Co-reporter:Baohua Yue, Guangbo Zeng, Yepei Zhang, Sheng Tao, Xiaoming Zhang, Liuming Yan
Solid State Ionics 2017 Volume 300(Volume 300) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.ssi.2016.11.011
•Phosphonic acid functionalized polysulfone (PSF-PA) and triazolyl functionalized polysulfone (PSF-Tri) are synthesized.•Synergetic proton conducting effect is observed in PSF-PA/PSF-Tri acid-base composite membrane.•The composite membrane shows reduced hydrophilicity, methanol permeability and improved tensile strength.The synergetic proton conducting effect was observed in an acid-base composite composed of phosphonic acid functionalized polysulfone (PSF-PA) and triazolyl functionalized polysulfone (PSF-CH2-iPrOHTri), with three orders of magnitude improvement compared to that of the acidic or basic component. The maximum proton conductivity of 42.71 mS cm− 1 at 100 °C and 90% RH was observed in PSF-PA/PSF-CH2-iPrOHTri composite membrane composed of 75.0% PSF-CH2-iPrOHTri. In addition, the acid-base composite membrane showed higher thermal and oxidative stability, reduced hydrophilicity and methanol permeability, significantly improved mechanical strength. For the hydrated membranes of PSF-PA or PSF-CH2-iPrOHTri, the tensile strengths showed significant degradation with values only about halves of the dry membranes. On the other side, the tensile strength of the acid-base composite composed of 75.0% PSF-CH2-iPrOHTri showed only slight drop from 35.52 MPa to 30.59 MPa.Download high-res image (136KB)Download full-size image
Co-reporter:Liuming Yan, Yi Lu and Xuejiao Li
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 7) pp:5529-5536
Publication Date(Web):07 Jan 2016
DOI:10.1039/C5CP06638G
A density functional theory (DFT) protocol for the calculation of redox potentials of copper complexes is developed based on 13 model copper complexes. The redox potentials are calculated in terms of Gibbs free energy change of the redox reaction at the theory level of CAM-B3LYP/6-31+G(d,p)/SMD, with the overall Gibbs free energy change being partitioned into the Gibbs free energy change of the gas phase reaction and the Gibbs free energy change of solvation. In addition, the calculated Gibbs free energy change of solvation is corrected by a unified correction factor of −0.258 eV as the second-layer Gibbs free energy change of solvation and other interactions for each redox reaction. And an empirical Gibbs free energy change of solvation at −0.348 eV is applied to each water molecule if the number of inner-sphere water molecule changes during the redox reaction. Satisfactory agreements between the DFT calculated and experimental results are obtained, with a maximum absolute error at 0.197 V, a mean absolute error at 0.114 V and a standard deviation at 0.133 V. Finally, it is concluded that the accurate prediction of redox potentials is dependent on the accurate prediction of geometrical structures as well as on geometrical conservation during the redox reaction.
Co-reporter:Xiaoming Zhang, Baohua Yue, Liuming Yan, Guangbo Zeng, Sheng Tao
International Journal of Hydrogen Energy 2016 Volume 41(Issue 8) pp:4740-4750
Publication Date(Web):2 March 2016
DOI:10.1016/j.ijhydene.2016.01.076
•Phosphonic acid functionalized poly(sulfone ether) is synthesized by polycondensation.•Triazolyl functionalized poly(sulfone ether) is synthesized by polycondensation.•Performances of phosphonic acid functionalized poly(sulfone ether) are improved.•Acid–base composite proton exchange membrane is prepared.The comprehensive performances of phosphonic acid functionalized poly(ether sulfone) (PES-PA) are improved by compositing with 1H-1,2,3-triazol-4-yl functionalized poly(ether sulfone) (PES-Tri). The PES-PA and PES-Tri are synthesized via the nucleophilic aromatic substitution (SNAR) polycondensation, respectively, of diethyl 2,5-dihydroxyphenylphosphonate and 4,4′-difuorodiphenylsulfone and of bisphenol A and 3,3′-diethynyl-4,4′-difluorodiphenylsulfone, and followed by hydrolysis or post-polymerization click reaction and deprotection reaction. In addition, two functionalized monomers, diethyl 2,5-dihydroxyphenylphosphonate and 3,3′-diethynyl-4,4′-difluorodiphenylsulfone are synthesized. Best comprehensive performances are achieved in the composite membrane consisting of 80% PES-PA and 20% PES-Tri by molar ratio of the functional structure units. At 100 °C and 90% RH (relative humidity), the proton conductivity of the composite membrane is 12.8 mS cm−1, about 1.8 times improvement compared with pristine PES-PA. In addition, the hydrophilicity and methanol permeability are alleviated, the water uptake is reduced from 31.3% to 16.1% at 100 °C and 90% RH and methanol permeability is reduced from 19.0 × 10−8 cm2 s−1 to 0.79 × 10−8 cm2 s−1 at 30 °C.The comprehensive performances of phosphonic acid functionalized poly(sulfone ether) are improved by compositing of 20% triazolyl functionalized poly(sulfone ether).
