phosphine ligands are used, a vacant coordination site on nickel creates an extra Ni–O bond in the five-centered C(aryl)–O cleavage transition state. The additional interaction energy between the catalyst and substrate makes C(aryl)–O activation favorable. In the case of aryl pivalates, nickel with bidentate phosphine ligands still favors the C(acyl)–O activation over the C(aryl)–O activation at the cleavage step. However, the subsequent decarbonylation generates a very unstable tBu-Ni(II) intermediate, and this unfavorable step greatly increases the overall barrier for generating the C(acyl)–O activation products. Instead, the subsequent C–H activation of azoles and C–C coupling in the C(aryl)–O activation pathway are much easier, leading to the observed C(aryl)–O activation products.
Co-reporter:Yu-hong Lam ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 27) pp:9556-9559
Publication Date(Web):June 26, 2014
DOI:10.1021/ja504714m
The origin of selectivity in the α-fluorination of cyclic ketones catalyzed by cinchona alkaloid-derived primary amines is determined with density functional calculations. The chair preference of a seven-membered ring at the fluorine transfer transition state is key in determining the sense and level of enantiofacial selectivity.
Co-reporter:Shan-Shui Meng ; Yong Liang ; Kou-Sen Cao ; Lufeng Zou ; Xing-Bang Lin ; Hui Yang ; K. N. Houk ;Wen-Hua Zheng
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12249-12252
Publication Date(Web):August 15, 2014
DOI:10.1021/ja507332x
A highly enantioselective catalytic protocol for the desymmetrization of a wide variety of 2-substituted and 2,2-disubstituted 1,3-diols is reported. This reaction proceeds through the formation of an “ortho ester” intermediate via oxidation of 1,3-diol benzylidene acetal by dimethyldioxirane (DMDO) and the subsequent proton transfer catalyzed by chiral phosphoric acid (CPA). The mechanism and origins of enantioselectivity of this reaction are identified using DFT calculations. The oxidation by DMDO is rate-determining, and the phosphoric acid significantly accelerates the proton transfer; the attractive interactions between the benzylidene part of the substrate and the 2,4,6-triisopropyl group of CPA are the key to high enantioselectivity.
Co-reporter:Jeffrey S. Cannon, Lufeng Zou, Peng Liu, Yu Lan, Daniel J. O’Leary, K. N. Houk, and Robert H. Grubbs
Journal of the American Chemical Society 2014 Volume 136(Issue 18) pp:6733-6743
Publication Date(Web):April 14, 2014
DOI:10.1021/ja5021958
The mechanism of C–H activation at metathesis-relevant ruthenium(II) benzylidene complexes was studied both experimentally and computationally. Synthesis of a ruthenium dicarboxylate at a low temperature allowed for direct observation of the C–H activation step, independent of the initial anionic ligand-exchange reactions. A first-order reaction supports an intramolecular concerted metalation–deprotonation mechanism with ΔG⧧298K = 22.2 ± 0.1 kcal·mol–1 for the parent N-adamantyl-N′-mesityl complex. An experimentally determined ΔS⧧ = −5.2 ± 2.6 eu supports a highly ordered transition state for carboxylate-assisted C(sp3)–H activation. Experimental results, including measurement of a large primary kinetic isotope effect (kH/kD = 8.1 ± 1.7), agree closely with a computed six-membered carboxylate-assisted C–H activation mechanism where the deprotonating carboxylate adopts a pseudo-apical geometry, displacing the aryl ether chelate. The rate of cyclometalation was found to be influenced by both the electronics of the assisting carboxylate and the ruthenium ligand environment.
Co-reporter:Matthew T. Richers ; Martin Breugst ; Alena Yu. Platonova ; Anja Ullrich ; Arne Dieckmann ; K. N. Houk ;Daniel Seidel
Journal of the American Chemical Society 2014 Volume 136(Issue 16) pp:6123-6135
Publication Date(Web):April 1, 2014
DOI:10.1021/ja501988b
Cyclic secondary amines and 2-hydroxybenzaldehydes or related ketones react to furnish benzo[e][1,3]oxazine structures in generally good yields. This overall redox-neutral amine α-C–H functionalization features a combined reductive N-alkylation/oxidative α-functionalization and is catalyzed by acetic acid. In contrast to previous reports, no external oxidants or metal catalysts are required. Reactions performed under modified conditions lead to an apparent reductive amination and the formation of o-hydroxybenzylamines in a process that involves the oxidation of a second equivalent of amine. A detailed computational study employing density functional theory compares different mechanistic pathways and is used to explain the observed experimental findings. Furthermore, these results also reveal the origin of the catalytic efficiency of acetic acid in these transformations.
Co-reporter:Xiao-Na Wang ; Elizabeth H. Krenske ; Ryne C. Johnston ; K. N. Houk ;Richard P. Hsung
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9802-9805
Publication Date(Web):July 3, 2014
DOI:10.1021/ja502252t
Electrocyclic ring opening of 4,6-fused cyclobutenamides 1 under thermal conditions leads to cis,trans-cyclooctadienones 2-E,E as transient intermediates, en route to 5,5-bicyclic products 3. Theoretical calculations predict that 4,5-fused cyclobutenamides should likewise undergo thermal ring opening, giving cis,trans-cycloheptadienones, but in this case conversion to 5,4-bicyclic products is thermodynamically disfavored, and these cyclobutenamides instead rearrange to vinyl cyclopentenones.
Co-reporter:Yang Cao ; Yong Liang ; Lei Zhang ; Sílvia Osuna ; Andra-Lisa M. Hoyt ; Alejandro L. Briseno ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 30) pp:10743-10751
Publication Date(Web):July 9, 2014
DOI:10.1021/ja505240e
The Diels–Alder (DA) reactions of pentacene (PT), 6,13-bis(2-trimethylsilylethynyl)pentacene (TMS-PT), bistetracene (BT), and 8,17-bis(2-trimethylsilylethynyl)bistetracene (TMS-BT) with the [6,6] double bond of [60]fullerene have been investigated by density functional theory calculations. Reaction barriers and free energies have been obtained to assess the effects of frameworks and substituent groups on the DA reactivity and product stability. Calculations indicate that TMS-BT is about 5 orders of magnitude less reactive than TMS-PT in the reactions with [60]fullerene. This accounts for the observed much higher stability of TIPS-BT than TIPS-PT when mixed with PCBM. Surprisingly, calculations predict that the bulky silylethynyl substituents of TMS-PT and TMS-BT have only a small influence on reaction barriers. However, the silylethynyl substituents significantly destabilize the corresponding products due to steric repulsions in the adducts. This is confirmed by experimental results here. Architectures of the polycyclic aromatic hydrocarbons (PAHs) play a crucial role in determining both the DA barrier and the adduct stability. The reactivities of different sites in various PAHs are related to the loss of aromaticity, which can be predicted using the simple Hückel molecular orbital localization energy calculations.
Co-reporter:Xin Hong, Yong Liang, Allison K. Griffith, Tristan H. Lambert and K. N. Houk
Chemical Science 2014 vol. 5(Issue 2) pp:471-475
Publication Date(Web):04 Nov 2013
DOI:10.1039/C3SC52882K
The mechanism of hydrazine-catalyzed carbonyl-olefin metathesis relying on a novel (3 + 2) strategy is studied by density functional theory (DFT) calculations. The origins of the special reactivity of cyclopropene in this transformation are revealed, and the reactivities of different alkenes in the (3 + 2) cycloadditions and cycloreversions are compared. It is found that the ease of distortion of reactants accelerates cycloadditions, and that the strain release is the controlling factor for cycloreversions.
Co-reporter:Hao Wang and K. N. Houk
Chemical Science 2014 vol. 5(Issue 2) pp:462-470
Publication Date(Web):20 Sep 2013
DOI:10.1039/C3SC52538D
Torsional effects control the π-facial stereoselectivities of a variety of synthetically important organic reactions. This review surveys theoretical calculations that have led to the understanding of the influence of the torsional effects on several types of stereoselective organic reactions, especially electrophilic additions and cycloadditions to alkenes.
Co-reporter:Claire L. Jarvis, Matthew T. Richers, Martin Breugst, K. N. Houk, and Daniel Seidel
Organic Letters 2014 Volume 16(Issue 13) pp:3556-3559
Publication Date(Web):June 13, 2014
DOI:10.1021/ol501509b
Secondary amines react with thiosalicylaldehydes in the presence of catalytic amounts of acetic acid to generate ring-fused N,S-acetals in redox-neutral fashion. A broad range of amines undergo α-sulfenylation, including challenging substrates such morpholine, thiomorpholine, and piperazines. Computational studies employing density functional theory indicate that acetic acid reduces the energy barriers of two separate steps, both of which involve proton transfer.
Co-reporter:Michael E. Jung, Gloria S. Lee, Hung V. Pham, and K. N. Houk
Organic Letters 2014 Volume 16(Issue 9) pp:2382-2385
Publication Date(Web):April 11, 2014
DOI:10.1021/ol500710v
The exomethylenes of 2,6-disubstituted bicyclo[3.3.1]nonan-9-ones 2 are readily isomerized over a palladium catalyst under an atmosphere of hydrogen to predominantly form the isomer 3 with C2 symmetry with very little formation of the analogous product with Cs symmetry. A hydrogen source is essential to effect the rearrangement.
Co-reporter:Xin Hong, Yong Liang, Matthias Brewer, and K. N. Houk
Organic Letters 2014 Volume 16(Issue 16) pp:4260-4263
Publication Date(Web):July 24, 2014
DOI:10.1021/ol501958s
Cationic 1-aza-2-azoniaallenes react intermolecularly with terminal alkenes to give 1,5-substituted (3 + 2)-cycloadducts, but intramolecular reactions lead to either 1,5- or 1,4-substituted (3 + 2)-cycloadducts or (4 + 2)-cycloadducts, depending on the tether length. DFT calculations and distortion/interaction analyses show that the (CH2)3 tether prevents the reacting partners from aligning efficiently to give 1,5-substituted (3 + 2)-cycloadducts, and the 1,4-regioselectivity dominates. With the (CH2)2 tether, the (3 + 2) cycloaddition is disfavored due to the forming four-membered ring in the transition state, and the (4 + 2) cycloaddition prevails.
Co-reporter:Ricardo A. Matute, Miguel A. Garcia-Garibay, and K. N. Houk
Organic Letters 2014 Volume 16(Issue 19) pp:5232-5234
Publication Date(Web):September 19, 2014
DOI:10.1021/ol5024823
The regioselectivities of the di-π-methane rearrangements of unsymmetrically substituted dibenzobarrelenes have been explored with DFT (UM06-2X). Regioselectivity depends on the intramolecular hydrogen bonding and originates from specific stabilization of the triplet biradical intermediates.
Co-reporter:Yong Liang, Xin Hong, Peiyuan Yu, and K. N. Houk
Organic Letters 2014 Volume 16(Issue 21) pp:5702-5705
Publication Date(Web):October 20, 2014
DOI:10.1021/ol502780w
The hexadehydro-Diels–Alder (HDDA) reactions between suitably substituted 1,3-diynes and alkynes produce highly reactive benzynes under thermal conditions without catalysts. DFT calculations and distortion/interaction analyses show that, for the activated substrates, the stepwise diradical pathway is more favorable than the concerted [4 + 2] process. One manifestation of this mechanism is that alkynyl substituents dramatically accelerate HDDA reactions, mainly by decreasing the distortion energy required to achieve the diradical transition state.
Co-reporter:Steven A. Lopez, Melika Pourati, Hans-Joachim Gais, and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 17) pp:8304-8312
Publication Date(Web):July 28, 2014
DOI:10.1021/jo501557z
Cycloadditions of 1,3-dipoles and related species to a cis-oxabicyclo[3.3.0]octenone occur on the more sterically crowded concave face. These cycloadditions were studied experimentally by Gais and co-workers in 1998 (Eur. J. Org. Chem. 1998, 257–273) and have now been studied computationally with density functional theory (DFT). Transition states have been computed for various types of (3 + 2) cycloadditions, including diazomethane 1,3-dipolar cycloadditions, a thermally promoted methylenecyclopropane acetal cycloaddition, and a Pd-catalyzed cycloaddition of methylenecyclopropane to an oxabicyclo[3.3.0]octenone. The concave stereoselectivities arise from alkene predistortion that leads to torsional steering in the transition states.
Co-reporter:Yun-Fang Yang, Lung Wa Chung, Xinhao Zhang, K. N. Houk, and Yun-Dong Wu
The Journal of Organic Chemistry 2014 Volume 79(Issue 18) pp:8856-8864
Publication Date(Web):August 26, 2014
DOI:10.1021/jo501730n
Density functional theory calculations with the M06 functional have been performed on the reactivity, selectivity, and mechanism of hydrosilylations of alkynes, ketones, and nitriles catalyzed by cationic ruthenium complexes [CpRu(L)(MeCN)2]+, with L = PiPr3 or MeCN. The hydrosilylation of alkynes with L = PiPr3 involves an initial silyl migration mechanism to generate the anti-Markovnikov product, in contrast to the Markovnikov product obtained with L = MeCN. The bulky phosphine ligand directs the silyl group to migrate to Cβ of the alkyne. This explains the anti-Markovnikov selectivity of the catalyst with L = PiPr3. By contrast, the silane additions to either ketone or nitrile proceed through an ionic SN2-Si outer-sphere mechanism, in which the substrate attacks the Si center. The PiPr3 ligand facilitates the activation of the Si–H bond to furnish a η2-silane complex, whereas a η1-silane complex is formed for the MeCN ligand. This property of the phosphine ligand enables the catalytic hydrosilylation of ketones and nitriles in addition to that of alkynes.
Co-reporter:Hung V. Pham and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 19) pp:8968-8976
Publication Date(Web):September 12, 2014
DOI:10.1021/jo502041f
Multiconfigurational complete active space methods (CASSCF and CASPT2) have been used to investigate the (4 + 2) cycloadditions of allene with butadiene and with benzene. Both concerted and stepwise radical pathways were examined to determine the mechanism of the Diels–Alder reactions with an allene dienophile. Reaction with butadiene occurs via a single ambimodal transition state that can lead to either the concerted or stepwise trajectories along the potential energy surface, while reaction with benzene involves two separate transition states and favors the concerted mechanism relative to the stepwise mechanism via a diradical intermediate.
Co-reporter:Seoung-ryoung Choi, Martin Breugst, Kendall N. Houk, and C. Dale Poulter
The Journal of Organic Chemistry 2014 Volume 79(Issue 8) pp:3572-3580
Publication Date(Web):March 25, 2014
DOI:10.1021/jo500394u
The biosynthetic pathways to isoprenoid compounds involve transfer of the prenyl moiety in allylic diphosphates to electron-rich (nucleophilic) acceptors. The acceptors can be many types of nucleophiles, while the allylic diphosphates only differ in the number of isoprene units and stereochemistry of the double bonds in the hydrocarbon moieties. Because of the wide range of nucleophilicities of naturally occurring acceptors, the mechanism for prenyltransfer reactions may be dissociative or associative with early to late transition states. We have measured δ-secondary kinetic isotope effects operating through four bonds for substitution reactions with dimethylallyl derivatives bearing deuterated methyl groups at the distal (C3) carbon atom in the double bond under dissociative and associative conditions. Computational studies with density functional theory indicate that the magnitudes of the isotope effects correlate with the extent of bond formation between the allylic moiety and the electron-rich acceptor in the transition state for alkylation and provide insights into the structures of the transition states for associative and dissociative alkylation reactions.
Co-reporter:Steven A. Lopez and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 13) pp:6189-6195
Publication Date(Web):June 2, 2014
DOI:10.1021/jo500919s
Transition structures for the conrotatory electrocyclic ring-opening reactions of N-substituted 2-azetines were computed with the density functional M06-2X/6-31+G(d,p). A wide range of substituents from π acceptors (e.g., CHO, CN) to π donors (NMe2, OMe) was explored. Acceptor substituents delocalize the nitrogen lone pair and stabilize the reactant state of 2-azetines, while donors destabilize the 2-azetine reactant state. The conrotatory ring-opening is torquoselective, and the transition state for the outward rotation of the N-substituent and inward rotation of the nitrogen lone pair is preferred. This transition structure is stabilized by an interaction between the nitrogen lone pair and the vacant π* orbital. The activation free energies are linearly related to the reaction free energies and the Taft σR0 parameter.
Co-reporter:Martin Breugst and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 13) pp:6302-6309
Publication Date(Web):June 13, 2014
DOI:10.1021/jo501227m
The Henry reaction between benzaldehyde and nitromethane catalyzed by a cyclophane-based bisthiourea has been studied with density functional theory [M06-2X-D3/def2-TZVPP/IEFPCM//TPSS-D2/6-31G(d)/IEFPCM]. The results of our study reveal that the transformation involves the reaction of a thiourea–nitronate complex with the uncoordinated aldehyde. On the basis of our calculations, the formation of the major stereoisomer is kinetically preferred. Employing smaller model systems, we show that the observed stereoselectivity arises primarily from differences in hydrogen bonding in diastereomeric transition states.
Co-reporter:Dr. Gonzalo Jiménez-Osés;Dr. Peng Liu;Dr. Ricardo A. Matute ;Dr. Kendall N. Houk
Angewandte Chemie International Edition 2014 Volume 53( Issue 33) pp:8664-8667
Publication Date(Web):
DOI:10.1002/anie.201310237
Abstract
The molecular dynamics of the triplet-state Zimmerman di-π-methane rearrangement of dibenzobarrelene were computed with B3LYP and M06-2X density functionals. All productive quasiclassical trajectories involve sequential formation and cleavage of CC bonds and an intermediate with lifetimes ranging from 13 to 1160 fs. Both dynamically concerted and stepwise trajectories are found. The average lifetime of this intermediate is significantly shorter than predicted by either transition-state theory or the Rice–Ramsperger–Kassel–Marcus model, thus indicating the non-statistical nature of the reaction mechanism.
Co-reporter:Dr. Jun Yang;Dr. Yong Liang;Dr. Jolita &x160;e&x10d;kut&x117;; K. N. Houk; Neal K. Devaraj
Chemistry - A European Journal 2014 Volume 20( Issue 12) pp:3365-3375
Publication Date(Web):
DOI:10.1002/chem.201304225
Abstract
Substituted cyclopropenes have recently attracted attention as stable “mini-tags” that are highly reactive dienophiles with the bioorthogonal tetrazine functional group. Despite this interest, the synthesis of stable cyclopropenes is not trivial and their reactivity patterns are poorly understood. Here, the synthesis and comparison of the reactivity of a series of 1-methyl-3-substituted cyclopropenes with different functional handles is described. The rates at which the various substituted cyclopropenes undergo Diels–Alder cycloadditions with 1,2,4,5-tetrazines were measured. Depending on the substituents, the rates of cycloadditions vary by over two orders of magnitude. The substituents also have a dramatic effect on aqueous stability. An outcome of these studies is the discovery of a novel 3-amidomethyl substituted methylcyclopropene tag that reacts twice as fast as the fastest previously disclosed 1-methyl-3-substituted cyclopropene while retaining excellent aqueous stability. Furthermore, this new cyclopropene is better suited for bioconjugation applications and this is demonstrated through using DNA templated tetrazine ligations. The effect of tetrazine structure on cyclopropene reaction rate was also studied. Surprisingly, 3-amidomethyl substituted methylcyclopropene reacts faster than trans-cyclooctenol with a sterically hindered and extremely stable tert-butyl substituted tetrazine. Density functional theory calculations and the distortion/interaction analysis of activation energies provide insights into the origins of these reactivity differences and a guide to the development of future tetrazine coupling partners. The newly disclosed cyclopropenes have kinetic and stability advantages compared to previously reported dienophiles and will be highly useful for applications in organic synthesis, bioorthogonal reactions, and materials science.
Co-reporter:Sharon R. Neufeldt, Gonzalo Jiménez-Osés, Daniel L. Comins, and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11609-11618
Publication Date(Web):November 5, 2014
DOI:10.1021/jo5022635
The role of twist-boat conformers of cyclohexanones in hydride reductions was explored. The hydride reductions of a cis-2,6-disubstituted N-acylpiperidone, an N-acyltropinone, and tert-butylcyclohexanone by lithium aluminum hydride and by a bulky borohydride reagent were investigated computationally and compared to experiment. Our results indicate that in certain cases, factors such as substrate conformation, nucleophile bulkiness, and remote steric features can affect stereoselectivity in ways that are difficult to predict by the general Felkin–Anh model. In particular, we have calculated that a twist-boat conformation is relevant to the reactivity and facial selectivity of hydride reduction of cis-2,6-disubstituted N-acylpiperidones with a small hydride reagent (LiAlH4) but not with a bulky hydride (lithium triisopropylborohydride).
Co-reporter:Ashay Patel and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11370-11377
Publication Date(Web):October 30, 2014
DOI:10.1021/jo5015728
M06-2X/6-31+G(d,p) computations are reported for the 8π–6π electrocyclization cascades of 1,3,5,7-tetraenes. The rate-determining step for these cascades is typically the second (6π) ring closure. According to experiment and theory, un- and monosubstituted tetraenes readily undergo 8π electrocyclic ring closure to form 1,3,5-cyclooctatrienes; however, the 6π electrocyclizations of these cyclooctatriene intermediates are slow and reversible, and mixtures of monocyclic and bicyclic products are formed. Computations indicate that di- and trisubstituted tetraenes undergo facile but less exergonic 8π electrocyclization due to a steric clash that destabilizes the 1,3,5-cyclooctatriene intermediates. Relief of this steric clash ensures the subsequent 6π ring closures of these intermediates are both kinetically facile and thermodynamically favorable, and only the bicyclic products are observed for the cascade reactions of naturally occurring tri- and tetrasubstituted tetraenes (in agreement with computations). The 6π electrocyclization step of these cascade electrocyclizations is also potentially diastereoselective, and di- and trisubstituted tetraenes often undergo cascade reactions with high diastereoselectivities. The exo mode of ring closure is favored for these 6π electrocyclizations due to a steric interaction that destabilizes the endo transition state. Thus, theory explains both the recalcitrance of the unsubstituted 1,3,5,7-octatetraene and 1-substituted tetraenes toward formation of the bicyclo[4.2.0]octa-2,4-diene products, as well as the ease and the stereoselectivity with which terminal di- and trisubstituted tetraenes are known to react biosynthetically.
Co-reporter:Gabrielle J. Dugas, Yu-hong Lam, K. N. Houk, and Isaac J. Krauss
The Journal of Organic Chemistry 2014 Volume 79(Issue 10) pp:4277-4284
Publication Date(Web):April 23, 2014
DOI:10.1021/jo500599h
Boron tris(trifluoroacetate) is identified as the first effective catalyst for the homoallyl- and homocrotylboration of aldehydes by cyclopropylcarbinylboronates. NMR spectroscopic studies and theoretical calculations of key intermediates and transition states both suggest that a ligand-exchange mechanism, akin to our previously reported PhBCl2-promoted homoallylations, is operative. Our experimental and theoretical results also suggest that the catalytic activity of boron tris(trifluoroacetate) might originate from more facile catalytic turnover of the trifluoroacetate ligands (in agreement with DFT calculations) or from a lower propensity for formation of off-pathway reservoir intermediates (as observed by 1H NMR). This work shows that carboxylates are viable catalytic ligands for homoallyl- and homocrotylations of carbonyl compounds and opens the door to the development of catalytic asymmetric versions of this transformation.
Co-reporter:Xin Hong, Dane Holte, Daniel C. G. Götz, Phil S. Baran, and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:12177-12184
Publication Date(Web):October 17, 2014
DOI:10.1021/jo502219d
Density functional theory (DFT) calculations with B3LYP and M06 functionals elucidated the reactivities of alkynes and Z/E selectivity of cyclodecatriene products in the Ni-catalyzed [4 + 4 + 2] cycloadditions of dienes and alkynes. The Ni-mediated oxidative cyclization of butadienes determines the Z/E selectivity. Only the oxidative cyclization of one s-cis to one s-trans butadiene is facile and exergonic, leading to the observed 1Z,4Z,8E-cyclodecatriene product. The same step with two s-cis or s-trans butadienes is either kinetically or thermodynamically unfavorable, and the 1Z,4E,8E- and 1Z,4Z,8Z-cyclodecatriene isomers are not observed in experiments. In addition, the competition between the desired cooligomerization and [2 + 2 + 2] cycloadditions of alkynes depends on the coordination of alkynes. With either electron-deficient alkynes or alkynes with free hydroxyl groups, the coordination of alkynes is stronger than that of dienes, and alkyne trimerization prevails. With alkyl-substituted alkynes, the generation of alkyne-coordinated nickel complex is much less favorable, and the [4 + 4 + 2] cycloaddition occurs.
Co-reporter:Dr. Gonzalo Jiménez-Osés;Dr. Peng Liu;Dr. Ricardo A. Matute ;Dr. Kendall N. Houk
Angewandte Chemie 2014 Volume 126( Issue 33) pp:8808-8811
Publication Date(Web):
DOI:10.1002/ange.201310237
Abstract
The molecular dynamics of the triplet-state Zimmerman di-π-methane rearrangement of dibenzobarrelene were computed with B3LYP and M06-2X density functionals. All productive quasiclassical trajectories involve sequential formation and cleavage of CC bonds and an intermediate with lifetimes ranging from 13 to 1160 fs. Both dynamically concerted and stepwise trajectories are found. The average lifetime of this intermediate is significantly shorter than predicted by either transition-state theory or the Rice–Ramsperger–Kassel–Marcus model, thus indicating the non-statistical nature of the reaction mechanism.
Co-reporter:Elizabeth H. Krenske and K. N. Houk
Accounts of Chemical Research 2013 Volume 46(Issue 4) pp:979
Publication Date(Web):July 24, 2012
DOI:10.1021/ar3000794
This Account describes how attractive interactions of aromatic rings with other groups can influence and control the stereoselectivity of many reactions. Recent developments in theory have improved the accuracy in the modeling of aromatic interactions. Quantum mechanical modeling can now provide insights into the roles of these interactions at a level of detail not previously accessible, both for ground-state species and for transition states of chemical reactions. In this Account, we show how transition-state modeling led to the discovery of the influence of aryl groups on the stereoselectivities of several types of organic reactions, including asymmetric dihydroxylations, transfer hydrogenations, hetero-Diels–Alder reactions, acyl transfers, and Claisen rearrangements.Our recent studies have also led to a novel mechanistic picture for two classes of (4 + 3) cycloadditions, both of which involve reactions of furans with oxyallyl intermediates. The first class of cycloadditions, developed by Hsung, features neutral oxyallyl intermediates that contain a chiral oxazolidinone auxiliary. Originally, it was thought that these cycloadditions relied on differential steric crowding of the two faces of a planar intermediate. Computations reveal a different picture and show that cycloaddition with furan takes place preferentially through the more crowded transition state: the furan adds on the same side as the Ph substituent of the oxazolidinone. The crowded transition state is stabilized by a CH–π interaction between furan and Ph worth approximately 2 kcal/mol.Attractive interactions with aromatic rings also control the stereoselectivity in a second class of (4+3) cycloadditions involving chiral alkoxy siloxyallyl cations. Alkoxy groups derived from chiral α-methylbenzyl alcohols favor crowded transition states, where a stabilizing CH–π interaction is present between the furan and the Ar group. The cationic cycloadditions are stepwise, while the Hsung cycloadditions are concerted. Our results suggest that this form of CH– π-directed stereocontrol is quite general and likely controls the stereoselectivities of other addition reactions in which one face of a planar intermediate bears a pendant aromatic substituent.
Co-reporter:Yvonne Schmidt ; Jonathan K. Lam ; Hung V. Pham ; K. N. Houk ;Christopher D. Vanderwal
Journal of the American Chemical Society 2013 Volume 135(Issue 19) pp:7339-7348
Publication Date(Web):May 1, 2013
DOI:10.1021/ja4025963
The unusual intramolecular arene/allene cycloaddition described 30 years ago by Himbert permits rapid access to strained polycyclic compounds that offer great potential for the synthesis of complex scaffolds. To more fully understand the mechanism of this cycloaddition reaction, and to guide efforts to extend its scope to new substrates, quantum mechanical computational methods were employed in concert with laboratory experiments. These studies indicated that the cycloadditions likely proceed via concerted processes; a stepwise biradical mechanism was shown to be higher in energy in the cases studied. The original Himbert cycloaddition chemistry is also extended from heterocyclic to carbocyclic systems, with computational guidance used to predict thermodynamically favorable cases. Complex polycyclic scaffolds result from the combination of the cycloaddition and subsequent ring-rearrangement metathesis reactions.
Co-reporter:Gui-Juan Cheng ; Yun-Fang Yang ; Peng Liu ; Ping Chen ; Tian-Yu Sun ; Gang Li ; Xinhao Zhang ; K. N. Houk ; Jin-Quan Yu ;Yun-Dong Wu
Journal of the American Chemical Society 2013 Volume 136(Issue 3) pp:894-897
Publication Date(Web):December 30, 2013
DOI:10.1021/ja411683n
A combined experimental/computational study on the amino acid ligand-assisted Pd-catalyzed C–H bond activation reveals a mechanism in which the amino acid acts as both a dianionic bidentate ligand and a proton acceptor. This new model explains the effects of amino acids on reactivity and selectivity and unveils the dual roles of amino acids: stabilizing monomeric Pd complexes and serving as the internal base for proton abstraction.
Co-reporter:Ashay Patel ; Gregg A. Barcan ; Ohyun Kwon ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 12) pp:4878-4883
Publication Date(Web):February 27, 2013
DOI:10.1021/ja400882y
A novel stereoselective electrocyclization developed for the total synthesis of reserpine has been explored by both experiment and theory. A stereocenter six atoms away from the newly forming chiral center is responsible for the diastereoselectivity of the ring closure. This stereogenic center, lying at the junction of two six-membered rings, defines the conformation of the substrates’ fused ring skeleton that ultimately distinguishes between the two allowed, disrotatory triene geometries at the transition state. The presence of allylic strain in the disfavored transition state results in a torquoselective ring closure (dr up to 15.7:1).
Co-reporter:Elizabeth H. Krenske ; Shuzhong He ; Jian Huang ; Yunfei Du ; K. N. Houk ;Richard P. Hsung
Journal of the American Chemical Society 2013 Volume 135(Issue 14) pp:5242-5245
Publication Date(Web):April 1, 2013
DOI:10.1021/ja312459b
Cycloadditions involving oxyallyl intermediates typically require an electron-rich diene or alkene, but we have discovered the first examples of the cycloaddition of heteroatom-stabilized oxyallyls onto carbonyl groups. An oxazolidinone-substituted oxyallyl undergoes chemoselective (3 + 2) cycloaddition onto the carbonyl group of a tethered dienone in preference to formation of the expected (4 + 3) cycloadduct. Density functional theory calculations indicated that the (3 + 2) cycloaddition takes place through a concerted, highly asynchronous mechanism. The transition state features simultaneous interactions of the oxyallyl LUMO with the carbonyl π and lone-pair orbitals, making this reaction “hemipseudopericyclic” (halfway between purely pericyclic and purely pseudopericyclic). Further (3 + 2) cycloadditions involving tethered phenyl ketones and a tethered enone were predicted theoretically and verified experimentally.
Co-reporter:Hiroshi Miyazaki ; Myles B. Herbert ; Peng Liu ; Xiaofei Dong ; Xiufang Xu ; Benjamin K. Keitz ; Thay Ung ; Garik Mkrtumyan ; K. N. Houk ;Robert H. Grubbs
Journal of the American Chemical Society 2013 Volume 135(Issue 15) pp:5848-5858
Publication Date(Web):April 2, 2013
DOI:10.1021/ja4010267
The Z-selective ethenolysis activity of chelated ruthenium metathesis catalysts was investigated with experiment and theory. A five-membered chelated catalyst that was successfully employed in Z-selective cross metathesis reactions has now been found to be highly active for Z-selective ethenolysis at low ethylene pressures, while tolerating a wide variety of functional groups. This phenomenon also affects its activity in cross metathesis reactions and prohibits crossover reactions of internal olefins via trisubstituted ruthenacyclobutane intermediates. In contrast, a related catalyst containing a six-membered chelated architecture is not active for ethenolysis and seems to react through different pathways more reminiscent of previous generations of ruthenium catalysts. Computational investigations of the effects of substitution on relevant transition states and ruthenacyclobutane intermediates revealed that the differences of activities are attributed to the steric repulsions of the anionic ligand with the chelating groups.
Co-reporter:Martin Breugst ; Albert Eschenmoser ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6658-6668
Publication Date(Web):April 3, 2013
DOI:10.1021/ja402099f
The cofactor riboflavin is biochemically synthesized by a constitutionally intricate process in which two molecules of 6,7-dimethyl-8-ribityllumazine react with each other to form one molecule of the cofactor and one molecule of 5-amino-6-(ribitylamino)uracil. Remarkably, this complex molecular transformation also proceeds non-enzymatically in boiling aqueous solution at pH 7.3. Four different mechanistic pathways for this transformation (nucleophilic catalysis, hydride transfer, hydrogen atom transfer, and a nucleophilic addition mechanism) have now been analyzed by density functional theory [M06-2X/def2-TZVPP/CPCM//M06-2X/6-31+G(d,p)/IEFPCM]. On the basis of these computational results, a so far unpublished nucleophilic addition mechanism is the lowest energy pathway yielding riboflavin. The previously proposed mechanism involving nucleophilic catalysis is higher in energy but is still a viable alternative for an enzyme-catalyzed process assisted by suitably positioned catalytic groups. Pathways involving the transfer of a hydride ion or of a hydrogen atom are predicted to proceed through higher energy transition states and intermediates.
Co-reporter:Gonzalo Jiménez-Osés ; Anthony J. Brockway ; Jared T. Shaw ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6633-6642
Publication Date(Web):March 26, 2013
DOI:10.1021/ja4015937
The mechanism of direct displacement of alkoxy groups in vinylogous and aromatic esters by Grignard reagents, a reaction that is not observed with expectedly better tosyloxy leaving groups, is elucidated computationally. The mechanism of this reaction has been determined to proceed through the inner-sphere attack of nucleophilic alkyl groups from magnesium to the reacting carbons via a metalaoxetane transition state. The formation of a strong magnesium chelate with the reacting alkoxy and carbonyl groups dictates the observed reactivity and selectivity. The influence of ester, ketone, and aldehyde substituents was investigated. In some cases, the calculations predicted the formation of products different than those previously reported; these predictions were then verified experimentally. The importance of studying the actual system, and not simplified models as computational systems, is demonstrated.
Co-reporter:Xin Hong ; Barry M. Trost ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6588-6600
Publication Date(Web):April 9, 2013
DOI:10.1021/ja4012657
The mechanism, solvent effects, and origins of selectivities in Ru(II)-catalyzed intramolecular (5+2) cycloaddition and ene reaction of vinylcyclopropanes (VCPs) and alkynes have been studied using density functional theory. B3LYP/6-31G(d)/LANL2DZ optimized structures were further evaluated with the M06 functional, 6-311+G(2d,p) and LANL2DZ basis sets, and the SMD solvent model. The favored mechanism involves an initial ene-yne oxidative cyclization to form a ruthenacyclopentene intermediate. This mechanism is different from that found earlier with rhodium catalysts. The subsequent β-hydride elimination and cyclopropane cleavage are competitive, determining the experimental selectivity. In trans-VCP, the cyclopropane cleavage is intrinsically favored and leads to the (5+2) cycloaddition product. Although the same intrinsic preferences occur with the cis-VCP, an unfavorable rotation is required in order to generate the cis-double bond in seven-membered ring product, which reverses the selectivity. Acetone solvent is found to facilitate the acetonitrile dissociation from the precatalyst, destabilizing the resting state of the catalyst and leading to a lower overall reaction barrier. In addition, the origins of diastereoselectivities when the allylic hydroxyl group is trans to the bridgehead hydrogen are found to be the electrostatic interactions. In the pathway that generates the favored diastereomer, the oxygen lone pairs from the substituent are closer to the cationic catalyst center and provide stabilizing electrostatic interactions. Similar pathways also determine the regioselectivities, that is, whether the more or less substituted C–C bond of cyclopropane is cleaved. In the trans-1,2-disubstitued cyclopropane substrate, the substituent from the cyclopropane is away from the reaction center in both pathways, and low regioselectivity is found. In contrast, the cleavage of the more substituted C–C bond of the cis-1,2-disubstituted cyclopropane has steric repulsions from the substituent, and thus higher regioselectivity is found.
Co-reporter:Arne Dieckmann ; Martin Breugst ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 8) pp:3237-3242
Publication Date(Web):January 27, 2013
DOI:10.1021/ja312043g
The Diels–Alder reactions of cyclic linear and cross-conjugated trienamines with oxindoles have been studied with density functional theory [M06-2X/def2-TZVPP/IEFPCM//B97D/6-31+G(d,p)/IEFPCM]. These reactions are found to proceed in a stepwise fashion. Computations revealed that these transformations involve complex mechanisms including zwitterionic intermediates and several unstable alternate cycloadducts arising from (2 + 2) cycloadditions and hetero-Diels–Alder reactions. The observed regio- and stereochemistry can be rationalized by a combination of kinetic and thermodynamic control.
Co-reporter:Yun-Fang Yang ; Gui-Juan Cheng ; Peng Liu ; Dasheng Leow ; Tian-Yu Sun ; Ping Chen ; Xinhao Zhang ; Jin-Quan Yu ; Yun-Dong Wu ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:344-355
Publication Date(Web):December 8, 2013
DOI:10.1021/ja410485g
Density functional theory investigations have elucidated the mechanism and origins of meta-regioselectivity of Pd(II)-catalyzed C–H olefinations of toluene derivatives that employ a nitrile-containing template. The reaction proceeds through four major steps: C–H activation, alkene insertion, β-hydride elimination, and reductive elimination. The C–H activation step, which proceeds via a concerted metalation–deprotonation (CMD) pathway, is found to be the rate- and regioselectivity-determining step. For the crucial C–H activation, four possible active catalytic species—monomeric Pd(OAc)2, dimeric Pd2(OAc)4, heterodimeric PdAg(OAc)3, and trimeric Pd3(OAc)6—have been investigated. The computations indicated that the C–H activation with the nitrile-containing template occurs via a Pd–Ag heterodimeric transition state. The nitrile directing group coordinates with Ag while the Pd is placed adjacent to the meta-C–H bond in the transition state, leading to the observed high meta-selectivity. The Pd2(OAc)4 dimeric mechanism also leads to the meta-C–H activation product but with higher activation energies than the Pd–Ag heterodimeric mechanism. The Pd monomeric and trimeric mechanisms require much higher activation free energies and are predicted to give ortho products. Structural and distortion energy analysis of the transition states revealed significant effects of distortions of the template on mechanism and regioselectivity, which provided hints for further developments of new templates.
Co-reporter:Peter K. Dornan ; Kevin G. M. Kou ; K. N. Houk ;Vy M. Dong
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:291-298
Publication Date(Web):December 18, 2013
DOI:10.1021/ja409824b
A dynamic kinetic resolution (DKR) of allylic sulfoxides has been demonstrated by combining the Mislow [2,3]-sigmatropic rearrangement with catalytic asymmetric hydrogenation. The efficiency of our DKR was optimized by using low pressures of hydrogen gas to decrease the rate of hydrogenation relative to the rate of sigmatropic rearrangement. Kinetic studies reveal that the rhodium complex acts as a dual-role catalyst and accelerates the substrate racemization while catalyzing olefin hydrogenation. Scrambling experiments and theoretical modeling support a novel mode of sulfoxide racemization which occurs via a rhodium π-allyl intermediate in polar solvents. In nonpolar solvents, however, the substrate racemization is primarily uncatalyzed. Computational studies suggest that the sulfoxide binds to rhodium via O-coordination throughout the catalytic cycle for hydrogenation.
Co-reporter:Xiufang Xu ; Peng Liu ; Xing-zhong Shu ; Weiping Tang ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9271-9274
Publication Date(Web):May 31, 2013
DOI:10.1021/ja4036785
The mechanism of Rh-catalyzed (5+2) cycloadditions of 3-acyloxy-1,4-enyne (ACE) and alkynes is investigated using density functional theory calculations. The catalytic cycle involves 1,2-acyloxy migration, alkyne insertion, and reductive elimination to form the cycloheptatriene product. In contrast to the (5+2) cycloadditions with vinylcyclopropanes (VCPs), in which alkyne inserts into a rhodium–allyl bond, alkyne insertion into a Rh–C(sp2) bond is preferred. The 1,2-acyloxy migration is found to be the rate-determining step of the catalytic cycle. The electron-rich p-dimethylaminobenzoate substrate promotes 1,2-acyloxy migration and significantly increases the reactivity. In the regioselectivity-determining alkyne insertion step, the alkyne substituent prefers to be distal to the forming C–C bond and thus distal to the OAc group in the product.
Co-reporter:Shudan Bian ; Amy M. Scott ; Yang Cao ; Yong Liang ; Sílvia Osuna ; K. N. Houk ;Adam B. Braunschweig
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9240-9243
Publication Date(Web):June 11, 2013
DOI:10.1021/ja4042077
Cyclopentadienes (CPs) with Raman and electrochemically active tags were patterned covalently onto graphene surfaces using force-accelerated Diels–Alder (DA) reactions that were induced by an array of elastomeric tips mounted onto the piezoelectric actuators of an atomic force microscope. These force-accelerated cycloadditions are a feasible route to locally alter the chemical composition of graphene defects and edge sites under ambient atmosphere and temperature over large areas (∼1 cm2).
Co-reporter:Jonathan K. Lam ; Hung V. Pham ; K. N. Houk ;Christopher D. Vanderwal
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17585-17594
Publication Date(Web):October 10, 2013
DOI:10.1021/ja409618p
Unusual observations in the ring-rearrangement metathesis of Himbert arene/allene cycloadducts to form fused polycylic lactams led to a more in-depth experimental study that yielded conflicting results. Differences in reactivity within related systems and unexpected changes in diastereoselectivity among other similar substrates were not readily explained on the basis of the experimental results. Computational investigations demonstrated substrate-dependent changes in reaction pathways (ring-opening metathesis/ring-closing metathesis [ROM/RCM] cascade vs ring-closing metathesis/ring-opening metathesis [RCM/ROM] cascade). Furthermore, some reactions were judged to be under thermodynamic control and others under kinetic control. The greater understanding of the most likely reaction pathways and their energetics provides a reasonable explanation for the previously irreconcilable results.
Co-reporter:Marie S. T. Morin ; Daniel J. St-Cyr ; Bruce A. Arndtsen ; Elizabeth H. Krenske ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17349-17358
Publication Date(Web):October 17, 2013
DOI:10.1021/ja406833q
1,3-Dipolar cycloadditions of mesoionic 1,3-dipoles (Münchnones, imino-Münchnones, and phospha-Münchnones) with alkynes offer versatile, modular synthetic routes to pyrroles. Reactivity and regioselectivity differ markedly for different members of this series, and we report here the first general rationale for differences in reactivity by means of a systematic investigation of 1,3-dipolar cycloadditions involving electron-poor and electron-rich alkynes. Competition kinetic measurements indicate that Münchnones and phospha-Münchnones are nucleophilic 1,3-dipoles that react most rapidly with electron-poor alkynes. However, the regioselectivities of cycloadditions are found to undergo an inversion as a function of alkyne ionization potential. The exact point at which this occurs is different for the two dipoles, allowing rational control of the pyrrole formed. The origins of these reactivities and regioselectivities are examined computationally. Frontier molecular orbital predictions are found not to be accurate for these reactions, but transition state calculations give correct predictions of reactivity and selectivity, the origins of which can be analyzed using the distortion/interaction model of reactivity. Cycloadditions with electron-poor alkynes are shown to favor the regioisomer that has either the most favorable TS interaction energy (Münchnones or imino-Münchnones) or the smallest TS distortion energy (phospha-Münchnones). Cycloadditions with more electron-rich aryl-substituted alkynes, on the other hand, generally favor the regioisomer that has the smaller TS distortion energy. These insights delineate the synthetically important distinctions between Münchnones and phospha-Münchnones: phospha-Münchnones undergo highly regioselective cycloadditions with electron-poor alkynes that do not react selectively with Münchnones, and the reverse is true for cycloadditions of Münchnones with electron-rich alkynes.
Co-reporter:David N. Kamber ; Lidia A. Nazarova ; Yong Liang ; Steven A. Lopez ; David M. Patterson ; Hui-Wen Shih ; K. N. Houk ;Jennifer A. Prescher
Journal of the American Chemical Society 2013 Volume 135(Issue 37) pp:13680-13683
Publication Date(Web):September 3, 2013
DOI:10.1021/ja407737d
Bioorthogonal chemistries have provided tremendous insight into biomolecule structure and function. However, many popular bioorthogonal transformations are incompatible with one another, limiting their utility for studies of multiple biomolecules in tandem. We identified two reactions that can be used concurrently to tag biomolecules in complex environments: the inverse electron-demand Diels–Alder reaction of tetrazines with 1,3-disubstituted cyclopropenes, and the 1,3-dipolar cycloaddition of nitrile imines with 3,3-disubstituted cyclopropenes. Remarkably, the cyclopropenes used in these transformations differ by the placement of a single methyl group. Such orthogonally reactive scaffolds will bolster efforts to monitor multicomponent processes in cells and organisms.
Co-reporter:Jeffrey C. Holder ; Lufeng Zou ; Alexander N. Marziale ; Peng Liu ; Yu Lan ; Michele Gatti ; Kotaro Kikushima ; K. N. Houk ;Brian M. Stoltz
Journal of the American Chemical Society 2013 Volume 135(Issue 40) pp:14996-15007
Publication Date(Web):September 12, 2013
DOI:10.1021/ja401713g
Enantioselective conjugate additions of arylboronic acids to β-substituted cyclic enones have been previously reported from our laboratories. Air- and moisture-tolerant conditions were achieved with a catalyst derived in situ from palladium(II) trifluoroacetate and the chiral ligand (S)-t-BuPyOx. We now report a combined experimental and computational investigation on the mechanism, the nature of the active catalyst, the origins of the enantioselectivity, and the stereoelectronic effects of the ligand and the substrates of this transformation. Enantioselectivity is controlled primarily by steric repulsions between the t-Bu group of the chiral ligand and the α-methylene hydrogens of the enone substrate in the enantiodetermining carbopalladation step. Computations indicate that the reaction occurs via formation of a cationic arylpalladium(II) species, and subsequent carbopalladation of the enone olefin forms the key carbon–carbon bond. Studies of nonlinear effects and stoichiometric and catalytic reactions of isolated (PyOx)Pd(Ph)I complexes show that a monomeric arylpalladium–ligand complex is the active species in the selectivity-determining step. The addition of water and ammonium hexafluorophosphate synergistically increases the rate of the reaction, corroborating the hypothesis that a cationic palladium species is involved in the reaction pathway. These additives also allow the reaction to be performed at 40 °C and facilitate an expanded substrate scope.
Co-reporter:Elizabeth H. Krenske ; Ashay Patel ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17638-17642
Publication Date(Web):November 6, 2013
DOI:10.1021/ja409928z
Biosynthetic 1,3-dipolar cycloadditions are rare. No enzymes have yet been identified whose function is to catalyze this class of reactions. Recently, however, a 1,3-dipolar cycloaddition was proposed as a key step in the biosynthesis of two Lycopodium alkaloids, lycojaponicumins A and B. The lycojaponicumins’ fused bicyclic tetrahydroisoxazole ring system was proposed to originate from a transannular 1,3-dipolar cycloaddition between a nitrone and an enone in a nine-membered macrocycle. We have used quantum mechanical calculations to predict whether this cycloaddition could constitute a feasible step in a biosynthetic pathway. Our calculations define a general computational approach for analyzing whether a putative biosynthetic reaction is likely to be enzyme-catalyzed. The quantum mechanically predicted rate of the uncatalyzed reaction in water is compared with the rate enhancement theoretically achievable when the reaction is catalyzed by a theozyme (theoretical enzyme). Density functional theory calculations (M06-2X) predict that the uncatalyzed transannular 1,3-dipolar cycloaddition of the putative lycojaponicumin precursor in water is moderately facile (ΔG⧧ = 21.5 kcal/mol, k = 10–3 s–1) and that an enzyme could accelerate the cycloaddition by placing hydrogen bond donors around the enone while maintaining an otherwise nonpolar active site. The theoretical enzyme-catalyzed process has ΔG⧧ ≈ 17 kcal/mol, corresponding to a 2000-fold rate enhancement, and the predicted kcat (2 s–1) is similar to those of known enzymes involved in secondary metabolic pathways. Thus, theory predicts that the proposed transannular 1,3-dipolar cycloaddition is a plausible step in a biosynthetic pathway leading to the lycojaponicumins and suggests that dipolar cycloadditions can be accelerated by enzyme catalysis.
Co-reporter:A. Dieckmann and K. N. Houk
Chemical Science 2013 vol. 4(Issue 9) pp:3591-3600
Publication Date(Web):27 Jun 2013
DOI:10.1039/C3SC51192H
The energetics of complex formation in a number of artificial self-replicating systems has been studied by density functional theory calculations. Complex stabilities were dissected into stabilizing contributions from hydrogen bonding, dispersion, electrostatics, and destabilizing distortions of monomers from their optimum geometries. Strong cooperative effects were identified in all systems, and dispersion and electrostatics contribute more to the overall energies of complex formation than hydrogen bonding in some systems.
Co-reporter:Christopher J. Rosenker, Elizabeth H. Krenske, K. N. Houk, and Peter Wipf
Organic Letters 2013 Volume 15(Issue 5) pp:1076-1079
Publication Date(Web):February 13, 2013
DOI:10.1021/ol400094k
The energetics of thiol addition and elimination reactions to bicyclic enones derived from an indole core structure were explored using 1H NMR and density functional theory (DFT) calculations. The agreement between experiment and theory is excellent, and the combined results reveal that even minor changes in the conformation of the enone, substituents on the scaffold, and the use of different bases have a signficant influence on product distribution. A potential application of these principles is in the rational design of new reversible covalent enzyme inhibitors.
Co-reporter:Sílvia Osuna;Seonah Kim;Guillaume Bollot
European Journal of Organic Chemistry 2013 Volume 2013( Issue 14) pp:2823-2831
Publication Date(Web):
DOI:10.1002/ejoc.201201738
Abstract
LynF, an enzyme from the TruF family, O-prenylates tyrosines in proteins; subsequent Claisen rearrangements give C-prenylated tyrosine products. These reactions in tyrosines and model phenolic systems have been explored with DFT and SCS-MP2 calculations. Various ab initio benchmarks have been used (CBS-QB3, MP2, SCS-MP2) to examine the accuracy of commonly used density functionals, such as B3LYP and M06-2X. Solvent effects from water were considered by using implicit and explicit models. Studies of the ortho-C-prenylation and Claisen rearrangement of tyrosine, and the Claisen rearrangement of α,α-dimethylallyl (prenyl) coumaryl ether establish the energetics of these reactions both in the gas phase and in aqueous solution.
Co-reporter:Lufeng Zou, Robert S. Paton, Albert Eschenmoser, Timothy R. Newhouse, Phil S. Baran, and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 8) pp:4037-4048
Publication Date(Web):March 5, 2013
DOI:10.1021/jo400350v
The site selectivities and stereoselectivities of C–H oxidations of substituted cyclohexanes and trans-decalins by dimethyldioxirane (DMDO) were investigated computationally with quantum mechanical density functional theory (DFT). The multiconfiguration CASPT2 method was employed on model systems to establish the preferred mechanism and transition state geometry. The reaction pathway involving a rebound step is established to account for the retention of stereochemistry. The oxidation of sclareolide with dioxirane reagents is reported, including the oxidation by the in situ generated tBu-TFDO, a new dioxirane that better discriminates between C–H bonds on the basis of steric effects. The release of 1,3-diaxial strain in the transition state contributes to the site selectivity and enhanced equatorial C–H bond reactivity for tertiary C–H bonds, a result of the lowering of distortion energy. In addition to this strain release factor, steric and inductive effects contribute to the rates of C–H oxidation by dioxiranes.