Co-reporter:Xuejiao Li, Liuming Yan and Baohua Yue
RSC Advances 2015 vol. 5(Issue 98) pp:80220-80227
Publication Date(Web):16 Sep 2015
DOI:10.1039/C5RA14272E
Ab initio molecular dynamics (AIMD) simulations are applied to the study of proton transport in solid state maleimide. The AIMD simulations reproduce correctly the structural and energetic characteristics. The simulations reveal the direct proton hopping between two maleimide molecules, and the proton hopping frequency is evaluated. The temperature dependence of proton hopping frequency obeys the Arrhenius activation process with an activation energy of 4.39 kcal mol−1. Finally, it is proposed that maleimide is a potential building block for the design of high-temperature proton exchange membranes.
Co-reporter:Chao Sun, Liuming Yan, Baohua Yue, Huiting Liu and Yanfeng Gao
Journal of Materials Chemistry A 2014 vol. 2(Issue 43) pp:9283-9293
Publication Date(Web):02 Sep 2014
DOI:10.1039/C4TC00778F
The modulation of metal–insulator transition (MIT) temperature and phase stability of thermochromic materials based on all the transition metal doped VO2 were systematically studied using density functional theory (DFT) calculations. The free energies, formation enthalpies, and Fermi energies of transition metal doped VO2 were evaluated from DFT calculations; the cell volumes and bulk moduli were obtained by fitting the free energies to the Birch–Murnaghan equation of states; and the decomposition enthalpies and entropies of the transition metal doped VO2 were calculated using both experimental data and DFT calculations. Based on these results, the MIT temperature was associated with lattice distortion of VO2 (M1) upon doping, the expansion of cell volume and the decrease in β-angle were associated with the decrease in MIT temperature, and the shrinkage of cell volume and the increase in β-angle were associated with the increase in MIT temperature. And it was also concluded that VO2 (M1) doped with high valence cations is more stable than those doped with low valence cations. These conclusions are consistent with experimental facts that W-, Mo-, and Re- are the most studied and the most effective dopants for the reduction of MIT temperature, and La-, Hg-, and Ag-doped VO2 undergoes phase separation. In addition, DFT calculations without spin-polarization were also carried out, and the influence of spin-polarization was evaluated. Finally, scandium was proposed as a potential dopant for VO2 in view of balanced comprehensive performance.
Co-reporter:Yepei Zhang, Baohua Yue, Shuaiyuan Han and Liuming Yan
RSC Advances 2014 vol. 4(Issue 64) pp:33702-33712
Publication Date(Web):17 Jul 2014
DOI:10.1039/C4RA04514A
The synergetic proton conducting effect with three orders of magnitude improvement in proton conductivity was observed in an acid–base composite composed of phosphonic acid functionalized polystyrene (PS-PA) and triazolyl functionalized polystyrene (PS-Tri). In addition, a new method for the development of proton conducting materials by the combination of different acidic and basic polymers is proposed. The PS-PA was synthesized by the bromination of polystyrene on the para-position of the phenyl ring followed by phosphonation and hydrolysis. The PS-Tri was synthesized by the chloromethylation of polystyrene on the para-position of the phenyl ring followed by azidation and 1,3-dipolar cycloaddition or ‘click’ reaction. A maximum proton conductivity of 11.2 mS cm−1, which is three orders of magnitude higher than that of pristine PS-PA or PS-Tri, a tensile strength of 16.3 MPa, and a minimum water uptake of 15.1% (90 °C, 90% RH) were observed in the PS-PA/PS-Tri composite composed of 66.7% PS-PA. Finally, a mosaic-like morphology model and space-charge effects were proposed to explain the synergetic proton conducting effect.
Co-reporter:Huiting Liu, Liuming Yan, Baohua Yue, and Aijun Li
The Journal of Physical Chemistry A 2014 Volume 118(Issue 25) pp:4405-4414
Publication Date(Web):June 3, 2014
DOI:10.1021/jp503872m
Density functional theory calculations have been successfully applied to investigate the formation of hydrocarbon radicals and hydrogen transfer pathways related to the chemical vapor infiltration process based on model molecules of phenanthrene, anthra[2,1,9,8-opqra]tetracene, dibenzo[a,ghi]perylene, benzo[uv]naphtho[2,1,8,7-defg]pentaphene, and dibenzo[bc,ef]ovalene. The hydrogen transfer reaction rate constants are calculated within the framework of the Rice–Ramsperger–Kassel–Marcus theory and the transition state theory by use of the density functional theory calculation results as input. From these calculations, it is concluded that the hydrogen transfer reaction between two bay sites can happen almost spontaneously with energy barrier as low as about 4.0 kcal mol–1, and the hydrogen transfer reactions between two armchair sites possess lower energy barrier than those between two zigzag sites.
Co-reporter:Liu-Ming Yan;Jun-Ming Su;Chao Sun;Bao-Hua Yue
Advances in Manufacturing 2014 Volume 2( Issue 4) pp:358-368
Publication Date(Web):2014 December
DOI:10.1007/s40436-014-0086-x
Cathode materials are the most critical challenge for the large scale application of Li-ion batteries in electric vehicles and for the storages of electricity. The first principles calculations play an important role in development and optimization of novel cathode materials. In this paper, we overview the first principles calculations of energy, volume change, band-gap, phase diagram, and Li-ion transport mechanism of cathode materials with an emphasis on the design of such materials. We also overview the recent progress of data mining techniques and the high-throughput first principles calculations for the design and development of cathode materials. Finally, we preview the challenges and opportunities of this rapidly developing field.
Co-reporter:Liu-Ming Yan;Chao Sun;Hui-Ting Liu
Advances in Manufacturing 2013 Volume 1( Issue 2) pp:160-165
Publication Date(Web):2013 June
DOI:10.1007/s40436-013-0024-3
An opposite phenomenon to the flying ice cube where kinetic energy is drained from the high frequency vibrational motion to the low frequency translational motion and rotational motion (Harvey et al., J Comput Chem 19:726–740, 1998) is reported in molecular dynamics simulations of the flexible TIP3P water. It is found that kinetic energy is drained from the low frequency translational motion and rotational motion to the high frequency vibrational motion of the flexible TIP3P water. In addition, the equipartition theorem is not applicable to the flexible TIP3P water, but applicable to the rigid TIP3P water. However, the Maxwell–Boltzmann velocity distribution is satisfied for cases even the equipartition theorem is not applicable.