Co-reporter:Dr. Sílvia Osuna;Dr. Alpay Dermenci; Scott J. Miller; K. N. Houk
Chemistry - A European Journal 2013 Volume 19( Issue 42) pp:14245-14253
Publication Date(Web):
DOI:10.1002/chem.201300745
Abstract
The stereoselective Rauhut–Currier (RC) reaction catalyzed by a cysteine derivative has been explored computationally with density functional theory (M06-2X). Both methanethiol and a chiral cysteine derivative were studied as nucleophiles. The complete reaction pathway involves rate-determining elimination of the thiol catalyst from the Michael addition product. The stereoselective Rauhut–Currier reaction, catalyzed by a cysteine derivative as a nucleophile, has also been studied in detail. This reaction was experimentally found to be extremely sensitive to the reaction conditions, such as the number of water equivalents and the effect of potassium counterion. The E1cB process for catalyst elimination has been explored computationally for the eight possible stereoisomers. The effect of explicit water solvation and the presence of counterion (either K+ or Na+) has been studied for the lowest energy enantiomer pair (1S, 2R, 3S)/(1R, 2S, 3R).
Co-reporter:Hao Wang;Fang Liu;Dr. Roger C. Helgeson ;Dr. Kendall N. Houk
Angewandte Chemie International Edition 2013 Volume 52( Issue 2) pp:655-659
Publication Date(Web):
DOI:10.1002/anie.201205376
Co-reporter:Steven A. Lopez, Morton E. Munk, and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 4) pp:1576-1582
Publication Date(Web):January 23, 2013
DOI:10.1021/jo302695n
The transition structures for the 1,3-dipolar cycloadditions of phenyl azide to enamines derived from acetophenone or phenylacetaldehyde and piperidine, morpholine, or pyrrolidine were located using quantum mechanical methods. These cycloadditions were studied experimentally in 1975 by Meilahn, Cox, and Munk (J. Org. Chem.1975, 40, 819–823). Calculations were carried out with M06-2X/6-311+G(d,p), SCS-MP2/6-311+G(d,p)//M06-2X/6-311+G(d,p), and B97D/6-311+G(d,p) methods with the IEF-PCM solvation model for chloroform and ethanol. The distortion/interaction model was utilized to understand mechanisms, reactivities, and selectivities.
Co-reporter:Hao Wang, Pankaj Jain, Jon C. Antilla, and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 3) pp:1208-1215
Publication Date(Web):January 8, 2013
DOI:10.1021/jo302787m
The chiral BINOL-phosphoric acid catalyzed allylboration and propargylation reactions are studied with density functional theory (B3LYP and B3LYP-D3). Two different models were recently proposed for these reactions by Goodman and our group, respectively. In Goodman’s model for allylborations, the catalyst interacts with the boronate pseudoaxial oxygen. By contrast, our model for propargylations predicts that the catalyst interacts with the boronate pseudoequatorial oxygen. In both models, the phosphoric acid stabilizes the transition state by forming a strong hydrogen bond with the oxygen of the boronate and is oriented by a formyl hydrogen bond (Goodman model) and by other electrostatic attractions in our model. Both of these models have now been reinvestigated for both allylborations and propargylations. For the most effective catalyst for these reactions, the lowest energy transition state corresponds to Goodman’s axial model, while the best transition state leading to the minor enantiomer involves the equatorial model. The high enantioselectivity observed with only the bulkiest catalyst arises from the steric interactions between the substrates and the bulky groups on the catalyst, and the resulting necessity for distortion of the catalyst in the disfavored transition state.
Co-reporter:Dr. Gert Kiss;Dr. Nihan Çelebi-Ölçüm;Dr. Rocco Moretti;Dr. David Baker;Dr.Dr. K. N. Houk
Angewandte Chemie International Edition 2013 Volume 52( Issue 22) pp:5700-5725
Publication Date(Web):
DOI:10.1002/anie.201204077
Abstract
Recent developments in computational chemistry and biology have come together in the “inside-out” approach to enzyme engineering. Proteins have been designed to catalyze reactions not previously accelerated in nature. Some of these proteins fold and act as catalysts, but the success rate is still low. The achievements and limitations of the current technology are highlighted and contrasted to other protein engineering techniques. On its own, computational “inside-out” design can lead to the production of catalytically active and selective proteins, but their kinetic performances fall short of natural enzymes. When combined with directed evolution, molecular dynamics simulations, and crowd-sourced structure-prediction approaches, however, computational designs can be significantly improved in terms of binding, turnover, and thermal stability.
Co-reporter:Hao Wang;Fang Liu;Dr. Roger C. Helgeson ;Dr. Kendall N. Houk
Angewandte Chemie 2013 Volume 125( Issue 2) pp:683-687
Publication Date(Web):
DOI:10.1002/ange.201205376
Co-reporter:Dr. Gert Kiss;Dr. Nihan Çelebi-Ölçüm;Dr. Rocco Moretti;Dr. David Baker;Dr.Dr. K. N. Houk
Angewandte Chemie 2013 Volume 125( Issue 22) pp:5810-5836
Publication Date(Web):
DOI:10.1002/ange.201204077
Abstract
Der “Inside-Out”-Ansatz des computerbasierten Enzymdesigns vereint die neuesten Entwicklungen im Bereich der Computerchemie und -biologie. In diesem Rahmen ist es möglich geworden, Proteine zur Katalyse von Reaktionen herzustellen, für die es in der Natur kein Pendant gibt. Die Erfolgsrate ist gegenwärtig gering, sodass nur ein Bruchteil dieser Konzeptproteine zum Schluss wie geplant funktioniert. Errungenschaften, aber auch Beschränkungen der gegenwärtigen Technologie werden hier behandelt und mit anderen Methoden verglichen. Auf sich alleine gestellt, ermöglicht der “Inside-Out”-Ansatz die Produktion von Proteinen mit katalytischer Aktivität und Selektivität – wenn auch mit bescheidenen kinetische Eigenschaften im Vergleich zu natürlich vorkommenden Enzymen. Gerichtete Evolution, Proteindynamiksimulationen und Crowd-Sourcing können jedoch zu erheblich verbesserten Varianten solcher “Inside-Out”-Proteine führen.
Co-reporter:Natalie C. James, Joann M. Um, Anne B. Padias, H. K. Hall Jr., and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 13) pp:6582-6592
Publication Date(Web):June 11, 2013
DOI:10.1021/jo400900x
The energetics of the Diels–Alder cycloaddition reactions of several 1,3-dienes with acrylonitrile, and the energetics of formation of diradicals, were investigated with density functional theory (B3LYP and M06–2X) and compared to experimental data (Hall et al., J. Org. Chem.1993, 58, 7049–7058). For the reaction of 2,3-dimethyl-1,3-butadiene with acrylonitrile, the concerted reaction is favored over the diradical pathway by 2.5 kcal/mol using B3LYP/6-31G(d); experimentally, this reaction gives both cycloadduct and copolymer. The concerted cycloaddition of cyclopentadiene with acrylonitrile is preferred computationally over the stepwise pathway by 5.9 kcal/mol; experimentally, only the Diels–Alder adduct is formed. For the reactions of (E)-1,3-pentadiene and acrylonitrile, both cycloaddition and copolymerization were observed experimentally; these trends were mimicked by the computational results, which showed only a 1.2 kcal/mol preference for the concerted pathway. For the reactions of (Z)-1,3-pentadiene and acrylonitrile, the stepwise pathway is preferred by 3.9 kcal/mol, in agreement with previous experimental findings that only polymerization occurs. M06–2X is known to give more accurate activation and reaction energetics (Pieniazek, et al., Angew. Chem. Int.2008, 47, 7746–7749), but the energies of diradicals are too high.
Co-reporter:Arne Dieckmann, Matthew T. Richers, Alena Yu. Platonova, Chen Zhang, Daniel Seidel, and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 8) pp:4132-4144
Publication Date(Web):March 21, 2013
DOI:10.1021/jo400483h
We have performed a combined computational and experimental study to elucidate the mechanism of a metal-free α-amination of secondary amines. Calculations predicted azaquinone methides and azomethine ylides as the reactive intermediates and showed that iminium ions are unlikely to participate in these transformations. These results were confirmed by experimental deuterium-labeling studies and the successful trapping of the postulated azomethine ylide and azaquinone methide intermediates. In addition, computed barrier heights for the rate-limiting step correlate qualitatively with experimental findings.
Co-reporter:Yunfei Du, Elizabeth H. Krenske, Jennifer E. Antoline, Andrew G. Lohse, K. N. Houk, and Richard P. Hsung
The Journal of Organic Chemistry 2013 Volume 78(Issue 5) pp:1753-1759
Publication Date(Web):July 31, 2012
DOI:10.1021/jo3011792
The regioselectivities and stereoselectivities of ZnCl2-catalyzed (4 + 3) cycloadditions between chiral oxazolidinone-substituted oxyallyls and unsymmetrical disubstituted furans have been determined. The substitution pattern on the furan is found to provide a valuable tool for controlling the stereochemistry (endo-I or endo-II) of the 7-membered cycloadduct. While cycloadditions with monosubstituted furans usually favor endo-I products, from addition of the furan to the more crowded face of the oxyallyl, cycloadditions with 2,3- and 2,5-disubstituted furans instead favor the endo-II stereochemistry. Density functional theory calculations are performed to account for the selectivities. For monosubstituted furans, the crowded transition state leading to the endo-I cycloadduct is stabilized by an edge-to-face interaction between the furan and the oxazolidinone 4-Ph group, but this stabilization is overcome by steric clashing if the furan bears a 2-CO2R group or is 2,3-disubstituted.
Co-reporter:Steven A. Lopez and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 5) pp:1778-1783
Publication Date(Web):July 5, 2012
DOI:10.1021/jo301267b
The transition structures for 1,3-dipolar cycloadditions of phenyl azide to norbornene derivatives were located with quantum mechanical methods. Calculations were carried out with M06-2X/6-311G(d,p) and SCS-MP2/6-311G(d,p)//M06-2X/6-311G(d,p) methods. The calculated activation barriers strongly correlate with transition state distortion energies (ΔEd⧧) but not with the reaction energies. Strain-promoted reactions are accelerated because it is easy to distort the strained reactants to a pyramidalized transition state geometry; a correlation of cycloaddition rates with substrate distortion was found for the bicyclic and tricyclic alkenes studied here. The stereoselectivities of reactions of norbornene derivatives are controlled primarily by torsional effects that also influence alkene pyramidalization. These reactions are distortion-accelerated.
Co-reporter:Martin Breugst, René Grée, and K. N. Houk
The Journal of Organic Chemistry 2013 Volume 78(Issue 19) pp:9892-9897
Publication Date(Web):August 28, 2013
DOI:10.1021/jo401628e
Brønsted and Lewis acids can catalyze the Prins cyclization, an efficient method for the synthesis of tetrahydropyrans from homoallylic alcohols and carbonyl compounds. Synergistic effects between weak Brønsted and Lewis acids in these reactions have been analyzed by density functional theory [M06-L/def2-QZVP/IEFPCM(CH2Cl2)//M06-L/6-311+G(2df,2p)]. In order to characterize the reactivities of the employed Lewis acids, methyl anion and hydroxide affinities were determined. On the basis of our calculations, we found that the coordination of Lewis acids to carboxylic and sulfonic acids results in a significant increase in the Brønsted acidities of the latter.
Co-reporter:Myles B. Herbert ; Yu Lan ; Benjamin K. Keitz ; Peng Liu ; Koji Endo ; Michael W. Day ; K. N. Houk ;Robert H. Grubbs
Journal of the American Chemical Society 2012 Volume 134(Issue 18) pp:7861-7866
Publication Date(Web):April 13, 2012
DOI:10.1021/ja301108m
The decomposition of a Z-selective ruthenium metathesis catalyst and structurally similar analogues has been investigated utilizing X-ray crystallography and density functional theory. Isolated X-ray crystal structures suggest that recently reported C–H activated catalysts undergo decomposition via insertion of the alkylidene moiety into the chelating ruthenium–carbon bond followed by hydride elimination, which is supported by theoretical calculations. The resulting ruthenium hydride intermediates have been implicated in previously observed olefin migration, and thus lead to unwanted byproducts in cross metathesis reactions. Preventing these decomposition modes will be essential in the design of more active and selective Z-selective catalysts.
Co-reporter:Yu-hong Lam ; K. N. Houk ; Ulf Scheffler ;Rainer Mahrwald
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6286-6295
Publication Date(Web):March 29, 2012
DOI:10.1021/ja2118392
Quantum mechanical calculations reveal the origin of diastereo- and enantioselectivities of aldol reactions between aldehydes catalyzed by histidine, and differences between related reactions catalyzed by proline. A stereochemical model that explains both the sense and the high levels of the experimentally observed stereoselectivity is proposed. The computations suggest that both the imidazolium and the carboxylic acid functionalities of histidine are viable hydrogen-bond donors that can stabilize the cyclic aldolization transition state. The stereoselectivity is proposed to arise from minimization of gauche interactions around the forming C–C bond.
Co-reporter:Elizabeth H. Krenske ; Edwin C. Davison ; Ian T. Forbes ; Jacqueline A. Warner ; Adrian L. Smith ; Andrew B. Holmes ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 4) pp:2434-2441
Publication Date(Web):January 17, 2012
DOI:10.1021/ja211568k
Quantum mechanical calculations have been used to study the intramolecular additions of hydroxylamines to alkenes and alkynes (“reverse Cope eliminations”). In intermolecular reverse Cope eliminations, alkynes are more reactive than alkenes. However, competition experiments have shown that tethering the hydroxylamine to the alkene or alkyne can reverse the reactivity order from that normally observed. The exact outcome depends on the length of the tether. In agreement with experiment, a range of density functional theory methods and CBS-QB3 calculations predict that the activation energies for intramolecular reverse Cope eliminations follow the order 6-exo-dig < 5-exo-trig < 5-exo-dig ≈ 7-exo-dig. The order of the barriers for the 5-, 6-, and 7-exo-dig reactions of alkynes arises mainly from differences in tether strain in the transition states (TSs), but is also influenced by the TS interaction between the hydroxylamine and alkyne. Cyclization onto an alkene in the 5-exo-trig fashion incurs slightly less tether strain than a 6-exo-dig alkyne cyclization, but its activation energy is higher because the hydroxylamine fragment must distort more before the TS is reached. If the alkene terminus is substituted with two methyl groups, the barrier becomes so much higher that it is also disfavored compared to the 5- and 7-exo-dig cyclizations.
Co-reporter:Peng Liu ; Xiufang Xu ; Xiaofei Dong ; Benjamin K. Keitz ; Myles B. Herbert ; Robert H. Grubbs ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 3) pp:1464-1467
Publication Date(Web):January 9, 2012
DOI:10.1021/ja2108728
The mechanism and origins of Z-selectivity in olefin metathesis with chelated Ru catalysts were explored using density functional theory. The olefin approaches from the “side” position of the chelated Ru catalysts, in contrast to reactions with previous unchelated Ru catalysts that favor the bottom-bound pathway. Steric repulsions between the substituents on the olefin and the N-substituent on the N-heterocyclic carbene ligand lead to highly selective formation of the Z product.
Co-reporter:Elizabeth H. Krenske ; Sesil Agopcan ; Viktorya Aviyente ; K. N. Houk ; Brian A. Johnson ;Andrew B. Holmes
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:12010-12015
Publication Date(Web):July 12, 2012
DOI:10.1021/ja300002k
The factors controlling chemo-, regio-, and stereoselectivity in a cascade of reactions starting from a bis(cyanoalkenyl)oxime and proceeding via nitrone cycloadditions have been unraveled through a series of density functional theory calculations with several different functionals. Both kinetic and thermodynamic control of the reaction cascade are important, depending upon the conditions. Kinetic control was analyzed by the distortion/interaction model and found to be dictated by differences in distortions of the cycloaddends in the transition states. A new mechanism competing with that originally proposed in the application of these reactions to the histrionicotoxin synthesis was discovered in these studies.
Co-reporter:Xiufang Xu ; Peng Liu ; Adam Lesser ; Lauren E. Sirois ; Paul A. Wender ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:11012-11025
Publication Date(Web):June 5, 2012
DOI:10.1021/ja3041724
The first theoretical study on the effects of ligands on the mechanism, reactivities, and regioselectivities of Rh(I)-catalyzed (5 + 2) cycloadditions of vinylcyclopropanes (VCPs) and alkynes has been performed using density functional theory (DFT) calculations. Highly efficient and selective intermolecular (5 + 2) cycloadditions of VCPs and alkynes have been achieved recently using two novel rhodium catalysts, [Rh(dnCOT)]+SbF6– and [Rh(COD)]+SbF6–, which provide superior reactivities and regioselectivities relative to that of the previously reported [Rh(CO)2Cl]2 catalyst. Computationally, the high reactivities of the dnCOT and COD ligands are attributed to the steric repulsions that destabilize the Rh-product complex, the catalyst resting state in the catalytic cycle. The regioselectivities of reactions with various alkynes and different Rh catalysts are investigated, and a predictive model is provided that describes substrate–substrate and ligand–substrate steric repulsions, electronic effects, and noncovalent π/π and C–H/π interactions. In the reactions with dnCOT or COD ligands, the first new C–C bond is formed proximal to the bulky substituent on the alkyne to avoid ligand–substrate steric repulsions. This regioselectivity is reversed either by employing the smaller [Rh(CO)2Cl]2 catalyst to diminish the ligand–substrate repulsions or by using aryl alkynes, for which the ligand–substrate interactions become stabilizing due to π/π and C–H/π dispersion interactions. Electron-withdrawing groups on the alkyne prefer to be proximal to the first new C–C bond to maximize metal–substrate back-bonding interactions. These steric, electronic, and dispersion effects can all be utilized in designing new ligands to provide regiochemical control over product formation with high selectivities. The computational studies reveal the potential of employing the dnCOT family of ligands to achieve unique regiochemical control due to the steric influences and dispersion interactions associated with the rigid aryl substituents on the ligand.
Co-reporter:Chelsea G. Gordon ; Joel L. Mackey ; John C. Jewett ; Ellen M. Sletten ; K. N. Houk ;Carolyn R. Bertozzi
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9199-9208
Publication Date(Web):May 3, 2012
DOI:10.1021/ja3000936
The 1,3-dipolar cycloaddition of cyclooctynes with azides, also called “copper-free click chemistry”, is a bioorthogonal reaction with widespread applications in biological discovery. The kinetics of this reaction are of paramount importance for studies of dynamic processes, particularly in living subjects. Here we performed a systematic analysis of the effects of strain and electronics on the reactivity of cyclooctynes with azides through both experimental measurements and computational studies using a density functional theory (DFT) distortion/interaction transition state model. In particular, we focused on biarylazacyclooctynone (BARAC) because it reacts with azides faster than any other reported cyclooctyne and its modular synthesis facilitated rapid access to analogues. We found that substituents on BARAC’s aryl rings can alter the calculated transition state interaction energy of the cycloaddition through electronic effects or the calculated distortion energy through steric effects. Experimental data confirmed that electronic perturbation of BARAC’s aryl rings has a modest effect on reaction rate, whereas steric hindrance in the transition state can significantly retard the reaction. Drawing on these results, we analyzed the relationship between alkyne bond angles, which we determined using X-ray crystallography, and reactivity, quantified by experimental second-order rate constants, for a range of cyclooctynes. Our results suggest a correlation between decreased alkyne bond angle and increased cyclooctyne reactivity. Finally, we obtained structural and computational data that revealed the relationship between the conformation of BARAC’s central lactam and compound reactivity. Collectively, these results indicate that the distortion/interaction model combined with bond angle analysis will enable predictions of cyclooctyne reactivity and the rational design of new reagents for copper-free click chemistry.
Co-reporter:Sarah M. Bronner ; Joel L. Mackey ; K. N. Houk ;Neil K. Garg
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:13966-13969
Publication Date(Web):August 9, 2012
DOI:10.1021/ja306723r
We report an experimental and computational study of 3-silylarynes. The addition of nucleophiles yield ortho-substituted products as a result of aryne distortion, but meta-substituted products form predominately when the nucleophile is large. Computations correctly predict the preferred site of attack observed in both nucleophilic addition and cycloaddition experiments. Nucleophilic additions to 3-tert-butylbenzyne, which is not significantly distorted, give meta-substituted products.
Co-reporter:Ramesh Giri ; Yu Lan ; Peng Liu ; K. N. Houk ;Jin-Quan Yu
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14118-14126
Publication Date(Web):July 25, 2012
DOI:10.1021/ja304643e
The origin of the high levels of reactivity and diastereoselectivity (>99:1 dr) observed in the oxazoline-directed, Pd(II)-catalyzed sp3 C–H bond iodination and acetoxylation reactions as reported in previous publications has been studied and explained on the basis of experimental and computational investigations. The characterization of a trinuclear chiral C–H insertion intermediate by X-ray paved the way for further investigations into C–H insertion step through the lens of stereochemistry. Computational investigations on reactivities and diastereoselectivities of C–H activation of t-Bu- and i-Pr-substituted oxazolines provided good agreement with the experimental results. Theoretical predictions with DFT calculations revealed that C–H activation occurs at the monomeric Pd center and that the most preferred transition state for C–H activation contains two sterically bulky t-Bu substituents in anti-positions due to steric repulsion and that this transition state leads to the major diastereomer, which is consistent with the structure of the newly characterized C–H insertion intermediate. The structural information about the transition state also suggests that a minimum dihedral angle between C–H bonds and Pd–OAc bonds is crucial for C–H bond cleavage. We have also utilized density functional theory (DFT) to calculate the energies of various potential intermediates and transition states with t-Bu- and i-Pr-substituted oxazolines and suggested a possible explanation for the substantial difference in reactivity between the t-Bu- and i-Pr-substituted oxazolines.
Co-reporter:Hao Wang ; Philipp Kohler ; Larry E. Overman ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 38) pp:16054-16058
Publication Date(Web):September 6, 2012
DOI:10.1021/ja3075538
Stereoselectivities of the dihydroxylations of cis-bicyclo[3.3.0]octene intermediates for a projected total synthesis of chromodorolide A have been explored experimentally. The reaction occurs unexpectedly on the apparently more hindered (concave) face; this result has been explained through computational studies using B3LYP and B3LYP-D3 methods. Torsional effects are largely responsible for the stereoselectivity encountered in the chromodorolide A synthesis. Many literature examples have been reported on related cases. QM calculations show that the stereoselectivities of dihydroxylations of fused cyclopentenes are influenced by the conformational rigidity or flexibility of the substrate. Torsional, electrostatic, and steric effects can all influence stereoselectivity, and the rigidity or flexibility of conformations of reactants provides a predictive guide to stereoselectivity.
Co-reporter:Mahboubeh Kheirabadi ; Nihan Çelebi-Ölçüm ; Matthew F. L. Parker ; Qingquan Zhao ; Gert Kiss ; K. N. Houk ;Christian E. Schafmeister
Journal of the American Chemical Society 2012 Volume 134(Issue 44) pp:18345-18353
Publication Date(Web):September 19, 2012
DOI:10.1021/ja3069648
Transesterification catalysts based on stereochemically defined, modular, functionalized ladder-molecules (named spiroligozymes) were designed, using the “inside-out” design strategy, and mutated synthetically to improve catalysis. A series of stereochemically and regiochemically diverse bifunctional spiroligozymes were first synthesized to identify the best arrangement of a pyridine as a general base catalyst and an alcohol nucleophile to accelerate attack on vinyl trifluoroacetate as an electrophile. The best bifunctional spiroligozyme reacted with vinyl trifluoroacetate to form an acyl-spiroligozyme conjugate 2.7 × 103-fold faster than the background reaction with a benzyl alcohol. Two trifunctional spiroligozymes were then synthesized that combined a urea with the pyridine and alcohol to act as an oxyanion hole and activate the bound acyl-spiroligozyme intermediate to enable acyl-transfer to methanol. The best trifunctional spiroligozyme carries out multiple turnovers and acts as a transesterification catalyst with k1/kuncat of 2.2 × 103 and k2/kuncat of 1.3 × 102. Quantum mechanical calculations identified the four transition states of the catalytic cycle and provided a detailed view of every stage of the transesterification reaction.
Co-reporter:Xing Yang ; Valentina D. Bumbu ; Peng Liu ; Ximin Li ; Hui Jiang ; Eric W. Uffman ; Lei Guo ; Wei Zhang ; Xuntian Jiang ; K. N. Houk ;Vladimir B. Birman
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17605-17612
Publication Date(Web):October 2, 2012
DOI:10.1021/ja306766n
In contrast to alcohols and amines, racemic lactams and thiolactams cannot be resolved directly via enzymatic acylation or classical resolution. Asymmetric N-acylation promoted by amidine-based catalysts, particularly Cl-PIQ 2 and BTM 3, provides a convenient method for the kinetic resolution of these valuable compounds and often achieves excellent levels of enantioselectivity in this process. Density functional theory calculations indicate that the reaction occurs via N-acylation of the lactim tautomer and that cation−π interactions play a key role in the chiral recognition of lactam substrates.
Co-reporter:Xin Hong ; Peng Liu ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 135(Issue 4) pp:1456-1462
Publication Date(Web):December 31, 2012
DOI:10.1021/ja309873z
The mechanism and origins of selectivities in [Ni(NHC)]-catalyzed intramolecular (5 + 2) cycloadditions and homo-ene reactions of vinylcyclopropanes (VCPs) and alkynes have been studied using density functional theory. The preferred mechanism involves oxidative alkyne–alkene cyclization to form a metallacyclopentene intermediate, in contrast to a cyclopropane cleavage pathway in the reaction with Rh(I) catalysts. The selectivity between the (5 + 2) and homo-ene products is determined in the subsequent competing reductive elimination and β-hydride elimination steps. Two similar-sized N-heterocyclic carbene (NHC) ligands, SIPr and ItBu, yielded reversed product selectivity, favoring the (5 + 2) and homo-ene products respectively. This is attributed to the anisotropic steric environment of these NHC ligands, which positions the bulky substituents on the ligand toward different directions and leads to distinct steric control in the reductive elimination and β-hydride elimination transition states.
Co-reporter:Yong Liang ; Joel L. Mackey ; Steven A. Lopez ; Fang Liu ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 43) pp:17904-17907
Publication Date(Web):October 13, 2012
DOI:10.1021/ja309241e
The azide–dibenzocyclooctyne and trans-cyclooctene–tetrazine cycloadditions are both bioorthogonal and mutually orthogonal: trans-cyclooctene derivatives greatly prefer to react with tetrazines rather than azides, while dibenzocyclooctyne derivatives react with azides but not with tetrazines under physiological conditions. DFT calculations used to identify the origins of this extraordinary selectivity are reported, and design principles to guide discovery of new orthogonal cycloadditions are proposed. Two new bioorthogonal reagents, methylcyclopropene and 3,3,6,6-tetramethylthiacycloheptyne, are predicted to be mutually orthogonal in azide and tetrazine cycloadditions.