Co-reporter:Baohua Yue, Liuming Yan, Shuaiyuan Han, and Liqing Xie
The Journal of Physical Chemistry B 2013 Volume 117(Issue 26) pp:7941-7949
Publication Date(Web):June 13, 2013
DOI:10.1021/jp404684e
The proton transport pathways in an acid–base complex consisting of a phosphonic acid group and a 1,2,3-triazolyl group were studied using density functional theory (DFT) calculations in terms of stable configurations and transition states of the molecular or ionic dimers and trimers and verified by proof-of-concept experiments including experimental measurements of overall conductivity and 1H NMR and FTIR spectroscopy of the methylphosphonic acid (MPA) and 1,2,3-triazole (Tri) complex as well as overall proton conductivity of polymeric blend of poly(vinylphosphonic acid) (PVPA) and poly(4-vinyl-1H-1,2,3-triazole) (PVTri). From the DFT calculations of dimers and trimers composed of ethylphosphonic acid (EPA), Tri, and their deprotonated counterparts, it was concluded that the intermolecular hydrogen bonds of the transition states corresponding to proton transport are much shorter than those of stable configurations, but the O–H and N–H bonds are much longer than those of stable configurations. The tautomerization activation energy decreases from 0.927–1.176 eV in Tri–Tri dimers to 0.336–0.444 eV in the EPA–Tri dimers. From the proof-of-concept experiments, about a 50 fold increase in overall conductivity was observed in the MPA–Tri complex consisting of 10% (molar ratio) MPA compared to pure Tri, and the calculated activation energy is consistent with the experimental activation energy evaluated from temperature dependence of proton conductivity of pure Tri and the MPA–Tri complex. In addition, the fast proton exchange between MPA and Tri, consistent with the DFT calculations, was verified by 1H NMR and FTIR spectroscopy. Finally, a polymeric blend of PVPA and PVTri was prepared, and its proton conductivity at about 2.1 mS·cm–1 in anhydrous state at 100 °C was observed to be significantly higher than that of PVPA or of poly(VPA-co-1-vinyl-1,2,4-triazole). The proton conductivity of the polymeric PVPA and PVTri blend in humidity state is in the same range as that of NAFION 117.
Co-reporter:Liqing Xie, Huiting Liu, Shuaiyuan Han, Baohua Yue, and Liuming Yan
The Journal of Physical Chemistry B 2013 Volume 117(Issue 50) pp:16345-16355
Publication Date(Web):November 22, 2013
DOI:10.1021/jp4094386
Intermolecular and intramolecular hydrogen bond (H-bond) and proton transport in acid–base complexes and amphoteric molecules consisting of phosphonic acid groups and nitrogenous heterocyclic rings are investigated by density functional theory calculations and 1H NMR and 31P NMR spectroscopy. It is concluded that a phosphonic acid group can act both as H-bond donor and H-bond acceptor, while an imine nitrogen atom can only act as H-bond acceptor and an amine group as H-bond donor. And the intramolecular H-bond is weaker than the intermolecular H-bond attributing to configurational restriction. In addition, the strongest H-bond interaction is observed between a phosphonic acid and a 1H-indazole because of the formation of double H-bonds. The 1H NMR and 31P NMR chemical shifts for the acid–base complexes are consistent with the density functional theory calculations. From the 1H NMR chemical shifts, fast proton exchange is observed between a phosphonic acid and 1H-benzimidazole or 1H-indazole. Finally, it is proposed that polymeric material tethered with 1H-benzimidazole or 1H-indazole rings is a favorable component for high-temperature proton exchange membranes based on acid–base complexes or acid–base amphoteric molecules.
Co-reporter:Suqing Di, Liuming Yan, Shuaiyuan Han, Baohua Yue, Qingxia Feng, Liqing Xie, Jin Chen, Dongfang Zhang, Chao Sun
Journal of Power Sources 2012 Volume 211() pp:161-168
Publication Date(Web):1 August 2012
DOI:10.1016/j.jpowsour.2012.03.091
The boron phosphate-poly(2,5-benzimidazole) (or BPO4–ABPBI) nanocomposite proton exchange membranes were prepared by preblending BPO4 nanoparticles to the 3,4-diaminobenzoic acid solution before its polycondensation. The phosphoric acid doped nanocomposite membrane possesses enhanced proton conductivity compared to the phosphoric acid doped pristine ABPBI membrane without BPO4 nanoparticles; and a maximum proton conductivity of 27.3 mS cm−1 was observed in the phosphoric acid doped nanocomposite membrane consisting of 25% BPO4 nanoparticles at 180 °C under anhydrous condition. The enhancement of proton conductivity is attributed to the dangling hydroxyl or geminal hydroxyl groups of the excess phosphoric acid molecules on surface of the BPO4 nanoparticles based on density functional theory calculations. In addition, the blending of BPO4 nanoparticles significantly decreases the methanol vapor permeability through the membrane by about two-fold.Graphical abstractThe sponge-like morphology of H3PO4 doped BPO4–ABPBI guarantees the formation of continuous H3PO4 subphase, maximum contact between H3PO4 and the membrane matrix.Highlights► Proton exchange membrane is based on H3PO4 doped BPO4–ABPBI nanocomposite. ► H3PO4 doped BPO4–ABPBI nanocomposite possesses 3-D network morphology. ► Blending BPO4 to ABPBI improves the proton conductivity. ► The maximum proton conductivity is 27.3 mS cm−1.