Co-reporter:Hongkun Lin ; Wenbo Pei ; Hao Wang ; Kendall N. Houk ;Isaac J. Krauss
Journal of the American Chemical Society 2012 Volume 135(Issue 1) pp:82-85
Publication Date(Web):December 20, 2012
DOI:10.1021/ja311061n
A practical route to optically pure syn-homocrotylation reagents is described, including highly diastereo- and enantioselective preparation of numerous syn-homocrotyl products, as well as several matched mismatched pairs. NMR experiments suggest that the active homocrotylating species is a cyclopropylcarbinyldichloroborane generated by chloride exchange from the PhBCl2 activator. Computational studies support the intermediacy of chloroboranes and suggest that homoallyl/homocrotyl transfers occur through Zimmerman–Traxler transition states.
Co-reporter:Hung V. Pham, David B. C. Martin, Christopher D. Vanderwal and K. N. Houk
Chemical Science 2012 vol. 3(Issue 5) pp:1650-1655
Publication Date(Web):02 Feb 2012
DOI:10.1039/C2SC01072K
Computational studies show that the base-mediated intramolecular Diels–Alder of tryptamine-derived Zincke aldehydes, used as a key step in the synthesis of the Strychnos alkaloids norfluorocurarine and strychnine, proceeds via a stepwise pathway. The experimentally determined importance of a potassium counterion in the base is explained by its ability to preorganize the Zincke aldehyde diene in an s-cis conformation suitable to bicyclization. Computation also supports the thermodynamic importance of the generation of a stable enolate in the final reaction step. The thermal cycloreversion reaction of the Diels–Alder products is also found to proceed in a stepwise manner.
Co-reporter:Yu Lan, Peng Liu, Stephen G. Newman, Mark Lautens and K. N. Houk
Chemical Science 2012 vol. 3(Issue 6) pp:1987-1995
Publication Date(Web):22 Feb 2012
DOI:10.1039/C2SC20103H
The mechanism of Pd(0)-catalyzed carbohalogenation of alkenes has been investigated with density functional theory. The catalytic cycle involves oxidative addition of the aryl halide, alkene insertion, and a novel C(sp3)–I reductive elimination. The C(sp3)–I reductive elimination leads to a weakly bonded Pd–product complex, which undergoes ligand exchange via a dissociative mechanism to regenerate the catalyst. In the rate-determining reductive elimination step, bromides and chlorides have higher barriers than iodides, because the stronger Pd–Br and Pd–Cl bonds are being cleaved in these transition states. Bulky ligands, such as P(t-Bu)3 and Q-Phos, facilitate the C(sp3)–I reductive elimination by preventing the formation of tetracoordinated intermediates. The mechanism of the competing β-hydrogen elimination pathway was also investigated. For reactions involving a syn-β-hydrogen atom in the alkyl Pd(II) iodide intermediate, β-hydrogen elimination is much more favorable, leading to Heck-type side products. Blocking β-elimination by the choice of substrates is the main reason why this example of carboiodination works.
Co-reporter:Peng Liu, Xing Yang, Vladimir B. Birman, and K. N. Houk
Organic Letters 2012 Volume 14(Issue 13) pp:3288-3291
Publication Date(Web):June 11, 2012
DOI:10.1021/ol301243f
Density functional theory (DFT) calculations were performed to investigate the origins of enantioselectivity in benzotetramisole (BTM)-catalyzed dynamic kinetic resolution of azlactones. The transition states of the fast-reacting enantiomer are stabilized by electrostatic interactions between the amide carbonyl group and the acetate anion bound to the nucleophile. The chiral BTM catalyst confines the conformation of the α-carbon and the facial selectivity of the nucleophilic attack to promote such electrostatic attractions.
Co-reporter:Elizabeth H. Krenske, Emma W. Perry, Steven V. Jerome, Thomas J. Maimone, Phil S. Baran, and K. N. Houk
Organic Letters 2012 Volume 14(Issue 12) pp:3016-3019
Publication Date(Web):May 25, 2012
DOI:10.1021/ol301083q
Unlike normal Diels–Alder reactions of acyclic alkadienes with alkenes, the vinylbicyclo[2.2.2]octene employed in the Baran total synthesis of vinigrol undergoes a quantitative Diels–Alder reaction with a tethered alkene at room temperature. Density functional theory calculations reveal that this unprecedented reactivity originates from a combination of preorganization, diene strain, and tether stabilization.
Co-reporter:Arne Dieckmann and K. N. Houk
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 12) pp:5064-5071
Publication Date(Web):September 12, 2012
DOI:10.1021/ct300655b
Cooperative effects caused by dispersion interactions between dienes and dienophiles in a so-called self-replicating system have been evaluated by density functional theory. A variety of functionals is tested for the elucidation of reactant complex stabilities. Dispersion interactions between dienes and dienophiles result in additional stabilizations of up to 10 kcal mol–1. These effects should be taken into account in future experimental studies and can be exploited to design more efficient systems.
Co-reporter:Geoffrey R. Nosrati and K. N. Houk
Biochemistry 2012 Volume 51(Issue 37) pp:
Publication Date(Web):August 21, 2012
DOI:10.1021/bi3008438
Catalytic atom maps (CAMs) are minimal models of enzyme active sites. The structures in the Protein Data Bank (PDB) were examined to determine if proteins with CAM-like geometries in their active sites all share the same catalytic function. We combined the CAM-based search protocol with a filter based on the weighted contact number (WCN) of the catalytic residues, a measure of the “crowdedness” of the microenvironment around a protein residue. Using this technique, a CAM based on the Ser-His-Asp catalytic triad of trypsin was able to correctly identify catalytic triads in other enzymes within 0.5 Å rmsd of the CAM with 96% accuracy. A CAM based on the Cys-Arg-(Asp/Glu) active site residues from the tyrosine phosphatase active site achieved 89% accuracy in identifying this type of catalytic functionality. Both of these CAMs were able to identify active sites across different fold types. Finally, the PDB was searched to locate proteins with catalytic functionality similar to that present in the active site of orotidine 5′-monophosphate decarboxylase (ODCase), whose mechanism is not known with certainty. A CAM, based on the conserved Lys-Asp-Lys-Asp tetrad in the ODCase active site, was used to search the PDB for enzymes with similar active sites. The ODCase active site has a geometry similar to that of Schiff base-forming Class I aldolases, with lowest aldolase rmsd to the ODCase CAM at 0.48 Å. The similarity between this CAM and the aldolase active site suggests that ODCase has the correct catalytic functionality present in its active site for the generation of a nucleophilic lysine.
Co-reporter:Yu-hong Lam;Janine Cossy;Domingo GomezPardo;Anne Cochi
Helvetica Chimica Acta 2012 Volume 95( Issue 11) pp:2265-2277
Publication Date(Web):
DOI:10.1002/hlca.201200461
Abstract
The origin of the variation in the regioselectivity of the nucleophilic ring opening of a series of bicyclic aziridinium ions derived from N-alkylprolinols was investigated by quantum-chemical computations (M06-2X/6-31+G(d,p)-SMD). These aziridiniums differ only in the degree and the configurations of F-substitution at C(4). With the azide ion as nucleophile, the ratio of the piperidine to the pyrrolidine product was computed. An electrostatic gauche effect influences the conformation of the adjoining five-membered ring in the fluorinated bicyclic aziridinium. This controls the regioselectivity of the aziridinium ring opening.
Co-reporter:Yu Lan, Rick L. Danheiser, and K. N. Houk
The Journal of Organic Chemistry 2012 Volume 77(Issue 3) pp:1533-1538
Publication Date(Web):December 21, 2011
DOI:10.1021/jo202424n
An intramolecular formal metal-free intramolecular [2 + 2 + 2] cycloaddition for the formation of pyridines has been investigated with M06-2X and B3LYP density functional methods, and compared to the experimentally established three-step mechanism that involves ene reaction–Diels–Alder reaction–hydrogen transfer. The ene reaction of two alkynes is the rate-determining step. This is considerably easier than other possible mechanisms, such as those involving an ene reaction of an alkyne with a nitrile, a one-step [2 + 2 + 2] cycloaddition, or a 1,4-diradical mechanism. The relative facilities of these processes are analyzed with the distortion-interaction model. A bimolecular hydrogen-transfer mechanism involving a radical-pair intermediate is proposed rather than a concerted intramolecular 1,5-hydrogen shift for the last step in the mechanism.
Co-reporter:Dr. Martin J. Schnermann;Nicholas L. Untiedt;Dr. Gonzalo Jiménez-Osés; Kendall N. Houk; Larry E. Overman
Angewandte Chemie 2012 Volume 124( Issue 38) pp:9719-9724
Publication Date(Web):
DOI:10.1002/ange.201205001
Co-reporter:Pankaj Jain;Hao Wang;Dr. Kendall N. Houk;Dr. Jon C. Antilla
Angewandte Chemie 2012 Volume 124( Issue 6) pp:1420-1423
Publication Date(Web):
DOI:10.1002/ange.201107407
Co-reporter:Daniel A. DiRocco;Elizabeth L. Noey; K. N. Houk; Tomislav Rovis
Angewandte Chemie 2012 Volume 124( Issue 10) pp:2441-2444
Publication Date(Web):
DOI:10.1002/ange.201107597
Co-reporter:Dr. Ana Bellomo;Dr. Nihan Celebi-Olcum;Dr. Xiaodong Bu;Dr. Nelo Rivera;Dr. Rebecca T. Ruck;Dr. Christopher J. Welch;Dr. Kendall N. Houk;Dr. Spencer D. Dreher
Angewandte Chemie 2012 Volume 124( Issue 28) pp:7018-7021
Publication Date(Web):
DOI:10.1002/ange.201201720
Co-reporter:Xing Yang;Peng Liu; K. N. Houk; Vladimir B. Birman
Angewandte Chemie 2012 Volume 124( Issue 38) pp:9776-9780
Publication Date(Web):
DOI:10.1002/ange.201203327
Co-reporter:Dr. Ricardo A. Matute ;Dr. Kendall N. Houk
Angewandte Chemie 2012 Volume 124( Issue 52) pp:13274-13277
Publication Date(Web):
DOI:10.1002/ange.201208002
Co-reporter:Dr. Ricardo A. Matute ;Dr. Kendall N. Houk
Angewandte Chemie 2012 Volume 124( Issue 52) pp:
Publication Date(Web):
DOI:10.1002/ange.201209048
Co-reporter:Dr. Martin J. Schnermann;Nicholas L. Untiedt;Dr. Gonzalo Jiménez-Osés; Kendall N. Houk; Larry E. Overman
Angewandte Chemie International Edition 2012 Volume 51( Issue 38) pp:9581-9586
Publication Date(Web):
DOI:10.1002/anie.201205001
Co-reporter:Pankaj Jain;Hao Wang;Dr. Kendall N. Houk;Dr. Jon C. Antilla
Angewandte Chemie International Edition 2012 Volume 51( Issue 6) pp:1391-1394
Publication Date(Web):
DOI:10.1002/anie.201107407
Co-reporter:Daniel A. DiRocco;Elizabeth L. Noey; K. N. Houk; Tomislav Rovis
Angewandte Chemie International Edition 2012 Volume 51( Issue 10) pp:2391-2394
Publication Date(Web):
DOI:10.1002/anie.201107597
Co-reporter:Dr. Ana Bellomo;Dr. Nihan Celebi-Olcum;Dr. Xiaodong Bu;Dr. Nelo Rivera;Dr. Rebecca T. Ruck;Dr. Christopher J. Welch;Dr. Kendall N. Houk;Dr. Spencer D. Dreher
Angewandte Chemie International Edition 2012 Volume 51( Issue 28) pp:6912-6915
Publication Date(Web):
DOI:10.1002/anie.201201720
Co-reporter:Xing Yang;Peng Liu; K. N. Houk; Vladimir B. Birman
Angewandte Chemie International Edition 2012 Volume 51( Issue 38) pp:9638-9642
Publication Date(Web):
DOI:10.1002/anie.201203327
Co-reporter:Dr. Ricardo A. Matute ;Dr. Kendall N. Houk
Angewandte Chemie International Edition 2012 Volume 51( Issue 52) pp:13097-13100
Publication Date(Web):
DOI:10.1002/anie.201208002
Co-reporter:Dr. Ricardo A. Matute ;Dr. Kendall N. Houk
Angewandte Chemie International Edition 2012 Volume 51( Issue 52) pp:
Publication Date(Web):
DOI:10.1002/anie.201209048
Co-reporter:C. Avery Sader and K. N. Houk
The Journal of Organic Chemistry 2012 Volume 77(Issue 11) pp:4939-4948
Publication Date(Web):April 26, 2012
DOI:10.1021/jo300314z
The mechanism of cyclohexyne insertion into a C(O)–Cα bond of cyclic ketones, explored experimentally by the Carreira group, has been investigated using density functional theory. B3LYP and M06–2X calculations were performed in both gas phase and THF (CPCM, UAKS radii). The reaction proceeds through a stepwise [2 + 2] cycloaddition of cyclohexyne to the enolate, followed by three disparate ring-opening possibilities of the cyclobutene alkoxide to give the product: (1) thermally allowed conrotatory electrocyclic ring-opening, (2) thermally forbidden disrotatory electrocyclic ring-opening, or (3) nonpericyclic C–C bond cleavage. Our computational results for the model alkoxide and potassium alkoxide systems show that the thermally allowed electrocyclic ring-opening pathway is favored by less than 1 kcal/mol. In more complex systems containing a potassium alkoxide (e–f), the barrier of the allowed conrotatory ring-opening is disfavored by 4–8 kcal/mol. This suggests that the thermodynamically more stable disrotatory product can be formed directly through a “forbidden” pathway. Analysis of geometrical parameters and atomic charges throughout the ring-opening pathways provides evidence for a nonpericyclic C–C bond cleavage, rather than a thermally forbidden disrotatory ring-opening. A true forbidden disrotatory ring-opening transition structure was computed for the cyclobutene alcohol; however, it was 19 kcal/mol higher in energy than the allowed conrotatory transition structure. An alternate mechanism in which the disrotatory product forms via isomerization of the conrotatory product was also explored for the alkoxide and potassium alkoxide systems.
Co-reporter:Andrej Petrič;Scott A. Johnson;Hung V. Pham;Ying Li;Simon Čeh;Amalija Golobič;Eric D. Agdeppa;Jie Liu;Gerald Timbol;Vladimir Kepe;Nagichettiar Satyamurthy;Gyochang Keum;Jorge R. Barrio
PNAS 2012 Volume 109 (Issue 41 ) pp:16492-16497
Publication Date(Web):2012-10-09
DOI:10.1073/pnas.1214134109
The positron-emission tomography (PET) probe 2-(1-[6-[(2-fluoroethyl)(methyl)amino]-2-naphthyl]ethylidene) (FDDNP) is used
for the noninvasive brain imaging of amyloid-β (Aβ) and other amyloid aggregates present in Alzheimer’s disease and other
neurodegenerative diseases. A series of FDDNP analogs has been synthesized and characterized using spectroscopic and computational
methods. The binding affinities of these molecules have been measured experimentally and explained through the use of a computational
model. The analogs were created by systematically modifying the donor and the acceptor sides of FDDNP to learn the structural
requirements for optimal binding to Aβ aggregates. FDDNP and its analogs are neutral, environmentally sensitive, fluorescent
molecules with high dipole moments, as evidenced by their spectroscopic properties and dipole moment calculations. The preferred
solution-state conformation of these compounds is directly related to the binding affinities. The extreme cases were a nonplanar
analog t-butyl-FDDNP, which shows low binding affinity for Aβ aggregates (520 nM Ki) in vitro and a nearly planar tricyclic analog cDDNP, which displayed the highest binding affinity (10 pM Ki). Using a previously published X-ray crystallographic model of 1,1-dicyano-2-[6-(dimethylamino)naphthalen-2-yl]propene (DDNP)
bound to an amyloidogenic Aβ peptide model, we show that the binding affinity is inversely related to the distortion energy
necessary to avoid steric clashes along the internal surface of the binding channel.
Co-reporter:Paul Ha-Yeon Cheong, Claude Y. Legault, Joann M. Um, Nihan Çelebi-Ölçüm, and K. N. Houk
Chemical Reviews 2011 Volume 111(Issue 8) pp:5042
Publication Date(Web):June 28, 2011
DOI:10.1021/cr100212h
Co-reporter:Robert S. Paton ; Sarah E. Steinhardt ; Christopher D. Vanderwal ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:3895-3905
Publication Date(Web):February 25, 2011
DOI:10.1021/ja107988b
The thermal pericyclic cascade rearrangement of Zincke aldehydes (5-(dialkylamino)-2,4-pentadienals) to afford Z-α,β,γ,δ-unsaturated amides discovered by the Vanderwal group has been studied in depth using quantum mechanical methods. Two mechanistic possibilities that had previously been put forth to explain this internal redox process, one that had been discounted by experiment and the other that had withstood experimental scrutiny, were evaluated. Both of these mechanisms suffered from energetic barriers that appeared too high to allow rearrangement to proceed under the conditions used; however, computational study of a third possibility that implicates the intermediacy of vinylketenes revealed that it is the most likely pathway of rearrangement. Further computational studies accounted for the relative rates of rearrangement in substituted Zincke aldehydes, predicted the feasibility of related processes for other donor−acceptor dienes, and provided insight into the rearrangement of allylamine-derived Zincke aldehydes that provide either dihydropyridones or polycyclic lactams by further pericyclic processes.
Co-reporter:Elizabeth L. Noey ; Yingdong Luo ; Liming Zhang ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 134(Issue 2) pp:1078-1084
Publication Date(Web):December 22, 2011
DOI:10.1021/ja208860x
Quantum mechanical studies of the mechanism of gold-catalyzed rearrangements of acetylenic amine-N-oxides to piperidinones or azepanones have revealed a new mechanism involving a concerted heteroretroene reaction, formally a 1,5 hydrogen shift from the N-alkyl groups to the vinyl position of a gold-coordinated methyleneisoxazolidinium or methyleneoxazinanium. Density functional calculations (B3LYP, B3LYP-D3) on the heteroretroene mechanism reproduce experimental regioselectivities and provide an explanation as to why the hydrogen is transferred from the smaller amine substituent. In support of the proposed mechanism, new experimental investigations show that the hydrogen shift is concerted and that gold carbenes are not involved as reaction intermediates.
Co-reporter:Lai Xu ; Charles E. Doubleday ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17848-17854
Publication Date(Web):September 30, 2011
DOI:10.1021/ja207051b
Quasiclassical trajectory calculations using quantum mechanical energies and forces generated by the Venus and Gaussian programs provide for the first time a detailed dynamical picture of singlet carbene, CCl2 and CF2, cycloadditions to alkenes on the B3LYP/6-31G* surface. For CF2, B3LYP/6-31G* with exact exchange reduced to 12% HF was also employed to better mimic the high accuracy surface. The range of geometries sampled in reactive trajectories and the timing of bond formation were explored. All trajectories follow the nonlinear approach proposed by Moore and Hoffmann. The reaction of CCl2 with ethylene is a dynamically concerted reaction, with an average time gap between formation of the two bonds of 50 fs. The reaction of CF2 with ethylene is dynamically complex with biexponential decay of the diradical species formed from the first bond formation. A general quantitative dynamical classification of cycloaddition mechanisms is proposed, based on the timing of bond formation.
Co-reporter:Andrew R. Bogdan, Steven V. Jerome, K. N. Houk, and Keith James
Journal of the American Chemical Society 2011 Volume 134(Issue 4) pp:2127-2138
Publication Date(Web):December 1, 2011
DOI:10.1021/ja208503y
The synthesis, X-ray crystal structures, and calculated strain energies are reported for a homologous series of 11- to 14-membered drug-like cyclophane macrocycles, representing an unusual region of chemical space that can be difficult to access synthetically. The ratio of macrocycle to dimer, generated via a copper catalyzed azide–alkyne cycloaddition macrocyclization in flow at elevated temperature, could be rationalized in terms of the strain energy in the macrocyclic product. The progressive increase in strain resulting from reduction in macrocycle ring size, or the introduction of additional conformational constraints, results in marked deviations from typical geometries. These strained cyclophane macrocyclic systems provide access to spatial orientations of functionality that would not be readily available in unstrained or acyclic analogs. The most strained system prepared represents the first report of an 11-membered cyclophane containing a 1,4-disubstituted 1,2,3-triazole ring and establishes a limit to the ring strain that can be generated using this macrocycle synthesis methodology.
Co-reporter:Nihan Çelebi-Ölçüm ; Ben W. Boal ; Alexander D. Huters ; Neil K. Garg ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 15) pp:5752-5755
Publication Date(Web):March 28, 2011
DOI:10.1021/ja201035b
The mechanisms of the Fischer indole synthesis and competing cleavage pathways were explored with SCS-MP2/6-31G(d) and aqueous solvation calculations. Electron-donating substituents divert the reaction pathway to heterolytic N−N bond cleavage and preclude the acid-promoted [3,3]-sigmatropic rearrangement.
Co-reporter:Peng Liu ; John Montgomery ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6956-6959
Publication Date(Web):April 20, 2011
DOI:10.1021/ja202007s
The regioselectivities of N-heterocyclic carbene (NHC) ligands in Ni-catalyzed alkyne–aldehyde reductive coupling reactions with silane reducing agents are investigated using density functional theory. Reversal of regioselectivity can be achieved by varying the steric bulkiness of the ligand. The steric influences of NHC ligands are highly anisotropic. Regioselectivity is primarily controlled by the steric hindrance at the region of the ligand close to the alkyne. Analysis of 2D contour maps of the NHC ligands indicates that the regioselectivities are directly affected by the shape and orientation of the N-substituents on the ligand.
Co-reporter:Joann M. Um ; Daniel A. DiRocco ; Elizabeth L. Noey ; Tomislav Rovis ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 29) pp:11249-11254
Publication Date(Web):June 15, 2011
DOI:10.1021/ja202444g
The asymmetric intermolecular Stetter reaction was investigated using the B3LYP and M06-2X functionals. Fluorination of a triazolium bicyclic catalyst had been found to significantly influence reaction yields and enantiomeric ratios. Computations indicate that the improved reactivity of the fluorinated catalyst is due to better electrostatic interactions between the nitroalkene and catalyst. Computational investigations of preferred conformations of the ground state catalyst and acyl anion equivalent, and the transition structures leading to both enantiomers of the products, are reported.
Co-reporter:Jennifer E. Antoline ; Elizabeth H. Krenske ; Andrew G. Lohse ; K. N. Houk ;Richard P. Hsung
Journal of the American Chemical Society 2011 Volume 133(Issue 36) pp:14443-14451
Publication Date(Web):August 18, 2011
DOI:10.1021/ja205700p
A systematic investigation of the regioselectivities and stereoselectivities of (4 + 3) cycloadditions between unsymmetrical furans and a chiral oxazolidinone-substituted oxyallyl is presented. Cycloadditions were performed using an oxyallyl containing a (R)-4-phenyl-2-oxazolidinone auxiliary (2Ph), under either thermal or ZnCl2-catalyzed conditions. Reactions of 2Ph with 2-substituted furans gave syn cycloadducts selectively, while cycloadditions with 3-substituted furans gave selectively anti cycloadducts. The stereoselectivities were in favor of a single diastereoisomer (I) in all but one case (2-CO2R). Density functional theory calculations were performed to explain the selectivities. The results support a mechanism in which all cycloadducts are formed from the E isomer of the oxyallyl (in which the oxazolidinone C═O and oxyallyl oxygen are anti to each other) or the corresponding (E)-ZnCl2 complex. The major diastereomer is derived from addition of the furan to the more crowded face of the oxyallyl. Crowded transition states are favored because they possess a stabilizing CH−π interaction between the furan and the Ph group.
Co-reporter:Kyle W. Quasdorf, Aurora Antoft-Finch, Peng Liu, Amanda L. Silberstein, Anna Komaromi, Tom Blackburn, Stephen D. Ramgren, K. N. Houk, Victor Snieckus, and Neil K. Garg
Journal of the American Chemical Society 2011 Volume 133(Issue 16) pp:6352-6363
Publication Date(Web):April 1, 2011
DOI:10.1021/ja200398c
The first Suzuki−Miyaura cross-coupling reactions of the synthetically versatile aryl O-carbamate and O-sulfamate groups are described. The transformations utilize the inexpensive, bench-stable catalyst NiCl2(PCy3)2 to furnish biaryls in good to excellent yields. A broad scope for this methodology has been demonstrated. Substrates with electron-donating and electron-withdrawing groups are tolerated, in addition to those that possess ortho substituents. Furthermore, heteroaryl substrates may be employed as coupling partners. A computational study providing the full catalytic cycles for these cross-coupling reactions is described. The oxidative addition with carbamates or sulfamates occurs via a five-centered transition state, resulting in the exclusive cleavage of the aryl C−O bond. Water is found to stabilize the Ni-carbamate catalyst resting state, which thus provides rationalization of the relative decreased rate of coupling of carbamates. Several synthetic applications are presented to showcase the utility of the methodology in the synthesis of polysubstituted aromatic compounds of natural product and bioactive molecule interest.
Co-reporter:Yang Cao and K. N. Houk
Journal of Materials Chemistry A 2011 vol. 21(Issue 5) pp:1503-1508
Publication Date(Web):22 Oct 2010
DOI:10.1039/C0JM02422H
The 1,3-dipolar cycloadditions of azomethine ylide and carbonyl ylide to models of graphene have been investigated with density functional theory. Reaction energetics have been obtained and show that edge areas of graphene are much more favourable reaction sites than the centre sites. Azomethine ylide cannot directly react at the centre area, while carbonyl ylides are promising reagents for functionalization of graphene. The influence of some 1,3-dipole substituents is also evaluated.
Co-reporter:Hao Wang, K. N. Houk, Damian A. Allen, and Michael E. Jung
Organic Letters 2011 Volume 13(Issue 12) pp:3238-3241
Publication Date(Web):May 13, 2011
DOI:10.1021/ol2011488
The non-aldol aldol reaction of the isomeric epoxy silyl ethers is controlled by the conformation of the transition states leading to an internal hydride shift. One isomer rearranges to the β-silyloxy ketone whereas the other isomer gives a β-elimination product. Theoretical calculations show that the substrates with substituents that favor the formation of the chairlike transition state rearrange normally while those that do not undergo elimination instead.
Co-reporter:Yu Lan, Steven E. Wheeler, and K. N. Houk
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 7) pp:2104-2111
Publication Date(Web):May 27, 2011
DOI:10.1021/ct200293w
Ozone and sulfur dioxide are valence isoelectronic yet show very different reactivity. While ozone is one of the most reactive 1,3-dipoles, SO2 does not react in this way at all. The activation energies of dipolar cycloadditions of sulfur dioxide with either ethylene or acetylene are predicted here by B3LYP, M06-2X, CBS-QB3, and CCSD(T) to be much higher than reactions of ozone. The dipolar cycloaddition of ozone is very exothermic, while that of than sulfur dioxide is endothermic. The prohibitive barriers in the case of SO2 arise from large distortion energies as well as unfavorable interaction energies in the transition states. This arises in part from the HOMO–LUMO gap of sulfur dioxide, which is larger than that of ozone. Valence bond calculations also show that while ozone has a high degree of diradical character, SO2 does not, and is better characterized as a dritterion.
Co-reporter:Elizabeth H. Krenske, K.N. Houk, Andrew B. Holmes, John Thompson
Tetrahedron Letters 2011 Volume 52(Issue 17) pp:2181-2184
Publication Date(Web):27 April 2011
DOI:10.1016/j.tetlet.2010.11.121
Intramolecular 1,3-dipolar cycloadditions of two N-alkenylnitrones are studied by means of density functional theory calculations. Cycloaddition of an acyclic 4-hexenylnitrone led to the expected isoxazolidine in 46% yield, but a 4-cycloheptenylnitrone did not react. Calculations of the transition states for cycloaddition indicate that although the cycloheptenyl nitrone has a more favorable activation entropy, the strain associated with distortion of the tethering groups into the required boat conformation disfavors the reaction of the cyclic substrate over the acyclic substrate by 8.7 kcal/mol.
Co-reporter:Peng Liu, K.N. Houk
Inorganica Chimica Acta 2011 Volume 369(Issue 1) pp:2-14
Publication Date(Web):15 April 2011
DOI:10.1016/j.ica.2010.12.042
This review highlights our theoretical studies of regioselectivities of Rh-catalyzed (5 + 2) cycloadditions, Ni-catalyzed reductive alkyne–aldehyde couplings, and Rh-catalyzed hydrogenative couplings of diynes and carbonyl partners. Factors that control the regioselectivities in these reactions are analyzed; these include steric repulsions involving the substrate and ligands, electronic effects dictated by metal–substrate interactions, and directing effects of conjugated alkenyl and alkynyl groups involving coordination to the metal.Graphical abstractWe honor Bob Bergman on this occasion with an account of our theoretical studies of regioselectivities of Ni- and Rh-catalyzed reductive coupling reactions and Rh-catalyzed (5 + 2) cycloadditions with unsymmetrical alkynes. The regioselectivities in these reactions are controlled by steric repulsions involving the substrate and the ligands, electronic effects, and directing effects of unsaturated substituents.Research highlights► The regioselectivities are controlled by both steric and electronic effects. ► Steric repulsions with the substrate and the ligands both affect regioselectivity. ► Conjugated enynes and diynes have strong directing effects in C–C couplings.
Co-reporter:Hong Ma;Alex K.-Y. Jen;Paul S. Weiss;Moonhee Kim;Yang Cao;J. Nathan Hohman
Science 2011 Volume 331(Issue 6022) pp:1312-1315
Publication Date(Web):11 Mar 2011
DOI:10.1126/science.1200830
Molecules align in molecular overlayers for photodimerization reactions that would be disfavored in solution.
Co-reporter:Dora Toledo Warshaviak, Laura Serbulea, K. N. Houk, and Wayne L. Hubbell
The Journal of Physical Chemistry B 2011 Volume 115(Issue 2) pp:397-405
Publication Date(Web):December 17, 2010
DOI:10.1021/jp108871m
In site directed spin labeling, a nitroxide side chain is introduced at a selected site in a protein; the most commonly used is a disulfide-linked side chain designated R1. The electron paramagnetic resonance (EPR) spectra of R1, and the interspin distance between pairs of R1 residues as determined by dipolar EPR spectroscopy, encode a wealth of information on the protein structure and dynamics. However, extracting this information requires structural and dynamical models of the R1 side chain, that is, the favored rotamers, the intraresidue interactions that stabilize them, and the internal modes of motion. X-ray crystal structures of R1 in proteins have revealed a set of preferred rotamers in the crystal lattice. To identify the intraresidue interactions that stabilize particular rotamers of R1 in the absence of interactions with nearby side chains in a helix, and to evaluate models for the internal motion of the side chain, quantum mechanical calculations were performed on a relevant fragment of R1 in a 10-residue α-helix. Relative rotamer energies were determined in the gas phase, and solvation energies were estimated from a continuum solvent model that includes both electrostatic and hydrophobic contributions. The results identified preferred rotamers that are in agreement with the X-ray crystallographic studies. The rotamers are apparently stabilized by intraresidue sulfur-backbone interactions, suggesting that the preferred rotamers may be the same at all solvent-exposed helix sites.
Co-reporter:Dr. Robert S. Paton;Dr. Seonah Kim;Audrey G. Ross; Samuel J. Danishefsky; K. N. Houk
Angewandte Chemie International Edition 2011 Volume 50( Issue 44) pp:10366-10368
Publication Date(Web):
DOI:10.1002/anie.201103998
Co-reporter:Dr. Nihan Çelebi-Ölçüm;Dr. Yu-hong Lam;Edward Richmond;Dr. Kenneth B. Ling;Dr. Andrew D. Smith;Dr.Dr. Kendall N. Houk
Angewandte Chemie International Edition 2011 Volume 50( Issue 48) pp:11478-11482
Publication Date(Web):
DOI:10.1002/anie.201105412
Co-reporter:Yu Lan, Lufeng Zou, Yang Cao, and K. N. Houk
The Journal of Physical Chemistry A 2011 Volume 115(Issue 47) pp:13906-13920
Publication Date(Web):October 3, 2011
DOI:10.1021/jp207563h
Theoretical calculations were performed on the 1,3-dipolar cycloaddition reactions of 24 1,3-dipoles with ethylene and acetylene. The 24 1,3-dipoles are of the formula X≡Y+—Z– (where X is HC or N, Y is N, and Z is CH2, NH, or O) or X═Y+—Z– (where X and Z are CH2, NH, or O and Y is NH, O, or S). The high-accuracy G3B3 method was employed as the reference. CBS-QB3, CCSD(T)//B3LYP, SCS-MP2//B3LYP, B3LYP, M06-2X, and B97-D methods were benchmarked to assess their accuracies and to determine an accurate method that is practical for large systems. Several basis sets were also evaluated. Compared to the G3B3 method, CBS-QB3 and CCSD(T)/maug-cc-pV(T+d)Z//B3LYP methods give similar results for both activation and reaction enthalpies (mean average deviation, MAD, < 1.5 kcal/mol). SCS-MP2//B3LYP and M06-2X give small errors for the activation enthalpies (MAD < 1.5 kcal/mol), while B3LYP has MAD = 2.3 kcal/mol. SCS-MP2//B3LYP and B3LYP give the reasonable reaction enthalpies (MAD < 5.0 kcal/mol). The B3LYP functional also gives good results for most 1,3-dipoles (MAD = 1.9 kcal/mol for 17 common 1,3-dipoles), but the activation and reaction enthalpies for ozone and sulfur dioxide are difficult to calculate by any of the density functional methods.
Co-reporter:Dr. Peng Liu;Dr. Michael J. Krische;Dr. Kendall N. Houk
Chemistry - A European Journal 2011 Volume 17( Issue 14) pp:4021-4029
Publication Date(Web):
DOI:10.1002/chem.201002741
Abstract
The mechanism of the rhodium-catalyzed reductive coupling of 1,3-diynes and vicinal dicarbonyl compounds employing H2 as reductant was investigated by density functional theory. Oxidative coupling through 1,4-addition of the RhI-bound dicarbonyl to the conjugated diyne via a seven-membered cyclic cumulene transition state leads to exclusive formation of linear adducts. Diyne 1,4-addition is much faster than the 1,2-addition to simple alkynes. The 1,2-dicarbonyl compound is bound to rhodium in a bidentate fashion during the oxidative coupling event. The chemo-, regio-, and enantioselectivities of this reaction were investigated and are attributed to this unique 1,4-addition pathway. The close proximity of the ligand and the alkyne substituent distal to the forming CC bond controls the regio- and enantioselectivity: coupling occurs at the sterically more demanding alkyne terminus, which minimizes nonbonded interaction with the ligand. A stereochemical model is proposed that accounts for preferential formation of the (R)-configurated coupling product when (R)-biaryl phosphine ligands are used.
Co-reporter:Dr. Le-Ping Liu;Deepika Malhotra;Zhuang Jin;Dr. Robert S. Paton;Dr. K. N. Houk;Dr. Gerald B. Hammond
Chemistry - A European Journal 2011 Volume 17( Issue 38) pp:10690-10699
Publication Date(Web):
DOI:10.1002/chem.201101448
Abstract
The gold-catalyzed intramolecular oxygen-transfer reactions of 2-alkynyl-1,5-diketones or 2-alkynyl-5-ketoesters—obtained from tetra-n-butylammonium fluoride mediated Michael addition of activated allenes to electron-deficient olefins—furnished cyclopentenyl ketones under very mild conditions. These reactions proceeded much easier and faster than similar reactions reported in literature, and the corresponding products were obtained in very good yields. Mechanistic investigations on the cycloisomerization were carried out by means of both 18O isotopic experiments and quantum chemical calculations. The results from both, the designed isotopic experiments and theoretical calculations, satisfactorily supported the novel proposed intramolecular [4+2] cycloaddition of a gold-containing furanium intermediate to a carbonyl group, instead of the previous well-accepted [2+2] pathway.
Co-reporter:Elizabeth L. Noey, Xiang Wang, and K. N. Houk
The Journal of Organic Chemistry 2011 Volume 76(Issue 9) pp:3477-3483
Publication Date(Web):March 23, 2011
DOI:10.1021/jo200556f
Several alkynylindoles undergo gold(I)-catalyzed cyclization reactions to form a single isomer in each case. Density functional theory shows why this reaction is favored over the many possible regio- and stereoisomeric reaction pathways. This transformation involves a two-step no-intermediate mechanism with surface bifurcations leading to two or three products. Such bifurcations could explain reactivity in many gold(I)-catalyzed enyne cyclization reactions.
Co-reporter:Yu Lan and K. N. Houk
The Journal of Organic Chemistry 2011 Volume 76(Issue 12) pp:4905-4909
Publication Date(Web):May 3, 2011
DOI:10.1021/jo2002903
The palladium(II)-catalyzed addition of arylboronic acids to β,β-disubstituted enones has been investigated with the BP86 density functional. The results show that the mechanism requires three steps: transmetalation, alkene insertion, and protonation. The alkene insertion is the rate-determining step. For unactivated alkenes, the Heck-type β-hydride elimination is more favored than protonation.
Co-reporter:Elizabeth H. Krenske, Russell C. Petter, Zhendong Zhu, and K. N. Houk
The Journal of Organic Chemistry 2011 Volume 76(Issue 12) pp:5074-5081
Publication Date(Web):May 16, 2011
DOI:10.1021/jo200761w
CBS-QB3 enthalpies of reaction have been computed for the conjugate additions of MeSH to six α,β-unsaturated ketones. Compared with addition to methyl vinyl ketone, the reaction becomes 1–3 kcal mol–1 less exothermic when an α-Me, β-Me, or β-Ph substituent is present on the C═C bond. The lower exothermicity for the substituted enones occurs because the substituted reactant is stabilized more by hyperconjugation or conjugation than the product is stabilized by branching. Substituent effects on the activation energies for the rate-determining step of the thiol addition (reaction of the enone with MeS–) were also computed. Loss of reactant stabilization, and not steric hindrance, is the main factor responsible for controlling the relative activation energies in the gas phase. The substituent effects are further magnified in solution; in water (simulated by CPCM calculations), the addition of MeS– to an enone is disfavored by 2–6 kcal mol–1 when one or two methyl groups are present on the C═C bond (ΔΔG⧧). The use of CBS-QB3 gas-phase energies in conjunction with CPCM solvation corrections provides kinetic data in good agreement with experimental substituent effects. When the energetics of the thiol additions were calculated with several popular density functional theory and ab initio methods (B3LYP, MPW1PW91, B1B95, PBE0, B2PLYP, and MP2), some substantial inaccuracies were noted. However, M06-2X (with a large basis set), B2PLYP-D, and SCS-MP2 gave results within 1 kcal mol–1 of the CBS-QB3 benchmark values.
Co-reporter:G-Yoon J. Im ; Sarah M. Bronner ; Adam E. Goetz ; Robert S. Paton ; Paul H.-Y. Cheong ; K. N. Houk ;Neil K. Garg
Journal of the American Chemical Society 2010 Volume 132(Issue 50) pp:17933-17944
Publication Date(Web):November 29, 2010
DOI:10.1021/ja1086485
Efficient syntheses of 4,5-, 5,6-, and 6,7-indolyne precursors beginning from commercially available hydroxyindole derivatives are reported. The synthetic routes are versatile and allow access to indolyne precursors that remain unsubstituted on the pyrrole ring. Indolynes can be generated under mild fluoride-mediated conditions, trapped by a variety of nucleophilic reagents, and used to access a number of novel substituted indoles. Nucleophilic addition reactions to indolynes proceed with varying degrees of regioselectivity; distortion energies control regioselectivity and provide a simple model to predict the regioselectivity in the nucleophilic additions to indolynes and other unsymmetrical arynes. This model has led to the design of a substituted 4,5-indolyne that exhibits enhanced nucleophilic regioselectivity.
Co-reporter:Yu Lan ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 50) pp:17921-17927
Publication Date(Web):December 1, 2010
DOI:10.1021/ja108432b
The 1,3-dipolar cycloadditions of a thiocarbonyl ylide to dimethyl 2,3-dicyanofumarate and 2,3-dicyanomaleate have been studied with DFT computations. The first C−C bond is formed via an anti attack to produce a very polar, zwitterionic diradical. The low stereoselectivity of the reaction was found to arise from rotations about single bonds in the intermediates that compete with cyclization. A distortion−interaction energy analysis was performed to compare the stepwise and concerted mechanisms, and to explain why the stepwise mechanism is favored in this unusual case.
Co-reporter:Joann M. Um ; Osvaldo Gutierrez ; Franziska Schoenebeck ; K. N. Houk ;David W. C. MacMillan
Journal of the American Chemical Society 2010 Volume 132(Issue 17) pp:6001-6005
Publication Date(Web):April 13, 2010
DOI:10.1021/ja9063074
The intramolecular α-arylation of aldehydes via organo-SOMO catalysis was investigated using density functional theory (B3LYP and M06-2X functionals). The geometries, spin densities, Mulliken charges, and molecular orbitals of the reacting enamine radical cations were analyzed, and the nature of the resulting cyclized radical cation intermediates was characterized. In agreement with experimental observations, the calculated 1,3-disubstituted aromatic system shows ortho selectivity, while the 1,3,4-trisubstituted systems show para, meta (instead of ortho, meta) selectivity. The selectivity change for the trisubstituted rings is attributed to a distortion of the ortho substituents in the ortho, meta cyclization transition structures that causes a destabilization of these isomers and therefore results in selectivity for the para, meta product.
Co-reporter:Robert S. Paton ; Joel L. Mackey ; Woo Han Kim ; Jun Hee Lee ; Samuel J. Danishefsky ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 27) pp:9335-9340
Publication Date(Web):June 17, 2010
DOI:10.1021/ja1009162
The regioselectivity and stereoselectivity aspects of the Diels−Alder/radical hydrodenitration reaction sequence leading to trans-fused ring systems have been investigated with density functional calculations. A continuum of transition structures representing Diels−Alder and hetero-Diels−Alder cycloadditions as well as a sigmatropic rearrangement have been located, and they all lie very close in energy on the potential energy surface. All three pathways are found to be important in the formation of the Diels−Alder adduct. Reported regioselectivities are reproduced by the calculations. The stereoselectivity of radical hydrodenitration of the cis-Diels−Alder adduct is found to be related to the relative conformational stabilities of bicyclic radical intermediates. Overall, the computations provide understanding of the regioselectivities and stereoselectivities of the trans-Diels−Alder paradigm.
Co-reporter:Gavin O. Jones ; Peng Liu ; K. N. Houk ;Stephen L. Buchwald
Journal of the American Chemical Society 2010 Volume 132(Issue 17) pp:6205-6213
Publication Date(Web):April 13, 2010
DOI:10.1021/ja100739h
Computational investigations of ligand-directed selectivities in Ullmann-type coupling reactions of methanol and methylamine with iodobenzene by β-diketone- and 1,10-phenanthroline-ligated CuI complexes are reported. Density functional theory calculations using several functionals were performed on both the nucleophile formation and aryl halide activation steps of these reactions. The origin of ligand-directed selectivities in N- versus O-arylation reactions as described in a previous publication (J. Am. Chem. Soc. 2007, 129, 3490−3491) were studied and explained. The selectivities observed experimentally are derived not from initial CuI(nucleophile) complex formation but from the subsequent steps involving aryl halide activation. The arylation may occur via single-electron transfer (SET) or iodine atom transfer (IAT), depending on the electron-donating abilities of the ligand and nucleophile. Mechanisms involving either oxidative addition/reductive elimination or σ-bond metathesis are disfavored. SET mechanisms are favored in reactions promoted by the β-diketone ligand; N-arylation is predicted to be favored in these cases, in agreement with experimental results. The phenanthroline ligand promotes O-arylation reactions via IAT mechanisms in preference to N-arylation reactions, which occur via SET mechanisms; this result is also in agreement with experimental results.
Co-reporter:Franziska Schoenebeck ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2496-2497
Publication Date(Web):February 1, 2010
DOI:10.1021/ja9077528
The reversal of regioselectivity in a Pd-catalyzed cross-coupling reaction of an aryl chloro triflate with different ligands, established by Fu and co-workers, has been studied computationally. C−O insertion was found to be controlled by Pd-HOMO ArO−LUMO interaction, where C−Cl insertion is facilitated by the lower C−Cl TS distortion energy. The greater reactivities of monoligated Pd species are due primarily to the lack of distortion of the Pd species.
Co-reporter:Lai Xu ; Charles E. Doubleday ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:3029-3037
Publication Date(Web):February 11, 2010
DOI:10.1021/ja909372f
The dynamics of 1,3-dipolar cycloadditions of nine 1,3-dipoles with ethylene and acetylene have been explored by quasiclassical trajectory and single trajectory calculations in the retro-cycloaddition direction to compute energy partitioning of reactants among relative translation, vibration, and rotation. The results are interpreted with an expanded version of Polanyi’s Rules for bimolecular reactions, and three trends are evident. (1) Relative translation of reactants is the main contributor to surmounting the barrier, since all transition states (TSs) are early with respect to σ bond formation. (2) Vibrational excitation in the 1,3-dipole bending modes required for reaction is related to the lateness of the TS with respect to dipole bending: diazonium betaines (late TS, dipole bending required) > nitrilium betaines > azomethine betaines (early TS, dipole bending least important). This is also the order of the activation barriers (high → low). (3) The previously reported linear correlation between activation barriers and the energy required to distort reactants to their TS geometries are understandable in terms of the requirements for vibrational excitation computed here. For the 1,3-dipolar cycloadditions, single trajectory calculations, which contain no zero point vibrational energy, give reasonable estimates of the mean energy partitioning of reactants derived from potential energy barrier release. The timing of bond formation and relative reactivities of different 1,3-dipoles are discussed.
Co-reporter:Steven E. Wheeler ; Anne J. McNeil ; Peter Müller ; Timothy M. Swager ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 10) pp:3304-3311
Publication Date(Web):February 16, 2010
DOI:10.1021/ja903653j
Stereoselective Diels−Alder cycloadditions that probe substituent effects in aryl−aryl sandwich complexes were studied experimentally and theoretically. Computations on model systems demonstrate that the stereoselectivity in these reactions is mediated by differential π-stacking interactions in competing transition states. This allows relative stacking free energies of substituted and unsubstituted sandwich complexes to be derived from measured product distributions. In contrast to gas-phase computations, dispersion effects do not appear to play a significant role in the substituent effects, in accord with previous experiments. The experimental π-stacking free energies are shown to correlate well with Hammett σm constants (r = 0.96). These substituent constants primarily provide a measure of the inductive electron-donating and -withdrawing character of the substituents, not donation into or out of the benzene π-system. The present experimental results are most readily explained using a recently proposed model of substituent effects in the benzene sandwich dimer in which the π-system of the substituted benzene is relatively unimportant and substituent effects arise from direct through-space interactions. Specifically, these results are the first experiments to clearly show that OMe enhances these π-stacking interactions, despite being a π-electron donor. This is in conflict with popular models in which substituent effects in aryl−aryl interactions are modulated by polarization of the aryl π-system.
Co-reporter:Peng Liu ; Patrick McCarren ; Paul Ha-Yeon Cheong ; Timothy F. Jamison ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 6) pp:2050-2057
Publication Date(Web):January 22, 2010
DOI:10.1021/ja909562y
The origins of reactivity and regioselectivity in nickel-catalyzed reductive coupling reactions of alkynes and aldehydes were investigated with density functional calculations. The regioselectivities of reactions of simple alkynes are controlled by steric effects, while conjugated enynes and diynes are predicted to have increased reactivity and very high regioselectivities, placing alkenyl or alkynyl groups distal to the forming C−C bond. The reactions of enynes and diynes involve 1,4-attack of the Ni−carbonyl complex on the conjugated enyne or diyne. The consequences of these conclusions on reaction design are discussed.
Co-reporter:Peng Liu ; Lauren E. Sirois ; Paul Ha-Yeon Cheong ; Zhi-Xiang Yu ; Ingo V. Hartung ; Heiko Rieck ◻; Paul A. Wender ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10127-10135
Publication Date(Web):June 30, 2010
DOI:10.1021/ja103253d
The first studies on the regioselectivity of Rh(I)-catalyzed (5 + 2) cycloadditions between vinylcyclopropanes (VCPs) and alkynes have been conducted experimentally and analyzed using density functional theory (DFT). The previously unexplored regiochemical consequences for this catalytic, intermolecular cycloaddition were determined by studying the reactions of several substituted VCPs with a range of unsymmetrical alkynes. Experimental trends were identified, and a predictive model was established. VCPs with terminal substitution on the alkene reacted with high regioselectivity (>20:1), as predicted by a theoretical model in which bulkier alkyne substituents prefer to be distal to the forming C−C bond to avoid steric repulsions. VCPs with substitution at the internal position of the alkene reacted with variable regioselectivity (ranging from >20:1 to a reversed 1:2.3), suggesting a refined model in which electron-withdrawing substituents on the alkyne decrease or reverse sterically controlled selectivity by stabilizing the transition state in which the substituent is proximal to the forming C−C bond.
Co-reporter:Elizabeth H. Krenske, K. N. Houk, Andrew G. Lohse, Jennifer E. Antoline and Richard P. Hsung
Chemical Science 2010 vol. 1(Issue 3) pp:387-392
Publication Date(Web):25 Jun 2010
DOI:10.1039/C0SC00280A
Chiral oxazolidinones were previously thought to control cycloaddition stereoselectivity by steric crowding of one face of the substrate. We have discovered that in (4 + 3) cycloaddition reactions of oxyallyls, the stereoinduction is caused instead by stabilising CH–π interactions that lead to reaction at the more crowded face of the oxyallyl. Density functional theory calculations on the (4 + 3) cycloadditions of oxazolidinone-substituted oxyallyls with furans establish unexpected transition state conformations and a new explanation of selectivity.
Co-reporter:Elizabeth H. Krenske, K. N. Houk and Michael Harmata
Organic Letters 2010 Volume 12(Issue 3) pp:444-447
Publication Date(Web):January 11, 2010
DOI:10.1021/ol902591k
The mechanisms and stereoselectivities of (4 + 3) cycloadditions between chiral alkoxy siloxyallyl cations and furan are examined using density functional theory calculations. These cycloadditions are predicted to take place via stepwise mechanisms. The stereoselectivities of cycloadditions involving siloxyallyl cations derived from chiral α-methyl benzylic alcohols are controlled by two effects: minimization of steric repulsion between the α-Me group and the allyl group and attractive CH−π interactions between the furan and the aryl group.
Co-reporter:Andrew G. Lohse, Elizabeth H. Krenske, Jennifer E. Antoline, K. N. Houk, and Richard P. Hsung
Organic Letters 2010 Volume 12(Issue 23) pp:5506-5509
Publication Date(Web):November 4, 2010
DOI:10.1021/ol1023745
The (4 + 3) cycloadditions of oxazolidinone-substituted oxyallyls and unsymmetrically substituted furans lead to syn regioselectivity when the furan has a 2-Me or 2-COOR substituent, while anti regioselectivity is obtained with a 3-Me or 3-COOR group. DFT calculations are performed to explain the selectivities. The reactivities and regioselectivities are consistent with the ambiphilic reactivity of amino-oxyallyls with furans.