Co-reporter:Jin Chen, Liuming Yan, Baohua Yue
Journal of Power Sources 2012 Volume 209() pp:7-14
Publication Date(Web):1 July 2012
DOI:10.1016/j.jpowsour.2012.02.072
A new type of nano-structured LiFePO4 particles were prepared exhibiting enhanced apparent lithium ion diffusion dynamics. The nano-structured LiFePO4 particles possess nano-layered morphology and were converted hydrothermally from the nano-layered templates composed of mainly ferrous metaphosphate and graphitic carbon. And the nano-layered templates were prepared by pyrolysis conversion of the nano-layered ferrous phenylphosphonate templates, or the raw nano-layered templates. Though the nano-layered LiFePO4 particles possess characteristics of nanostructured LiFePO4, the overall dimensions are still in micro-size and the tap density is about 1.36 g cm−3 comparable to the micro-sized LiFePO4 particles. The apparent lithium ion diffusion coefficients are 1.5 × 10−11 and 3.1 × 10−13 cm2 s−1 evaluated using the cyclic voltammetry and electrical impedance spectroscopy, respectively. In addition, the organic moiety from the raw nano-layered templates was converted into tiny carbon particles with abundance of ordered graphitic structure well dispersed in the nano-layered LiFePO4 particles; and the nano-layered LiFePO4 particles possess an electronic conductivity as high as 3.28 mS cm−1.Graphical abstractThe apparent Li+ diffusion coefficient D is enhanced by the nano-layered morphology of the LiFePO4 because of the abundant phase boundaries between the nano-layers.Highlights► Nano-layered LiFePO4 particles are fabricated. ► The nano-layered LiFePO4 particles are converted from nano-templates. ► The lithium ion diffusion dynamics is enhanced. ► The lithium diffusion coefficient is 3.1 × 10−13 cm2 s−1.
Co-reporter:Liqing Xie, Liuming Yan, Chao Sun, Xinluo Zhao
Computational and Theoretical Chemistry 2012 Volume 997() pp:14-18
Publication Date(Web):1 October 2012
DOI:10.1016/j.comptc.2012.07.034
The molecular force field model for polyynes is developed by fitting the empirical force field potential function into intermolecular interaction potentials of various configurations including side-by-side, parallel, crossover, and head-to-head configurations calculated using the Møller–Plesset perturbation theory at MP2/6-311G(d, p) level of theory. And the force field model is applied to the molecular dynamics simulation of polyyne–methanol mixture. The calculations reveal that the polyyne molecules aggregate in bundles, and hydrogen bonds are formed between the polyyne hydrogen and methanol oxygen.Graphical abstractMD simulations based on molecular force field developed are consistent with density functional theory calculations at B3LYP/6-311G(d, p) level of theory.Highlights► The interactions of 420 configurations of polyynes were calculated. ► The molecular force field model for polyynes was developed. ► The force field model was compared with the MP2 potentials. ► Force field model was applied to MD simulation of polyyne–methanol mixture. ► Our results were consistent with literatures.