Co-reporter:KendallN. Houk
Helvetica Chimica Acta 2010 Volume 93( Issue 7) pp:1241-1260
Publication Date(Web):
DOI:10.1002/hlca.201000209
Co-reporter:Christophe Allemann, Joann M. Um, K.N. Houk
Journal of Molecular Catalysis A: Chemical 2010 324(1–2) pp: 31-38
Publication Date(Web):
DOI:10.1016/j.molcata.2010.03.020
Co-reporter:Dr. Le-Ping Liu;Deepika Malhotra;Dr. Robert S. Paton;Dr. K. N. Houk;Dr. Gerald B. Hammond
Angewandte Chemie International Edition 2010 Volume 49( Issue 48) pp:9132-9135
Publication Date(Web):
DOI:10.1002/anie.201005514
Co-reporter:JoannM. Um;NaeemS. Kaka Dr.;DavidM. Hodgson ;K.N. Houk
Chemistry - A European Journal 2010 Volume 16( Issue 21) pp:6310-6316
Publication Date(Web):
DOI:10.1002/chem.201000046
Abstract
The asymmetric C-alkylation of chiral enamines derived from terminal epoxides and lithium 2,2,6-trialkylpiperidides has previously been shown to provide α-alkylated aldehydes by intermolecular nucleophilic substitution in good levels of asymmetric induction. We now report a computational study of the origins of asymmetric induction in these reactions. Computational modeling with density functional theory (B3LYP/6-31G(d)) agrees closely with the experimental observations. This stereoselectivity is attributed to a preferential conformation of the enamine and the piperidine ring that places the C-6 alkyl substituent in an axial position due to A1, 3 strain. Preferential attack occurs away from the axial group, for steric reasons. The effects of changing the C-6 substituent from methyl to isopropyl were studied, and twist transition states were found to contribute significantly in the latter alkylations.
Co-reporter:Steven E. Wheeler and K. N. Houk
The Journal of Physical Chemistry A 2010 Volume 114(Issue 33) pp:8658-8664
Publication Date(Web):April 30, 2010
DOI:10.1021/jp1010549
Substituent effects in Cl−···C6H6−nXn complexes, models for anion/π interactions, have been examined using density functional theory and robust ab initio methods paired with large basis sets. Predicted interaction energies for 83 model Cl−···C6H6−nXn complexes span almost 40 kcal mol−1 and show an excellent correlation (r = 0.99) with computed electrostatic potentials. In contrast to prevailing models of anion/π interactions, which rely on substituent-induced changes in the aryl π-system, it is shown that substituent effects in these systems are due mostly to direct interactions between the anion and the substituents. Specifically, interaction energies for Cl−···C6H6−nXn complexes are recovered using a model system in which the substituents are isolated from the aromatic ring and π-resonance effects are impossible. Additionally, accurate potential energy curves for Cl− interacting with prototypical anion-binding arenes can be qualitatively reproduced by adding a classical charge−dipole interaction to the Cl−···C6H6 interaction potential. In substituted benzenes, binding of anions arises primarily from interactions of the anion with the local dipoles induced by the substituents, not changes in the interaction with the aromatic ring itself. When designing anion-binding motifs, phenyl rings should be viewed as a scaffold upon which appropriate substituents can be placed, because there are no attractive interactions between anions and the aryl π-system of substituted benzenes.
Co-reporter:Roger C. Helgeson, Amy E. Hayden and K. N. Houk
The Journal of Organic Chemistry 2010 Volume 75(Issue 3) pp:570-575
Publication Date(Web):December 29, 2009
DOI:10.1021/jo9012496
Introduction of a disulfide unit into the linker of a hemicarcerand creates a new way to control the entry and exit of guests. When the disulfide bond is reduced to two thiols, the “gate” opens, and guests can freely enter the hydrophobic core of the hemicarcerand. However, when the gate is closed, the host must be heated in the presense of excess guest in order for complexation to result. Several novel hemicarceplexes of this type have been synthesized. Molecular mechanics calculations are employed to explore the differing stabilities and ease of complexation of these host−guest complexes.
Co-reporter:Michael E. Jung, Ting-Hu Zhang, Rebecca M. Lui, Osvaldo Gutierrez, and K. N. Houk
The Journal of Organic Chemistry 2010 Volume 75(Issue 20) pp:6933-6940
Publication Date(Web):September 15, 2010
DOI:10.1021/jo101533h
While thermolysis of the macrobicyclic triene lactone 12 did not produce the expected bicyclic transannular Diels−Alder (BTADA) product 13, heating the corresponding ether 18 to 110 °C for 4 h afforded a quantitative yield of the desired cycloadduct 19, which could be easily reduced to the perhydrophenanthrene, an ABC ring analogue of fusidic acid 1. Theoretical calculations with hybrid density functional theory (B3LYP/6-31G(d)) help rationalize why the lactone does not cyclize whereas the ether does.
Co-reporter:Elizabeth H. Krenske, K. N. Houk, Daniel Lim, Sarah E. Wengryniuk, and Don M. Coltart
The Journal of Organic Chemistry 2010 Volume 75(Issue 24) pp:8578-8584
Publication Date(Web):November 11, 2010
DOI:10.1021/jo1019877
Density functional theory calculations and experiment reveal the origin of stereoselectivity in the deprotonation−alkylation of chiral N-amino cyclic carbamate (ACC) hydrazones. When the ACC is a rigid, camphor-derived carbamate, the two conformations of the azaenolate intermediate differ in energy due to conformational effects within the oxazolidinone ring and steric interactions between the ACC and the azaenolate. An electrophile adds selectively to the less-hindered π-face of the azaenolate. Although it was earlier reported that use of ACC auxiliaries led to α-alkylated ketones with er values of 82:18 to 98:2, B3LYP calculations predict higher stereoselectivity. Direct measurement of the dr of an alkylated hydrazone prior to removal of the auxiliary confirms this prediction; the removal of the auxiliary under the reported conditions can compromise the overall stereoselectivity of the process.
Co-reporter:Amy E. Hayden, Robert S. Paton, Jochen Becker, Yee Hwee Lim, K. C. Nicolaou and K. N. Houk
The Journal of Organic Chemistry 2010 Volume 75(Issue 3) pp:922-928
Publication Date(Web):December 22, 2009
DOI:10.1021/jo902572y
The regioselectivities of several Diels−Alder reactions utilized en route to bisanthraquinone antibiotic BE-43472B are examined using density functional theory calculations. These reactions involve highly substituted dienes and juglone dienophiles, and there is an opposite regiochemical outcome for Diels−Alder reactions with β-aryl substituted juglones when compared to reactions of unsubstituted juglone. In this article, the effect of an aromatic conjugating group bonded to juglone is explored.
Co-reporter:Şeref Gül, Franziska Schoenebeck, Viktorya Aviyente and K. N. Houk
The Journal of Organic Chemistry 2010 Volume 75(Issue 6) pp:2115-2118
Publication Date(Web):February 18, 2010
DOI:10.1021/jo100033d
The origins of the boat transition state preference in the Ireland−Claisen rearrangements studied experimentally by Kishi and co-workers have been explored computationally with Density Functional Theory. Steric interactions in the chair transition states were identified as the principal reason for the boat transition state preference.
Co-reporter:Joann M. Um ; Huadong Xu ; K. N. Houk ;Weiping Tang
Journal of the American Chemical Society 2009 Volume 131(Issue 19) pp:6664-6665
Publication Date(Web):April 29, 2009
DOI:10.1021/ja9016446
The thermal ring-opening reactions of 4-phenyl-1,3,3-triethoxycarbonylcyclobutene and 4-methyl-1,3,3-triethoxycarbonylcyclobutene yield dienes that result from an unexpected selectivity for “inward” rotation of the phenyl and methyl groups. With 1-ethoxycarbonyl-4-phenylcyclobutene, “outward” rotation of the phenyl group occurs exclusively. Density functional theory was used to investigate the role of the 3,3-geminal diester groups and the origin of torquoselectivity in these electrocyclic reactions. The rules of torquoselectivity still hold, with a calculated 6−8 kcal/mol preference for outward rotation of the methyl and phenyl substituents. However, cyclization of the “out” dienes to pyran intermediates allows for isomerization and thermodynamic control of stereoselectivity.
Co-reporter:P. R. McCarren ; Peng Liu ; Paul Ha-Yeon Cheong ; Timothy F. Jamison ;K. N. Houk
Journal of the American Chemical Society 2009 Volume 131(Issue 19) pp:6654-6655
Publication Date(Web):April 28, 2009
DOI:10.1021/ja900701g
The mechanism of nickel-catalyzed reductive alkyne−aldehyde coupling reactions has been investigated using density functional theory. The preferred mechanism involves oxidative cyclization to form the nickeladihydrofuran intermediate followed by transmetalation and reductive elimination. The rate- and selectivity-determining oxidative cyclization transition state is analyzed in detail. The d → π*⊥ back-donation stabilizes the transition state and leads to higher reactivity for alkynes than alkenes. Strong Lewis acids accelerate the couplings with both alkynes and alkenes by coordinating with the aldehyde oxygen in the transition state.
Co-reporter:Franziska Schoenebeck ; Daniel H. Ess ; Gavin O. Jones ;K. N. Houk
Journal of the American Chemical Society 2009 Volume 131(Issue 23) pp:8121-8133
Publication Date(Web):May 21, 2009
DOI:10.1021/ja9003624
The transition states and activation barriers of the 1,3-dipolar cycloadditions of azides with cycloalkynes and cycloalkenes were explored using B3LYP density functional theory (DFT) and spin component scaled SCS-MP2 methods. A survey of benzyl azide cycloadditions to substituted cyclooctynes (OMe, Cl, F, CN) showed that fluorine substitution has the most dramatic effect on reactivity. Azide cycloadditions to 3-substituted cyclooctynes prefer 1,5-addition regiochemistry in the gas phase, but CPCM solvation abolishes the regioselectivity preference, in accord with experiments in solution. The activation energies for phenyl azide addition to cycloalkynes decrease considerably as the ring size is decreased (cyclononyne ΔG⧧ = 29.2 kcal/mol, cyclohexyne ΔG⧧ = 14.1 kcal/mol). The origin of this trend is explained by the distortion/interaction model. Cycloalkynes are predicted to be significantly more reactive dipolarophiles than cycloalkenes. The activation barriers for the cycloadditions of phenyl azide and picryl azide (2,4,6-trinitrophenyl azide) to five- through nine-membered cycloalkenes were also studied and compared to experiment. Picryl azide has considerably lower activation barriers than phenyl azide. Dissection of the transition state energies into distortion and interaction energies revealed that “strain-promoted” cycloalkyne and cycloalkene cycloaddition transition states must still pay an energetic penalty to achieve their transition state geometries, and the differences in reactivity are more closely related to differences in distortion energies than the amount of strain released in the product. Trans-cycloalkene dipolarophiles have much lower barriers than cis-cycloalkenes.
Co-reporter:Osvaldo Gutierrez, Robert G. Iafe and K. N. Houk
Organic Letters 2009 Volume 11(Issue 19) pp:4298-4301
Publication Date(Web):September 1, 2009
DOI:10.1021/ol901586t
The organocatalytic transfer hydrogenation reactions of 3-phenyl-2-cyclopentenone with imidazolidinone catalysts are evaluated using the hybrid density functional (B3LYP/6-31G(d)) theory. The origin of the preference for the (E) iminium transition state is determined, and the stereoselectivity of hydride transfer is investigated.
Co-reporter:Adam J. T. Smith ; Xiyun Zhang ; Andrew G. Leach ;K. N. Houk
Journal of Medicinal Chemistry 2009 Volume 52(Issue 2) pp:225-233
Publication Date(Web):November 21, 2008
DOI:10.1021/jm800498e
Co-reporter:Steven E. Wheeler and K. N. Houk
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 9) pp:2301-2312
Publication Date(Web):July 28, 2009
DOI:10.1021/ct900344g
Model systems have been studied using density functional theory to assess the contributions of π-resonance and through-space effects on electrostatic potentials (ESPs) of substituted arenes. The results contradict the widespread assumption that changes in molecular ESPs reflect only local changes in the electron density. Substituent effects on the ESP above the molecular plane are commonly attributed to changes in the aryl π-system. We show that ESP changes for a collection of substituted benzenes and more complex aromatic systems can be accounted for mostly by through-space effects, with no change in the aryl π-electron density. Only when π-resonance effects are substantial do they influence changes to any extent in the ESP above the aromatic ring. Examples of substituted arenes studied here are taken from the fields of drug design, host−guest chemistry, and crystal engineering. These findings emphasize the potential pitfalls of assuming ESP changes reflect changes in the local electron density. Since ESP changes are frequently used to rationalize and predict intermolecular interactions, these findings have profound implications for our understanding of substituent effects in countless areas of chemistry and molecular biology. Specifically, in many noncovalent interactions there are significant, often neglected, through-space interactions with the substituents. Finally, the present results explain the good performance of many molecular mechanics force-fields when applied to supramolecular assembly phenomena, despite the neglect of the polarization of the aryl π-system by substituents.
Co-reporter:Adam J. T. Smith, Ying Li and K. N. Houk
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 13) pp:2716-2724
Publication Date(Web):06 May 2009
DOI:10.1039/B901429B
The mechanisms of enzyme-catalyzed phosphate transfer and hydrolysis reactions are of great interest due to their importance and abundance in biochemistry. The reaction may proceed in a stepwise fashion, with either a pentavalent phosphorane or a metaphosphate anion intermediate, or by a concerted SN2 mechanism. Despite much theoretical work focused on a few key enzymes, a consensus for the mechanism has not been reached, and examples of all three possibilities have been demonstrated. We have investigated the mechanism of human uridine-cytidine kinase 2 (UCK2, EC 2.7.1.48), which catalyzes the transfer of a phosphate group from ATP to the ribose 5′-hydroxyl of cytidine and uridine. UCK2 is normally expressed in human placenta, but is overexpressed in certain cancer cells, where it is responsible for activating a class of antitumor prodrugs. The UCK2 mechanism was investigated by generating a 2D potential energy surface as a function of the P–O bonds forming and breaking, with energies calculated using a quantum mechanics/molecular mechanics potential (B3LYP/6-31G(d):AMBER). The mechanism of phosphate transfer is shown to be concerted, and is accompanied by concerted proton transfer from the 5′-hydroxyl to a conserved active site aspartic acid that serves as a catalytic base. The calculated barrier for this reaction is 15.1 kcal/mol, in relatively good agreement with the experimental barrier of 17.5 kcal/mol. The interactions of the enzyme active site with the reactant, transition state, and product are examined for their implications on the design of anticancer prodrugs or positron emission tomography (PET) reporter probes for this enzyme.
Co-reporter:Hakan Gunaydin and K. N. Houk
Chemical Research in Toxicology 2009 Volume 22(Issue 5) pp:894
Publication Date(Web):April 17, 2009
DOI:10.1021/tx800463y
The mechanisms of tyrosine nitration by peroxynitrous acid or nitrosoperoxycarbonate were investigated with the CBS-QB3 method. Either the protonation of peroxynitrite or a reaction with carbon dioxide gives a reactive peroxide intermediate. Peroxynitrous acid-mediated nitration of phenol occurs via unimolecular decomposition to give nitrogen dioxide and hydroxyl radicals. Nitrosoperoxycarbonate also undergoes unimolecular decomposition to give carbonate and nitrogen dioxide radicals. The reactions of tyrosine with the hydroxyl or carbonate radicals give a phenoxy radical intermediate. The reaction of the nitrogen dioxide with this radical intermediate followed by tautomerization gives nitrated tyrosine in both cases. According to CBS-QB3 calculations, the rate-limiting step for the nitration of phenol is the decomposition of peroxynitrous acid or nitrosoperoxycarbonate.
Co-reporter:Steven E. Wheeler, Antonio Moran, Susan N. Pieniazek and K. N. Houk
The Journal of Physical Chemistry A 2009 Volume 113(Issue 38) pp:10376-10384
Publication Date(Web):August 27, 2009
DOI:10.1021/jp9058565
Enthalpies for bond-forming reactions that are subject to organocatalysis have been predicted using the high-accuracy CBS-QB3 model chemistry and six DFT functionals. Reaction enthalpies were decomposed into contributions from changes in bonding and other intramolecular effects via the hierarchy of homodesmotic reactions. The order of the reaction exothermicities (aldol < Mannich ≈ α-aminoxylation) arises primarily from changes in formal bond types mediated by contributions from secondary intramolecular interactions. In each of these reaction types, methyl substitution at the β- and γ-positions stabilizes the products relative to the unsubstituted case. The performance of six DFT functionals (B3LYP, B3PW91, B1B95, MPW1PW91, PBE1PBE, and M06-2X), MP2, and SCS-MP2 has been assessed for the prediction of these reaction enthalpies. Even though the PBE1PBE and M06-2X functionals perform well for the aldol and Mannich reactions, errors roughly double when these functionals are applied to the α-aminoxylation reactions. B3PW91 and B1B95, which offer modest accuracy for the aldol and Mannich reactions, yield reliable predictions for the two α-aminoxylation reactions. The excellent performance of the M06-2X and PBE1PBE functionals for aldol and Mannich reactions stems from the cancellation of sizable errors arising from inadequate descriptions of the underlying bond transformations and intramolecular interactions. SCS-MP2/cc-pVTZ performs most consistently across these three classes of reactions, although the reaction exothermicities are systematically underestimated by 1−3 kcal mol−1. Conventional MP2, when paired with the cc-pVTZ basis set, performs somewhat better than SCS-MP2 for some of these reactions, particularly the α-aminoxylations. Finally, the merits of benchmarking DFT functionals for the set of simple chemically meaningful transformations underlying all bond-forming reactions are discussed.
Co-reporter:Lai Xu;CharlesE. Doubleday Dr.;K.N. Houk Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 15) pp:2746-2748
Publication Date(Web):
DOI:10.1002/anie.200805906
Co-reporter:Nihan Çelebi-Ölçüm, Viktorya Aviyente and K. N. Houk
The Journal of Organic Chemistry 2009 Volume 74(Issue 18) pp:6944-6952
Publication Date(Web):August 18, 2009
DOI:10.1021/jo901109s
Density functional theory calculations were used to investigate the [3,3]- and [1,3]-shifts of O-allylic trichloroacetimidates in the presence of cinchona alkaloids. Thermal [1,3]- and [3,3]-rearrangements proceed through concerted pseudopericyclic transition states to give the corresponding rearranged products. [1,3]-Rearrangement is catalyzed via a double SN2′ mechanism in which syn addition of the nucleophile is exclusively preferred in both steps. The catalyzed mechanism is favored by a 6.3 kcal/mol free energy difference compared to the alternative [3,3]-rearrangement pathway. The fast-reacting enantiomer is predicted to be determined by the availability of the H-bonding interaction between the catalyst and the substrate.
Co-reporter:Sílvia Osuna;KendallN. Houk
Chemistry - A European Journal 2009 Volume 15( Issue 47) pp:13219-13231
Publication Date(Web):
DOI:10.1002/chem.200901761
Abstract
Diels–Alder cycloadditions of butadiene and 1,3-dipolar cycloadditions of azomethine ylide, fulminic acid, and the parent nitrone to polyacenes, fullerenes, and nanotubes have been investigated with density functional theory and ONIOM methods. Activation barriers obtained for cycloaddition reactions on planar and curved systems have been shown to be highly correlated to the energy needed to distort the reactants to the geometry of the transition state (TS).
Co-reporter:Courtney L. Stanton and Kendall N. Houk
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 6) pp:951-966
Publication Date(Web):May 9, 2008
DOI:10.1021/ct8000014
Methods for estimation of pKa values of residues in proteins were tested on a set of benchmark proteins with experimentally known pKa values. The benchmark set includes 80 different residues (20 each for Asp, Glu, Lys, and His), half of which consists of significantly variant cases (ΔpKa ≥ 1 pKa unit from the amino acid in solution). The method introduced by Case and co-workers [J. Am. Chem. Soc. 2004, 126, 4167−4180], referred to as the molecular dynamics/generalized-Born/thermodynamic integration (MD/GB/TI) technique, gives a root-mean-square deviation (rmsd) of 1.4 pKa units on the benchmark set. The use of explicit waters in the immediate region surrounding the residue was shown to generally reduce high errors for this method. Longer simulation time was also shown to increase the accuracy of this method. The empirical approach developed by Jensen and co-workers [Proteins 2005, 61, 704−721], PROPKA, also gives an overall rmsd of 1.4 pKa units and is more or less accurate based on residue type—the method does very well for Lys and Glu, but less so for Asp and His. Likewise, the absolute deviation is quite similar for the two methods—5.2 for PROPKA and 5.1 for MD/GB/TI. A comparison of these results with several prediction methods from the literature is presented. The error in pKa prediction is analyzed as a function of variation of the pKa from that in water and the solvent accessible surface area (SASA) of the residue. A case study of the catalytic lysine residue in 2-deoxyribose-5-phosphate aldolase (DERA) is also presented.
Co-reporter:Peng Liu;PaulHa-Yeon Cheong Dr.;Zhi-Xiang Yu Dr.;PaulA. Wender Dr.;KendallN. Houk Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 21) pp:3939-3941
Publication Date(Web):
DOI:10.1002/anie.200800420
Co-reporter:PhilipJ. Pye Dr.;Yong-Li Zhong Dr.;GavinO. Jones Dr.;RobertA. Reamer;KendallN. Houk ;David Askin Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 22) pp:4134-4136
Publication Date(Web):
DOI:10.1002/anie.200703681
Co-reporter:DanielH. Ess Dr.;StevenE. Wheeler Dr.;RobertG. Iafe;Lai Xu;Nihan Çelebi-Ölçüm ;KendallN. Houk
Angewandte Chemie International Edition 2008 Volume 47( Issue 40) pp:7592-7601
Publication Date(Web):
DOI:10.1002/anie.200800918
Abstract
A single transition state may lead to multiple intermediates or products if there is a post-transition-state reaction pathway bifurcation. These bifurcations arise when there are sequential transition states with no intervening energy minimum. For such systems, the shape of the potential energy surface and dynamic effects, rather than transition-state energetics, control selectivity. This Minireview covers recent investigations of organic reactions exhibiting reaction pathway bifurcations. Such phenomena are surprisingly general and affect experimental observables such as kinetic isotope effects and product distributions.
Co-reporter:Peng Liu;PaulHa-Yeon Cheong Dr.;Zhi-Xiang Yu Dr.;PaulA. Wender Dr.;KendallN. Houk Dr.
Angewandte Chemie 2008 Volume 120( Issue 21) pp:4003-4005
Publication Date(Web):
DOI:10.1002/ange.200800420
Co-reporter:SusanN. Pieniazek Dr.;FernoR. Clemente Dr. ;KendallN. Houk Dr.
Angewandte Chemie 2008 Volume 120( Issue 40) pp:7860-7863
Publication Date(Web):
DOI:10.1002/ange.200801843
Co-reporter:DanielH. Ess Dr.;StevenE. Wheeler Dr.;RobertG. Iafe;Lai Xu;Nihan Çelebi-Ölçüm ;KendallN. Houk
Angewandte Chemie 2008 Volume 120( Issue 40) pp:7704-7713
Publication Date(Web):
DOI:10.1002/ange.200800918
Abstract
Ein einziger Übergangszustand kann zu mehreren Zwischenstufen oder Produkten führen, wenn sich der Reaktionsweg nach dem Übergangszustand gabelt. Solche Gabelungen (Bifurkationen) entstehen beim Vorliegen sequenzieller Übergangszustände ohne dazwischenliegendem Energieminimum. Die Selektivität dieser Reaktionen wird nicht von der Energetik des Übergangszustands, sondern von der Form der Potentialenergiehyperfläche und von dynamischen Effekten gesteuert. In diesem Kurzaufsatz werden jüngste Untersuchungen von organischen Reaktionen mit Reaktionswegbifurkationen vorgestellt. Diese Bifurkationen sind überraschend häufig und wirken sich auf experimentell bestimmbare Größen wie kinetische Isotopeneffekte und Produktverteilungen aus.
Co-reporter:SusanN. Pieniazek Dr.;FernoR. Clemente Dr. ;KendallN. Houk Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 40) pp:7746-7749
Publication Date(Web):
DOI:10.1002/anie.200801843
Co-reporter:Daniel H. Ess;Gavin O. Jones;K. N. Houk
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 16-17) pp:
Publication Date(Web):27 NOV 2006
DOI:10.1002/adsc.200600431
The application and performance of conceptual and qualitative theories and quantitative quantum mechanical methods to the study of mechanism, reactivity, and selectivity of 1,3-dipolar and Diels–Alder cycloadditions are reviewed. This review emphasizes the application of conceptual density functional theory (DFT) for predicting reactivity and regioselectivity, and highly accurate quantum mechanical methods for predicting barrier heights and reaction energetics. Applications of computations to solvation effects, metal and organocatalysis, are also described.
Co-reporter:Susan N. Pieniazek Dr.
Angewandte Chemie 2006 Volume 118(Issue 9) pp:
Publication Date(Web):20 JAN 2006
DOI:10.1002/ange.200502677
Ursache und Allgemeingültigkeit des Halogeneffekts in Diels-Alder-Reaktionen wurden mit hoch präzisen CBS-QB3-Rechnungen untersucht. Ein Halogensubstituent erhöht die Exergonie einer Cycloaddition und senkt ihre Aktivierungsbarriere. Dieses Verhalten beruht darauf, dass die Bindung des elektronegativen Halogensubstituenten an ein höher alkyliertes, elektropositiveres Kohlenstoffgerüst energetisch begünstigt ist.
Co-reporter:Brian H. Northrop;Daniel P. O'Malley;Alexros L. Zografos Dr.;Phil S. Baran ;K. N. Houk
Angewandte Chemie International Edition 2006 Volume 45(Issue 25) pp:
Publication Date(Web):16 MAY 2006
DOI:10.1002/anie.200600514
Naturally radical: The microwave-induced rearrangement of sceptrin to natural products ageliferin and nagelamide E results in approximately the same ratio as they are isolated from natural sources. Computational investigations show that the vinylcyclobutane–cyclohexene rearrangement occurs via diradical intermediates and support the involvement of this rearrangement in the biosynthesis of the natural products.
Co-reporter:Brian H. Northrop;Daniel P. O'Malley;Alexros L. Zografos Dr.;Phil S. Baran ;K. N. Houk
Angewandte Chemie 2006 Volume 118(Issue 25) pp:
Publication Date(Web):16 MAY 2006
DOI:10.1002/ange.200600514
Natürlich radikal: Die Mikrowellen-induzierte Umlagerung von Sceptrin in die Naturstoffe Ageliferin und Nagelamid E liefert die Verbindungen etwa im gleichen Verhältnis, wie sie aus natürlichen Quellen isoliert werden. Nach Berechnungen verläuft die Vinylcyclobutan-Cyclohexen-Umlagerung über Diradikalintermediate, und es spricht alles dafür, dass diese Umlagerung auch an der Biosynthese der Naturstoffe beteiligt ist.
Co-reporter:Jennifer A.R. Luft, Zhi-Xiang Yu, David L. Hughes, Guy C. Lloyd-Jones, Shane W. Krska, Kendall N. Houk
Tetrahedron: Asymmetry 2006 Volume 17(Issue 4) pp:716-724
Publication Date(Web):20 February 2006
DOI:10.1016/j.tetasy.2006.01.037
Density functional theory calculations were performed on the apparent η3-π-allyl molybdenum intermediate that is observed during molybdenum-catalyzed asymmetric allylation [Proc. Natl. Acad. Sci. U.S.A.2004, 101, 5379]. The relative stabilities of geometric isomers, diastereoisomers, and π-allyl rotamers of the π-allyl molybdenum intermediate were investigated. Calculations show that the observed intermediate is the most stable because of two factors: (1) the observed π-allyl molybdenum intermediate maximizes the bonding and back-bonding interactions between molybdenum and the π-allyl ligand; and (2) the observed π-allyl molybdenum intermediate minimizes the steric interactions between the chiral ligand and the π-allyl group.
Co-reporter:Susan N. Pieniazek,Kendall N. Houk
Angewandte Chemie International Edition 2006 45(9) pp:1442-1445
Publication Date(Web):
DOI:10.1002/anie.200502677
Co-reporter:Daniel H. Ess;Gavin O. Jones;Kendall N. Houk
Helvetica Chimica Acta 2005 Volume 88(Issue 7) pp:1702-1710
Publication Date(Web):20 JUL 2005
DOI:10.1002/hlca.200590134
The reactions of hydrazoic acid (HN3) with ethene, acetylene, formaldimine (H2CNH), and HCN were explored with the high-accuracy CBS-QB3 method, as well as with the B3LYP and mPW1K density functionals. CBS-QB3 predicts that the activation energies for the reactions of hydrazoic acid with ethylene, acetylene, formaldimine, and HCN have remarkably similar activation enthalpies of 19.0, 19.0, 21.6, and 25.2 kcal/mol, respectively. The reactions are calculated to have reaction enthalpies of −21.5 for triazoline formation from ethene, and −63.7 kcal/mol for formation of the aromatic triazole from acetylene. The reaction to form tetrazoline from formaldimine has a reaction enthalpy of −8 kcal/mol (ΔGrxn=+5.6 kcal/mol), and the formation of tetrazole from HCN has a reaction enthalpy of −23.0 kcal/mol. The trends in the energetics of these processes are rationalized by differences in σ-bond energies in the transition states and adducts, and the energy required to distort hydrazoic acid to its transition-state geometry. The density functionals predict activation enthalpies that are in relatively good agreement with CBS-QB3, the results differing from CBS-QB3 results by ca. 1–2 kcal/mol. Significant errors are revealed for mPW1K in predicting the reaction enthalpies for all reactions.
Co-reporter:Xiyun Zhang Dr. Dr.;James L. Leighton Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 6) pp:
Publication Date(Web):28 DEC 2004
DOI:10.1002/anie.200462130
Explaining the experiment: The transition states for a concerted asymmetric allylation (see scheme) were studied by theoretical methods. The apical attack of the aldehyde oxygen atom on Si accompanied by minimization of electrostatic repulsion in the trigonal-bipyramidal transition state leads to high stereoselectivity.
Co-reporter:Christopher P. Suhrada;Cenk Selçuki Dr.;Maja Nendel Dr.;Carina Cannizzaro Dr.;K. N. Houk Dr.;Peter-Jürgen Rissing Dr.;Dirk Baumann Dr.;Dieter Hasselmann Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 23) pp:
Publication Date(Web):28 APR 2005
DOI:10.1002/anie.200500027
Deuterium labeling experiments on the rearrangement of methylenebicycloalkene 1 to 3 show stereoselectivity despite the intermediacy of a stabilized diradical structure 2. A theoretical model built from computational studies explains the observed product ratios. Stereoselectivity among [3,3] (minor) products implicates an unusual bifurcation of reaction trajectories after the initial bond-breaking transition state.
Co-reporter:Xiyun Zhang Dr. Dr.;James L. Leighton Dr.
Angewandte Chemie 2005 Volume 117(Issue 6) pp:
Publication Date(Web):28 DEC 2004
DOI:10.1002/ange.200462130
Das Experiment erklärt: Die Übergangszustände einer konzertierten asymmetrischen Allylierung (siehe Schema) wurden mit theoretischen Methoden untersucht. Der apikale Angriff des Aldehyd-Sauerstoffatoms auf Si, begleitet von einer Minimierung der elektrostatischen Abstoßung im trigonal-bipyramidalen Übergangszustand, führt zu hoher Stereoselektivität.
Co-reporter:Christopher P. Suhrada;Cenk Selçuki Dr.;Maja Nendel Dr.;Carina Cannizzaro Dr.;K. N. Houk Dr.;Peter-Jürgen Rissing Dr.;Dirk Baumann Dr.;Dieter Hasselmann Dr.
Angewandte Chemie 2005 Volume 117(Issue 23) pp:
Publication Date(Web):28 APR 2005
DOI:10.1002/ange.200500027
Deuterium-Markierungsexperimente belegen, dass das Methylenbicycloalken 1 stereoselektiv zu 3 umlagert, obwohl intermediär das stabilisierte Diradikal 2 auftritt. Ein theoretisches Modell auf der Grundlage von Rechnungen erklärt die beobachteten Produktverhältnisse. Die Stereoselektivität bei den [3,3]-(Neben-)Produkten legt eine ungewöhnliche Gabelung der Reaktionstrajektorien nach dem Übergangszustand der ersten Bindungsspaltung nahe.
Co-reporter:Ruth Gordillo;Jennifer Carter;K. N. Houk
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 9-10) pp:
Publication Date(Web):21 SEP 2004
DOI:10.1002/adsc.200404107
The transition structures for MacMillan's alkylations of N-methylpyrrole by aldehydes catalyzed by chiral amine salts were explored with B3LYP/6-31G(d) density functional theory. These results provide an explanation of the enantioselectivities observed with these two catalysts.
Co-reporter:Paul Ha-Yeon Cheong;K. N. Houk;Jayakumar S. Warrier;Stephen Hanessian
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 9-10) pp:
Publication Date(Web):21 SEP 2004
DOI:10.1002/adsc.200404127
Methanoprolines are found to be catalysts for the Hajos–Parrish–Eder–Sauer–Wiechert reaction.[1]cis-4,5-Methanoproline exhibits catalytic ability similar to proline (86% yield, 93% ee), whereas the trans-4,5-methanoproline is less selective (67% yield, 83% ee) and shows less acceleration. The reaction was also studied with hybrid density functional theory (B3LYP). The nearly planar cis-4,5-methanoproline amine better reflects the planar iminium of the transition states than the pyramidalized trans-4,5-methanoproline. This difference in conformation is responsible for the observed higher enantioselectivity and enhanced catalytic behavior of the cis-4,5-methanoproline.
Co-reporter:Avril R. Williams Dr.;Brian H. Northrop ;J. Fraser Stoddart ;David J. Williams
Chemistry - A European Journal 2004 Volume 10(Issue 21) pp:
Publication Date(Web):16 SEP 2004
DOI:10.1002/chem.200400221
Three constitutionally isomeric bis(naphthylmethyl)ammonium ions, in which the two naphthyl groups are substituted 1) both at their 1-positions, 2) one at its 1-position and the other at its 2-position, and 3) both at their 2-positions, have been investigated separately in solution for their propensities to undergo spontaneous self-assembly with three different [24]crown-8 derivatives, namely, pyrido[24]crown-8 (P24C8), dipyrido[24]crown-8 (DP24C8) and dibenzo[24]crown-8 (DB24C8), in turn to form [2]pseudorotaxanes. The strengths of the 1:1 complexes depend on the composition of the secondary dialkylammonium ions and on the nature of the crown ether hosts; generally, as far as the guest cation is concerned, the 1/1- and 2/2-isomers form stronger complexes, as indicated by stability constant measurements, than the 1/2-isomer and, as far as the crown ethers are concerned, the more flexible P24C8 is a much more efficient host than either DP24C8 or DB24C8. The rates of formation of the [2]pseudorotaxanes are fast (i.e., taking no more than a few minutes) in solution with the exception of one case, that is, in which the crown ether host is DB24C8 and the guest cation is the 1/1-isomer, when it can take upwards of one month for the complexation–decomplexation equilibrium to be established at room temperature. In all cases, the equilibrium between complexed and uncomplexed species is slow on the NMR timescale, allowing the determination of stability constants to be made readily using the single-point method. X-ray crystallography and molecular modeling have been used to gain insight into ground and transition state interactions, respectively, in some of the [2]pseudorotaxanes. The relative stabilities of the three [2]pseudorotaxanes formed by each guest cation in the presence of the three crown ether hosts were also evaluated in solution by competition experiments that were monitored by 1H NMR spectroscopy. By and large the results of the competition experiments could be predicted on the basis of the derived stability constants for the individual [2]pseudorotaxanes.
Co-reporter:Audrey Auffrant Dr.;Bernhard Jaun Dr.;Peter D. Jarowski Dr.;François Diederich Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 12) pp:
Publication Date(Web):26 APR 2004
DOI:10.1002/chem.200400218
A variety of 1,1,4,4-tetraal kynylbutatrienes and 1,4-dialkynylbutatrienes was synthezized by dimerization of the corresponding gem-dibromoolefins. Both 1H and 13C NMR spectroscopy indicated that the di- and tetraalkynylated butatrienes are formed as a mixture of cis and trans isomers. Variable temperature NMR studies evidenced a facile cis–trans isomerization, thus preventing the separation of these isomers by gravity or high-performance liquid chromatography (HPLC). For 1,1,4,4-tetraalkynylbutatrienes, the activation barrier ΔG≠ was measured by magnetization transfer to be around 20 kcal mol−1, in the range of the barrier for internal rotation about a peptide bond. Unlike the tetraalkynylated [3]cumulenes, 1,4-dialkynylbutatrienes are more difficult to isomerize and could, in one case, be obtained isomerically pure. Based on experimental data, the rotational barrier ΔG≠ for 1,4-dialkynylbutatrienes is estimated to be around 25 kcal mol−1. The hypothesis of a stabilizing effect of the four alkynyl substituents on the proposed but-2-yne-1,4-diyl singlet diradical transition state of this cis–trans isomerization is further supported by a computational study.
Co-reporter:Ferno R. Clemente Dr. and;K. N. Houk Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 43) pp:
Publication Date(Web):14 OCT 2004
DOI:10.1002/anie.200460916
A comparison of previously proposed models of the CC bond-forming step of the title reaction with density functional methods indicate that the most favored one involves an enamine intermediate undergoing a concerted aldol cyclization with proton transfer from the proline carboxylic acid group (see structure). This step is equal in energy to the intramolecular deprotonation leading to the enamine, and both are partially rate-determining steps.
Co-reporter:Ferno R. Clemente Dr. and;K. N. Houk Dr.
Angewandte Chemie 2004 Volume 116(Issue 43) pp:
Publication Date(Web):14 OCT 2004
DOI:10.1002/ange.200460916
Ein Vergleich von bekannten Modellen für den C-C-verknüpfenden Schritt der Titelreaktion mithilfe von Dichtefunktional-Methoden zeigt, dass der günstigste Weg eine Enamin-Zwischenstufe einschließt, deren Aldolcyclisierung unter Protonenübertragung von der Carboxygruppe des Prolins konzertiert verläuft (siehe Bild). Dieser Schritt und die intramolekulare Deprotonierung, die zum Enamin führt, sind energetisch ähnlich, sodass beide die Reaktionsgeschwindigkeit bestimmen.
Co-reporter:Travis Dudding
PNAS 2004 Volume 101 (Issue 16 ) pp:5770-5775
Publication Date(Web):2004-04-20
DOI:10.1073/pnas.0307256101
The catalytic asymmetric thiazolium- and triazolium-catalyzed benzoin condensations of aldehydes and ketones were studied
with computational methods. Transition-state geometries were optimized by using Morokuma's IMOMO [integrated MO (molecular
orbital) + MO method] variation of ONIOM (n-layered integrated molecular orbital method) with a combination of B3LYP/6–31G(d) and AM1 levels of theory, and final transition-state
energies were computed with single-point B3LYP/6–31G(d) calculations. Correlations between experiment and theory were found,
and the origins of stereoselection were identified. Thiazolium catalysts were predicted to be less selective then triazolium
catalysts, a trend also found experimentally.
Co-reporter:Andrew G. Leach and K. N. Houk
Organic & Biomolecular Chemistry 2003 vol. 1(Issue 8) pp:1389-1403
Publication Date(Web):26 Mar 2003
DOI:10.1039/B300285C
A theoretical study of the mechanisms of ene reactions of nitroso compounds has been completed, using UB3LYP, CASPT2, UCCSD(T) and UQCISD(T) methods. Stepwise paths through polarized diradical intermediates are always preferred. These intermediates have unusual properties, involving high rotational barriers about formally single bonds, which permit them to maintain stereochemical relationships. The diradicals may exchange the RNO moiety between the two ends of the alkene via an aziridine N-oxide. The aziridine N-oxide cannot be accessed directly from reactants and cannot lead directly to ene products. It is therefore an innocent by-stander in the way proposed by Singleton for the aziridinium imide in the ene reactions of triazolinediones. A detailed analysis of the electronic structure of the polarized diradicals is given. The kinetic isotope effects measured in a Stephenson isotope effect test have been reproduced. These kinetic isotope effects are consistent with a mechanism in which partitioning of the polarized diradical between cyclization to an aziridine N-oxide and H-abstraction to ene product takes place, and in which the formation of the polarized diradical is to some extent reversible. Finally, calculated regioselectivities reproduce those observed experimentally.
Co-reporter:Kendall N. Houk Dr.;Andrew G. Leach Dr.;Susanna P. Kim;Xiyun Zhang
Angewandte Chemie 2003 Volume 115(Issue 40) pp:
Publication Date(Web):15 OCT 2003
DOI:10.1002/ange.200200565
Die Affinitäten von Wirten – ausgehend von kleinen synthetischen Cavitanten bis hin zu großen Proteinen – für organische Moleküle sind gut dokumentiert. Die mittleren Assoziationskonstanten für die Bindung organischer Moleküle durch Cyclodextrine, synthetische Wirte und Albumine in Wasser sowie von katalytischen Antikörpern oder Enzymen für Substrate betragen im Allgemeinen 103.5±2.5 M−1. Die Bindungsaffinitäten steigen bei der Komplexierung von Übergangszuständen und biologischen Antigenen durch Antikörper oder von Enzymen durch Inhibitoren auf 108±2 M−1 und auf 1016±4 M−1 für Enzym-Übergangszustands-Komplexe. Die Gründe für die unterschiedlichen Stabilitäten dieser Wirt-Gast-Systeme sollen hier untersucht werden, und wir beschreiben Ansätze zur computergestützten Analyse der Wirt-Gast-Komplexbildung in Lösung. Bei vielen Komplexklassen besteht eine ungefähre Korrelation der Bindungsaffinität mit der Größe der Oberfläche, die bei der Komplexierung vergraben wird. Enzyme folgen dieser Korrelation nicht, sondern binden Übergangszustände sehr viel stärker als es anhand der Oberfläche zu erwarten wäre.
Co-reporter:K. N. Houk Dr.;Andrew G. Leach Dr.;Susanna P. Kim;Xiyun Zhang
Angewandte Chemie International Edition 2003 Volume 42(Issue 40) pp:
Publication Date(Web):15 OCT 2003
DOI:10.1002/anie.200200565
The affinities of hosts—ranging from small synthetic cavitands to large proteins—for organic molecules are well documented. The average association constants for the binding of organic molecules by cyclodextrins, synthetic hosts, and albumins in water, as well as of catalytic antibodies or enzymes for substrates are 103.5±2.5 M−1. Binding affinities are elevated to 108±2 M−1 for the complexation of transition states and biological antigens by antibodies or inhibitors by enzymes, and to 1016±4 M−1 for transition states with enzymes. The origins of the distributions of association constants observed for the broad range of host–guest systems are explored in this Review, and typical approaches to compute and analyze host–guest binding in solution are discussed. In many classes of complexes a rough correlation is found between the binding affinity and the surface area that is buried upon complexation. Enzymes transcend this effect and achieve transition-state binding much greater than is expected from the surface areas.
Co-reporter:Zhi-Xiang Yu Dr.;K N. Houk Dr.
Angewandte Chemie 2003 Volume 115(Issue 7) pp:
Publication Date(Web):17 FEB 2003
DOI:10.1002/ange.200390184
Denn nur dann kann eine relativ schnelle, durch agostische Wechselwirkung unterstützte Wasserstoffverschiebung die Produktbildung unterstützen. Das ist die Antwort auf die Frage im Titel, und sie folgt aus einer theoretischen Analyse des gezeigten Katalysezyklus sowie seiner Alternativen für die Dimerisierung, Tetramerisierung und höhere Oligomerisierung.
Co-reporter:Emanuel Vogel Dr.;Martin Michels Dr.;Lars Zer Dr.;Johann Lex Dr.;Nurcan S. Tuzun Dr.
Angewandte Chemie 2003 Volume 115(Issue 25) pp:
Publication Date(Web):24 JUN 2003
DOI:10.1002/ange.200250690
Die überraschende Bildung des ersten Spirodiporphyrins in Form eines Metallkomplexes, des Spirodicorrol-Dinickelkomplexes 1, ist ein Ergebnis der vielseitigen Chemie des „Figure-Eight“-Cyclooctapyrrols Octaphyrin-(1.1.1.0.1.1.1.0). Die Umwandlung des Cyclooctapyrrols in 1 beruht auf einer Folge von Oxidation und einer durch Nickel(II)-acetat ausgelösten Umlagerungskaskade. Hervorstechendes strukturelles Merkmal von 1 ist die Orthogonalität der über ein gemeinsames Spirozentrum verknüpften Nickelcorrolat-Chromophore.
Co-reporter:Emanuel Vogel Dr.;Martin Michels Dr.;Lars Zer Dr.;Johann Lex Dr.;Nurcan S. Tuzun Dr.
Angewandte Chemie 2003 Volume 115(Issue 25) pp:
Publication Date(Web):24 JUN 2003
DOI:10.1002/ange.200390534
Co-reporter:Zhi-Xiang Yu Dr.;K N. Houk Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 7) pp:
Publication Date(Web):17 FEB 2003
DOI:10.1002/anie.200390215
Because only then a relatively rapid agostic-assisted hydrogen shift can help to form the product. This is the answer to the question in the title and it results from a computational analysis of the catalytic cycle shown and its alternatives for dimerization, tetramerization, and higher oligomerization.
Co-reporter:Emanuel Vogel Dr.;Martin Michels Dr.;Lars Zer Dr.;Johann Lex Dr.;Nurcan S. Tuzun Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 25) pp:
Publication Date(Web):24 JUN 2003
DOI:10.1002/anie.200250690
Figures of eight correlate with corrolates: The surprising formation of the spirodicorrole dinickel complex 1, the first spirodiporphyrin as a metal complex, is the result of the varied chemistry of the “figure-eight” cyclooctapyrrole octaphyrin-(1.1.1.0.1.1.1.0). The conversion of the cyclooctapyrrole into 1 is brought about by a sequence of oxidation and a nickel(II) acetate induced cascade of rearrangement reactions. In structural respects, 1 stands out because of the orthogonal arrangement of the two nickelcorrolate chromophores linked through a common spiro carbon atom.
Co-reporter:Emanuel Vogel Dr.;Martin Michels Dr.;Lars Zer Dr.;Johann Lex Dr.;Nurcan S. Tuzun Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 25) pp:
Publication Date(Web):24 JUN 2003
DOI:10.1002/anie.200390499
Co-reporter:Sarah Wilsey;K. N. Houk
Photochemistry and Photobiology 2002 Volume 76(Issue 6) pp:616-621
Publication Date(Web):1 MAY 2007
DOI:10.1562/0031-8655(2002)0760616HVCIFD2.0.CO2
The potential role of “H–vinyl” conical intersections in a photochemical process known as the hula twist has been evaluated. The H–vinyl conical intersections of butadiene are explored with complete active space self-consistent field calculations, and proposals about the geometries involved in the hula-twist mechanism are discussed and contrasted with the conventional one-bond flip mechanism of cis–trans isomerization.
Co-reporter:Andrew G. Leach Dr.;Saron Catak;K. N. Houk Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 6) pp:
Publication Date(Web):6 MAR 2002
DOI:10.1002/1521-3765(20020315)8:6<1290::AID-CHEM1290>3.0.CO;2-K
The mechanisms of [3s,5s]-sigmatropic shifts of octa-1,3,7-triene and 7-methylenenona-1,3,8-triene have been elaborated using B3LYP and BPW91 density functional theory and CASPT2 methods. These orbital symmetry forbidden rearrangements are stepwise, involving diradical intermediates. A comparison with several [3,3]-sigmatropic shifts of substituted hexadienes and of [5,5]-sigmatropic shifts that are allowed, but nevertheless follow stepwise paths, shows that the activation barrier for the disallowed [3,5] shift is significantly larger than that for the stepwise reactions that are orbital symmetry allowed. Cyclic diradicals that have an aromatic circuit of electrons including the two radical centers and conjugated π or σ bonds are stabilized as compared to cyclic diradicals with an antiaromatic circuit of electrons. This applies to the transition states leading to and from the diradicals and influences the activation energies of stepwise sigmatropic shifts. The magnitudes of these effects are small but will have a significant influence on the rates of competing processes. This series of calculations has been used to assess the relative capabilities of the two functionals. We find that BPW91 underestimates the endothermicity of diradical formation and the barrier to diradical formation whereas B3LYP overestimates these quantities.
Co-reporter:Michael D. Bartberger;Wei Liu;Eleonora Ford;Katrina M. Miranda;Christopher Switzer;Jon M. Fukuto;Patrick J. Farmer;David A. Wink
PNAS 2002 Volume 99 (Issue 17 ) pp:10958-10963
Publication Date(Web):2002-08-20
DOI:10.1073/pnas.162095599
A potential of about −0.8 (±0.2) V (at 1 M versus normal hydrogen electrode) for the reduction of nitric oxide (NO) to its
one-electron reduced species, nitroxyl anion (3NO−) has been determined by a combination of quantum mechanical calculations, cyclic voltammetry measurements, and chemical reduction
experiments. This value is in accord with some, but not the most commonly accepted, previous electrochemical measurements
involving NO. Reduction of NO to 1NO− is highly unfavorable, with a predicted reduction potential of about −1.7 (±0.2) V at 1 M versus normal hydrogen electrode.
These results represent a substantial revision of the derived and widely cited values of +0.39 V and −0.35 V for the NO/3NO− and NO/1NO− couples, respectively, and provide support for previous measurements obtained by electrochemical and photoelectrochemical
means. With such highly negative reduction potentials, NO is inert to reduction compared with physiological events that reduce
molecular oxygen to superoxide. From these reduction potentials, the pKa of 3NO− has been reevaluated as 11.6 (±3.4). Thus, nitroxyl exists almost exclusively in its protonated form, HNO, under physiological
conditions. The singlet state of nitroxyl anion, 1NO−, is physiologically inaccessible. The significance of these potentials to physiological and pathophysiological processes
involving NO and O2 under reductive conditions is discussed.
Co-reporter:Gregori Ujaque;Patrick S. Lee;K. N. Houk ;Martin F. Hentemann;Samuel J. Danishefsky
Chemistry - A European Journal 2002 Volume 8(Issue 15) pp:
Publication Date(Web):26 JUL 2002
DOI:10.1002/1521-3765(20020802)8:15<3423::AID-CHEM3423>3.0.CO;2-X
Theoretical studies of stereoselectivity have been carried out with B3LYP and MP2 calculations. The high endo selectivity of hetero-Diels–Alder reactions of ortho-xylylenes with acetaldehydes is shown to result from attractive CH–π interactions between alkyl groups of the aldehyde and the aromatic ring in the transition states of the reaction. For the hetero-Diels–Alder reactions of ortho-xylylenes with benzaldehyde, the stereoselectivity is shown to be mainly governed by the attractive π–π interactions between the phenyl rings of the benzaldehyde and the ortho-xylylene. MP2 calculations are necessary to reproduce the stabilizing dispersion interactions.
Co-reporter:Yves Rubin ;Thibaut Jarrosson;Guan-Wu Wang Dr.;Michael D. Bartberger Dr.;K. N. Houk ;Georg Schick Dr.;Martin Saunders ;R. James Cross
Angewandte Chemie 2001 Volume 113(Issue 8) pp:
Publication Date(Web):17 APR 2001
DOI:10.1002/1521-3757(20010417)113:8<1361::AID-ANGE1361>3.0.CO;2-C
Das Titelbild zeigt den Vorgang der Wasserstoff- und Heliuminsertion/abgabe, der erstmals mit einem offenen Fullerenderivat (im Hintergrund angedeutet) gelang. Die experimentelle Aktivierungsbarriere für die Heliumdekomplexierung konnte bestimmt werden; sie ist sehr gut mit dem berechneten Wert (Dichtefunktionaltheorie) in Einklang. Die Barriere für die Komplexierung/Dekomplexierung von H2 ist interessanterweise fast doppelt so groß wie die bei Helium, wie das Energiediagramm im Vordergrund illustriert. Der Grund für den Unterschied ist die größere, gestreckte Oberfläche von H2, das stärkere van-der-Waals-Wechselwirkungen im Übergangszustand eingeht als Helium, obwohl beide Atome denselben Radius haben. Mehr über diesen Prozess erfahren Sie in dem Beitrag von Rubin, Houk, Saunders, Cross et al. auf S. 1591 ff.
Co-reporter:Kendall N. Houk ;Jeehiun K. Lee ;Dean J. Tantillo;Sogole Bahmanyar;Bruce N. Hietbrink
ChemBioChem 2001 Volume 2(Issue 2) pp:
Publication Date(Web):26 JAN 2001
DOI:10.1002/1439-7633(20010202)2:2<113::AID-CBIC113>3.0.CO;2-T
The mystery of proficiency remains: The recently solved crystal structures of free orotidine monophosphate decarboxylase (ODCase) and of the enzyme in complex with inhibitors reveal unusual structural features. However, despite the similarities among the structures, several startling new mechanisms of action have been proposed to explain how ODCase achieves its spectacular catalytic proficiency in catalyzing the decarboxylation of OMP (1) to UMP (2). R = ribose 5′-monophosphate.
Co-reporter:Ilyas Washington;K. N. Houk Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 23) pp:
Publication Date(Web):28 NOV 2001
DOI:10.1002/1521-3773(20011203)40:23<4485::AID-ANIE4485>3.0.CO;2-N
Peracids prefer to attack syn to an alkyl group as shown by the most stable transition state for the epoxidation of 3-methylcyclohexene (see section of transition state). Computational and experimental evidence explain the origin of this effect and how it might be exploited in synthesis.
Co-reporter:Ilyas Washington;K. N. Houk Dr.
Angewandte Chemie 2001 Volume 113(Issue 23) pp:
Publication Date(Web):28 NOV 2001
DOI:10.1002/1521-3757(20011203)113:23<4617::AID-ANGE4617>3.0.CO;2-V
Persäuren greifen bevorzugt syn zu einer Alkylgruppe an, wie der stabilste Übergangszustand (siehe Ausschnitt) für die Epoxidierung von 3-Methylcyclohexen zeigt. Rechnergestützte und experimentelle Befunde erklären den Ursprung dieses Effekts, was eine gezielte Anwendung in der Synthese ermöglichen sollte.
Co-reporter:Michael D. Bartberger;K. N. Houk;Jon M. Fukuto
PNAS 2001 Volume 98 (Issue 5 ) pp:2194-2198
Publication Date(Web):2001-02-27
DOI:10.1073/pnas.041481598
The gas phase and aqueous thermochemistry and reactivity of
nitroxyl (nitrosyl hydride, HNO) were elucidated with
multiconfigurational self-consistent field and hybrid density
functional theory calculations and continuum solvation methods. The
pKa of HNO is predicted to be 7.2 ± 1.0, considerably
different from the value of 4.7 reported from pulse radiolysis
experiments. The ground-state triplet nature of NO−
affects the rates of acid-base chemistry of the HNO/NO−
couple. HNO is highly reactive toward dimerization and addition of soft
nucleophiles but is predicted to undergo negligible hydration
(Keq = 6.9 × 10−5).
HNO is predicted to exist as a discrete species in solution and is a
viable participant in the chemical biology of nitric oxide and
derivatives.
Co-reporter:Jiangang Chen;Qiaolin Deng;Renxiao Wang;Kendall N. Houk ;Donald Hilvert
ChemBioChem 2000 Volume 1(Issue 4) pp:
Publication Date(Web):14 NOV 2000
DOI:10.1002/1439-7633(20001117)1:4<255::AID-CBIC255>3.0.CO;2-S
Antibody 1E9 is a protein catalyst for the Diels–Alder reaction between tetrachlorothiophene dioxide and N-ethylmaleimide. Quantum mechanical calculations have been employed to study the 1E9-catalyzed Diels–Alder reaction in the gas phase. The transition states and intermediates were all determined at the B3LYP/6-31G*//HF/6-31G* level. The cycloaddition step is predicted to be rate-determining, and the endo reaction pathway is strongly favored. Binding of the reactants and the transition states to antibody 1E9 was investigated by docking and molecular dynamics simulations. The linear interaction energy (LIE) method was adopted to estimate the free energy barrier of the 1E9-catalyzed Diels–Alder reaction. The catalytic efficiency of antibody 1E9 is achieved by enthalpic stabilization of the transition state, near-perfect shape complementarity of the hydrophobic binding site for the transition state, and a strategically placed hydrogen-bonding interaction.
Co-reporter:Jiangang Chen;Qiaolin Deng;Renxiao Wang;Kendall N. Houk ;Donald Hilvert
ChemBioChem 2000 Volume 1(Issue 4) pp:
Publication Date(Web):14 NOV 2000
DOI:10.1002/1439-7633(20001117)1:4<207::AID-CBIC207>3.0.CO;2-3
The cover picture shows the active site of the catalytic antibody 1E9, which promotes the bimolecular Diels–Alder reaction between tetrachlorothiophene dioxide and N-ethylmaleimide. Docking and molecular dynamics simulations of the reactant complex (green) and the endo-transition state (yellow) bound at the antibody active site illustrate the excellent shape complementarity between ligand and protein. Residues Trp H50 and Asn H35, which interact with the dienophile through π-stacking and hydrogen bonding, respectively, are seen at the bottom of the active site beneath the transparent protein surface. More about shape complementarity, binding site dynamics, and transition state stabilization can be found in the theoretical study by K. N. Houk, D. Hilvert et al. on p. 255 ff. (The cover image was produced by Kinya Hotta.)
Co-reporter:K. N. Houk;Kensuke Nakamura;Chimin Sheu;Amy E. Keating
Science 1996 Vol 273(5275) pp:627-629
Publication Date(Web):02 Aug 1996
DOI:10.1126/science.273.5275.627
Abstract
Theoretical modeling of the dynamics of complexation and decomplexation of guest molecules by container molecules reveals that gating has a critical influence on the ease of formation and stability of host-guest complexes. Hosts equipped with gates can form very stable complexes with a variety of guests under readily achievable conditions. Gating involves conformational processes of the host molecule that alter the size of the portals through which guest molecules pass. “French door” and “sliding door” mechanisms of gate opening are identified.
Co-reporter:Paul H.-Y. Cheong ; Robert S. Paton ; Sarah M. Bronner ; G-Yoon J. Im ; Neil K. Garg ;K. N. Houk
Journal of the American Chemical Society () pp:
Publication Date(Web):January 8, 2010
DOI:10.1021/ja9098643
Density functional theory computations reproduce the surprisingly high regioselectivities in nucleophilic additions and cycloadditions to 4,5-indolynes and the low regioselectivities in the reactions of 5,6-indolynes. Transition-state distortion energies control the regioselectivities, activating the 5 and 6 positions over the 4 and 7 positions, leading to high preferences for 5- and 6-substituted products from 4,5- and 6,7-indolynes, respectively. Orbital and electrostatic interactions have only minor effects, producing low regioselectivities in the reactions of 5,6-indolynes. The distortion model predicts high regioselectivities with 6,7-indolynes; these have been verified experimentally. The regioselectivities found with other arynes are explained on the basis of distortion energies that are reflected in reactant geometries.
Co-reporter:Fang Liu ; Robert S. Paton ; Seonah Kim ; Yong Liang ;K. N. Houk
Journal of the American Chemical Society () pp:
Publication Date(Web):September 17, 2013
DOI:10.1021/ja408437u
The Diels–Alder reactions of the cycloalkenes, cyclohexene through cyclopropene, with a series of dienes—1,3-dimethoxybutadiene, cyclopentadiene, 3,6-dimethyltetrazine, and 3,6-bis(trifluoromethyl)tetrazine—were studied with quantum mechanical calculations and compared with experimental values when available. The reactivities of cycloalkenes as dienophiles were found by a distortion/interaction analysis to be distortion controlled. The energies required for cycloalkenes to be distorted into the Diels–Alder transition states increase as the ring size of cycloalkenes increases from cyclopropene to cyclohexene, resulting in an increase in activation barriers. The reactivities of the dienes are controlled by both distortion and interaction energies. In normal Diels–Alder reactions with cycloalkenes, the electron-rich 1,3-dimethoxybutadiene exhibits stronger interaction energies than cyclopentadiene, but the high distortion energies required for 1,3-dimethoxybutadiene to achieve transition-state geometries overtake the favorable interaction, resulting in higher activation barriers. In inverse-electron-demand Diels–Alder reactions of 3,6-dimethyltetrazine and 3,6-bis(trifluoromethyl)tetrazine, the reactivities are mainly controlled by interaction energies.
Co-reporter:Ilhan Yavuz; Blanton N. Martin; Jiyong Park;K. N. Houk
Journal of the American Chemical Society () pp:
Publication Date(Web):February 6, 2015
DOI:10.1021/ja5076376
Molecular ordering and charge transport have been studied computationally for 22 conjugated oligomers fabricated as crystal or thin-film semiconductors. Molecular dynamics (MD) simulations are employed to equilibrate crystal morphologies at 300 K. The paracrystalline order parameter, g, is calculated to characterize structural order in the materials. Charge-transport dynamics are predicted using kinetic Monte Carlo methods based on a charge-hopping mechanism described by the Marcus theory of electron transfer to calculate charge-transfer rates using the VOTCA package. We introduce an error function to assess the reliability of our computed values to reproduce experimental hole mobilities in both crystalline and thin-film morphologies of the 22 conjugated oligomers. For each of the oligomers, we compute hole mobility with three different theoretical models incorporating increasing measures of disorder: (1) a perfect crystal, based on the experimentally derived crystal structure, with no disorder, (2) an MD-equilibrated structure incorporating thermal disorder into the crystal structure, and (3) model 2 above but also incorporating energetic disorder arising from variations in site energies. For the series of known crystals with long-range order, we find that the perfect crystal model produces hole mobilities giving the best fit to experimental data. For the series of thin-film morphologies with short-range order, we observe that the presence of both thermal and energetic disorder is essential for accurate calculation. We also discuss the interplay between hole mobility and other charge-transport parameters in these morphologies, such as reorganization energy and energetic disorder.
Co-reporter:Fang Liu ; Yong Liang ;K. N. Houk
Journal of the American Chemical Society () pp:
Publication Date(Web):July 19, 2014
DOI:10.1021/ja505569a
The Diels–Alder reactions of seven 1,2,4,5-tetrazines with unstrained and strained alkenes and alkynes were studied with quantum mechanical calculations (M06-2X density functional theory) and analyzed with the distortion/interaction model. The higher reactivities of alkenes compared to alkynes in the Diels–Alder reactions with tetrazines arise from the differences in both interaction and distortion energies. Alkenes have HOMO energies higher than those of alkynes and therefore stronger interaction energies in inverse-electron-demand Diels–Alder reactions with tetrazines. We have also found that the energies to distort alkenes into the Diels–Alder transition-state geometries are smaller than for alkynes in these reactions. The strained dienophiles, trans-cyclooctene and cyclooctyne, are much more reactive than unstrained trans-2-butene and 2-butyne, because they are predistorted toward the Diels–Alder transition structures. The reactivities of substituted tetrazines correlate with the electron-withdrawing abilities of the substituents. Electron-withdrawing groups lower the LUMO+1 of tetrazines, resulting in stronger interactions with the HOMO of dienophiles. Moreover, electron-withdrawing substituents destabilize the tetrazines, and this leads to smaller distortion energies in the Diels–Alder transition states.
Co-reporter:Adam J. T. Smith, Ying Li and K. N. Houk
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 13) pp:NaN2724-2724
Publication Date(Web):2009/05/06
DOI:10.1039/B901429B
The mechanisms of enzyme-catalyzed phosphate transfer and hydrolysis reactions are of great interest due to their importance and abundance in biochemistry. The reaction may proceed in a stepwise fashion, with either a pentavalent phosphorane or a metaphosphate anion intermediate, or by a concerted SN2 mechanism. Despite much theoretical work focused on a few key enzymes, a consensus for the mechanism has not been reached, and examples of all three possibilities have been demonstrated. We have investigated the mechanism of human uridine-cytidine kinase 2 (UCK2, EC 2.7.1.48), which catalyzes the transfer of a phosphate group from ATP to the ribose 5′-hydroxyl of cytidine and uridine. UCK2 is normally expressed in human placenta, but is overexpressed in certain cancer cells, where it is responsible for activating a class of antitumor prodrugs. The UCK2 mechanism was investigated by generating a 2D potential energy surface as a function of the P–O bonds forming and breaking, with energies calculated using a quantum mechanics/molecular mechanics potential (B3LYP/6-31G(d):AMBER). The mechanism of phosphate transfer is shown to be concerted, and is accompanied by concerted proton transfer from the 5′-hydroxyl to a conserved active site aspartic acid that serves as a catalytic base. The calculated barrier for this reaction is 15.1 kcal/mol, in relatively good agreement with the experimental barrier of 17.5 kcal/mol. The interactions of the enzyme active site with the reactant, transition state, and product are examined for their implications on the design of anticancer prodrugs or positron emission tomography (PET) reporter probes for this enzyme.
Co-reporter:Hung V. Pham, David B. C. Martin, Christopher D. Vanderwal and K. N. Houk
Chemical Science (2010-Present) 2012 - vol. 3(Issue 5) pp:NaN1655-1655
Publication Date(Web):2012/02/02
DOI:10.1039/C2SC01072K
Computational studies show that the base-mediated intramolecular Diels–Alder of tryptamine-derived Zincke aldehydes, used as a key step in the synthesis of the Strychnos alkaloids norfluorocurarine and strychnine, proceeds via a stepwise pathway. The experimentally determined importance of a potassium counterion in the base is explained by its ability to preorganize the Zincke aldehyde diene in an s-cis conformation suitable to bicyclization. Computation also supports the thermodynamic importance of the generation of a stable enolate in the final reaction step. The thermal cycloreversion reaction of the Diels–Alder products is also found to proceed in a stepwise manner.
Co-reporter:C. Thiehoff, M. C. Holland, C. Daniliuc, K. N. Houk and R. Gilmour
Chemical Science (2010-Present) 2015 - vol. 6(Issue 6) pp:NaN3571-3571
Publication Date(Web):2015/04/17
DOI:10.1039/C5SC00871A
The gauche conformation of the 1,2-difluoroethane motif is known to involve stabilising hyperconjugative interactions between donor (bonding, σC–H) and acceptor (antibonding, σ*C–F) orbitals. This model rationalises the generic conformational preference of F–Cβ–Cα–X systems (ϕFCCX ≈ 60°), where X is an electron deficient substituent containing a Period 2 atom. Little is known about the corresponding Period 3 systems, such as sulfur and phosphorus, where multiple oxidation states are possible. Conformational analyses of β-fluorosulfides, -sulfoxides and -sulfones are disclosed here, thus extending the scope of the fluorine gauche effect to the 3rd Period (F–C–C–S(O)n; ϕFCCS ≈ 60°). Synergy between experiment and computation has revealed that the gauche effect is only pronounced in structures bearing an electropositive vicinal sulfur atom (S+–O−, SO2).
Co-reporter:Maruthi Kumar Narayanam, Yong Liang, K. N. Houk and Jennifer M. Murphy
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:NaN1261-1261
Publication Date(Web):2015/11/11
DOI:10.1039/C5SC03259H
Density functional theory (DFT) calculations and experiments in tandem led to discoveries of new reactivities and selectivities involving bioorthogonal sydnone cycloadditions. Dibenzocyclooctyne derivatives (DIBAC and BARAC) were identified to be especially reactive dipolarophiles, which undergo the (3 + 2) cycloadditions with N-phenyl sydnone with the rate constant of up to 1.46 M−1 s−1. Most significantly, the sydnone-dibenzocyclooctyne and norbornene-tetrazine cycloadditions were predicted to be mutually orthogonal. This was validated experimentally and used for highly selective fluorescence labeling of two proteins simultaneously.
Co-reporter:Hao Wang and K. N. Houk
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN470-470
Publication Date(Web):2013/09/20
DOI:10.1039/C3SC52538D
Torsional effects control the π-facial stereoselectivities of a variety of synthetically important organic reactions. This review surveys theoretical calculations that have led to the understanding of the influence of the torsional effects on several types of stereoselective organic reactions, especially electrophilic additions and cycloadditions to alkenes.
Co-reporter:Xin Hong, Yong Liang, Allison K. Griffith, Tristan H. Lambert and K. N. Houk
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN475-475
Publication Date(Web):2013/11/04
DOI:10.1039/C3SC52882K
The mechanism of hydrazine-catalyzed carbonyl-olefin metathesis relying on a novel (3 + 2) strategy is studied by density functional theory (DFT) calculations. The origins of the special reactivity of cyclopropene in this transformation are revealed, and the reactivities of different alkenes in the (3 + 2) cycloadditions and cycloreversions are compared. It is found that the ease of distortion of reactants accelerates cycloadditions, and that the strain release is the controlling factor for cycloreversions.
Co-reporter:A. Dieckmann and K. N. Houk
Chemical Science (2010-Present) 2013 - vol. 4(Issue 9) pp:NaN3600-3600
Publication Date(Web):2013/06/27
DOI:10.1039/C3SC51192H
The energetics of complex formation in a number of artificial self-replicating systems has been studied by density functional theory calculations. Complex stabilities were dissected into stabilizing contributions from hydrogen bonding, dispersion, electrostatics, and destabilizing distortions of monomers from their optimum geometries. Strong cooperative effects were identified in all systems, and dispersion and electrostatics contribute more to the overall energies of complex formation than hydrogen bonding in some systems.
Co-reporter:Yu Lan, Peng Liu, Stephen G. Newman, Mark Lautens and K. N. Houk
Chemical Science (2010-Present) 2012 - vol. 3(Issue 6) pp:NaN1995-1995
Publication Date(Web):2012/02/22
DOI:10.1039/C2SC20103H
The mechanism of Pd(0)-catalyzed carbohalogenation of alkenes has been investigated with density functional theory. The catalytic cycle involves oxidative addition of the aryl halide, alkene insertion, and a novel C(sp3)–I reductive elimination. The C(sp3)–I reductive elimination leads to a weakly bonded Pd–product complex, which undergoes ligand exchange via a dissociative mechanism to regenerate the catalyst. In the rate-determining reductive elimination step, bromides and chlorides have higher barriers than iodides, because the stronger Pd–Br and Pd–Cl bonds are being cleaved in these transition states. Bulky ligands, such as P(t-Bu)3 and Q-Phos, facilitate the C(sp3)–I reductive elimination by preventing the formation of tetracoordinated intermediates. The mechanism of the competing β-hydrogen elimination pathway was also investigated. For reactions involving a syn-β-hydrogen atom in the alkyl Pd(II) iodide intermediate, β-hydrogen elimination is much more favorable, leading to Heck-type side products. Blocking β-elimination by the choice of substrates is the main reason why this example of carboiodination works.
Co-reporter:Elizabeth H. Krenske, K. N. Houk, Andrew G. Lohse, Jennifer E. Antoline and Richard P. Hsung
Chemical Science (2010-Present) 2010 - vol. 1(Issue 3) pp:NaN392-392
Publication Date(Web):2010/06/25
DOI:10.1039/C0SC00280A
Chiral oxazolidinones were previously thought to control cycloaddition stereoselectivity by steric crowding of one face of the substrate. We have discovered that in (4 + 3) cycloaddition reactions of oxyallyls, the stereoinduction is caused instead by stabilising CH–π interactions that lead to reaction at the more crowded face of the oxyallyl. Density functional theory calculations on the (4 + 3) cycloadditions of oxazolidinone-substituted oxyallyls with furans establish unexpected transition state conformations and a new explanation of selectivity.
Co-reporter:Yang Cao and K. N. Houk
Journal of Materials Chemistry A 2011 - vol. 21(Issue 5) pp:NaN1508-1508
Publication Date(Web):2010/10/22
DOI:10.1039/C0JM02422H
The 1,3-dipolar cycloadditions of azomethine ylide and carbonyl ylide to models of graphene have been investigated with density functional theory. Reaction energetics have been obtained and show that edge areas of graphene are much more favourable reaction sites than the centre sites. Azomethine ylide cannot directly react at the centre area, while carbonyl ylides are promising reagents for functionalization of graphene. The influence of some 1,3-dipole substituents is also evaluated.
Co-reporter:Edward Richmond, Kenneth B. Ling, Nicolas Duguet, Lois B. Manton, Nihan Çelebi-Ölçüm, Yu-Hong Lam, Sezen Alsancak, Alexandra M. Z. Slawin, K. N. Houk and Andrew D. Smith
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 6) pp:NaN1817-1817
Publication Date(Web):2014/12/12
DOI:10.1039/C4OB02526A
The reaction of L-serine derived N-arylnitrones with alkylarylketenes generates asymmetric 3-alkyl-3-aryloxindoles in good to excellent yields (up to 93%) and excellent enantioselectivity (up to 98% ee) via a pericyclic cascade process. The optimization, scope and applications of this transformation are reported, alongside further synthetic and computational investigations. The preparation of the enantiomer of a Roche anti-cancer agent (RO4999200) 1 (96% ee) in three steps demonstrates the potential utility of this methodology.
Co-reporter:Ilhan Yavuz, Steven A. Lopez, Janice B. Lin and K. N. Houk
Journal of Materials Chemistry A 2016 - vol. 4(Issue 47) pp:NaN11243-11243
Publication Date(Web):2016/11/10
DOI:10.1039/C6TC03823A
The morphologies and electron mobilities for 20 single-crystal and 21 thin-film organic n-type semiconductors are predicted using a multi-mode methodology previously applied by our group for p-type materials [I. Yavuz, et al., J. Am. Chem. Soc., 2015, 137, 2856–2866]. The calculations simulate Marcus charge-hopping with a kinetic Monte Carlo method using the VOTCA package of Andrienko et al. The calculations assume perfect order for single crystal morphologies, but structural disorder is incorporated into thin-film calculations using molecular dynamics to simulate the energetic disorder of thin-film morphologies. Predicted electron mobilities for both morphologies are typically within one order of magnitude.
Co-reporter:Xu Han, Jiyong Park, Wei Wu, Andres Malagon, Lingyu Wang, Edgar Vargas, Athula Wikramanayake, K. N. Houk and Roger M. Leblanc
Chemical Science (2010-Present) 2017 - vol. 8(Issue 3) pp:NaN2009-2009
Publication Date(Web):2016/11/17
DOI:10.1039/C6SC04854D
Amyloid-β peptides (Aβ) fibrillation is the hallmark of Alzheimer's disease (AD). However, it has been challenging to discover potent agents in order to inhibit Aβ fibrillation. Herein, we demonstrated the effect of resorcinarene on inhibiting Aβ fibrillation in vitro via experimental and computational methods. Aβ were incubated with different concentrations of resorcinarene so as to monitor the kinetics by using thioflavin T binding assay. The results, which were further confirmed by far-UV CD spectroscopy and atomic force microscopy, strongly indicated that the higher concentration of resorcinarene, the more effective the inhibition of Aβ fibrillation. A cytotoxicity study showed that when sea urchin embryos were exposed to the resorcinarene, the majority survived due to the resorcinarene low toxicity. In addition, when the resorcinarene was added, the formation of toxic Aβ 42 species was delayed. Computational studies of Aβ fibrillation, including docking simulations and MD simulations, illustrated that the interaction between inhibitor resorcinarene and Aβ is driven by the non-polar interactions. These studies display a novel strategy for the exploration of promising antiamyloiddogenic agents for AD treatments.
.jpg)
![4-Thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid,6-[(1R)-1-hydroxyethyl]-7-oxo-3-[[(1R,3S)-tetrahydro-1-oxido-3-thienyl]thio]-,(5R,6S)-](http://img.cochemist.com/ccimg/120800/120788-07-0.png)
![4-Thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid,6-[(1R)-1-hydroxyethyl]-7-oxo-3-[[(1R,3S)-tetrahydro-1-oxido-3-thienyl]thio]-,(5R,6S)-](http://img.cochemist.com/ccimg/120800/120788-07-0_b.png)
![6(13H)-Pentacenone, 13-hydroxy-13-[2-[tris(1-methylethyl)silyl]ethynyl]-](http://img.cochemist.com/ccimg/917100/917015-45-3.png)
![6(13H)-Pentacenone, 13-hydroxy-13-[2-[tris(1-methylethyl)silyl]ethynyl]-](http://img.cochemist.com/ccimg/917100/917015-45-3_b.png)



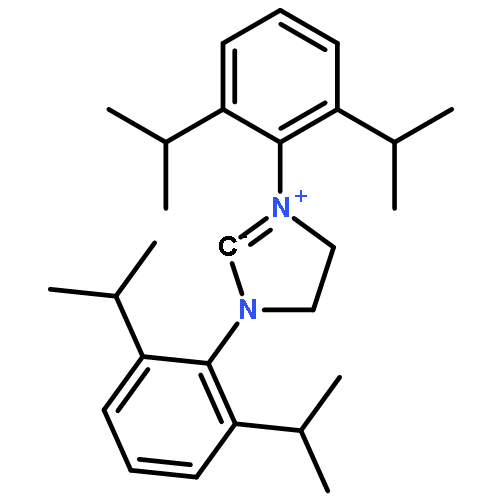












































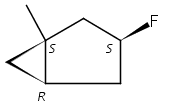
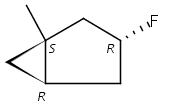
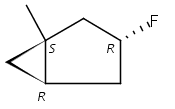



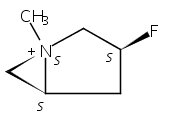
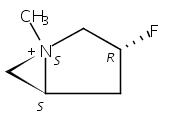
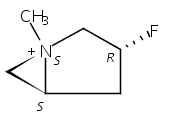

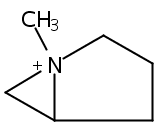








































![2-Allyl-6-nitro-benzo[de]isoquinoline-1,3-dione](http://img.cochemist.com/ccimg/189200/189183-96-8.png)
![2-Allyl-6-nitro-benzo[de]isoquinoline-1,3-dione](http://img.cochemist.com/ccimg/189200/189183-96-8_b.png)
![Phosphine,1,1'-[(1R)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-diphenyl-](/data/chemimg/110700/185913-97-7.png)
![Phosphine,1,1'-[(1R)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-diphenyl-](/data/chemimg/110700/185913-97-7_b.png)








![2H-1-Benzopyran-2-one, 7-[(1,1-dimethyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/27500/27421-19-8.png)
![2H-1-Benzopyran-2-one, 7-[(1,1-dimethyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/27500/27421-19-8_b.png)


![L-Serine, N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/17200/17136-46-8.png)
![L-Serine, N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/17200/17136-46-8_b.png)
![1-methylbicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/4700/4625-24-5.png)
![1-methylbicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/4700/4625-24-5_b.png)






![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)


![Quinazolino[3,2-a][1,4]benzodiazepine-5,13-dione,6,7-dihydro-7-(1H-indol-3-ylmethyl)-, (7S)-](http://img.cochemist.com/ccimg/93500/93413-06-0.png)
![Quinazolino[3,2-a][1,4]benzodiazepine-5,13-dione,6,7-dihydro-7-(1H-indol-3-ylmethyl)-, (7S)-](http://img.cochemist.com/ccimg/93500/93413-06-0_b.png)