Co-reporter:Liuming Yan, Qingxia Feng, Liqing Xie, Dongfang Zhang
Solid State Ionics 2011 190(1) pp: 8-17
Publication Date(Web):
DOI:10.1016/j.ssi.2011.03.010
Co-reporter:Suhua Zhu, Liuming Yan, Dongfang Zhang, Qingxia Feng
Polymer 2011 Volume 52(Issue 3) pp:881-892
Publication Date(Web):3 February 2011
DOI:10.1016/j.polymer.2010.12.037
The microscopic structure and hydrogen bonding characteristics of the pristine and phosphoric acid (PA) doped polybenzimidazoles (poly[2,2′-(meta-phenylene)-5,5′-benzimidazole] or m-PBI, and poly[2,2′-(para-phenylene)-5,5′-benzimidazole] or p-PBI) were studied by molecular dynamics simulations based on a united-atom force field model. In all the simulated systems, a benzimidazole group and an adjacent phenylene group are almost in coplanar configuration, while two adjacent benzimidazole groups are in twisted configuration. In pristine PBIs, the p-PBI is more ordered and stretched than the m-PBI. In PA doped PBIs, hydrogen bonding network is formed by donating phosphoric acid proton to imine nitrogen, phosphoric acid proton to phosphoric acid oxygen, and amine proton to phosphoric acid oxygen. In addition, the structural transport of proton is attributed to the formation of hydrogen bonding network in PA doped PBIs.
Co-reporter:Suhua Zhu, Liuming Yan, Xiaobo Ji, Wencong Lu
Journal of Molecular Structure: THEOCHEM 2010 Volume 951(1–3) pp:60-68
Publication Date(Web):15 July 2010
DOI:10.1016/j.theochem.2010.04.008
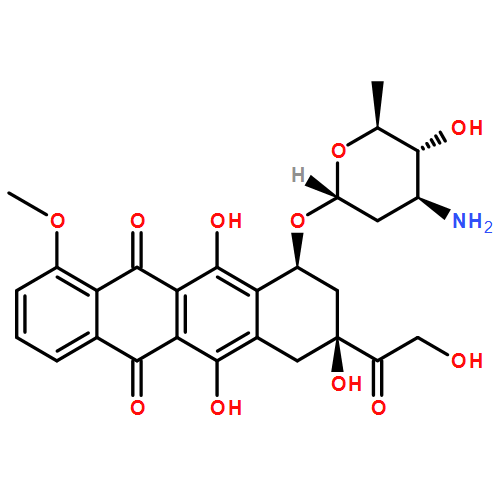
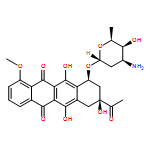
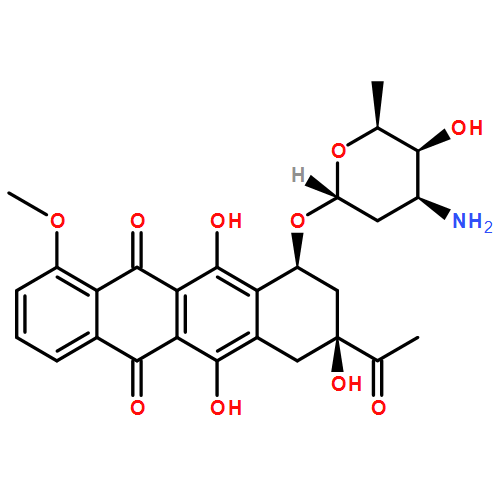
The conformational diversity of doxorubicin, daunorubicin, epirubicin, and idarubicin, is studied based on density functional theory calculations at the B3LYP/6-31G(d,p) level of theory. The calculations identified three conformational domains: the anthracycline quinone–hydroquinone backbone, the anchor, and the daunosamine. The backbone exists in three conformations and three prototropic tautomerizations, the anchor in four conformations relating to the orientations of the C8 and C9 atoms, and the daunosamine also in four conformations according to the distance between the amino nitrogen and the quinone oxygen. The overall molecular conformation is determined by combination of the conformational types of all the three conformational domains. Finally, conformations of intercalated drug molecules reported in the literatures are classified according to these criteria.
Co-reporter:Dongfang Zhang and Liuming Yan
The Journal of Physical Chemistry B 2010 Volume 114(Issue 38) pp:12234-12241
Publication Date(Web):September 1, 2010
DOI:10.1021/jp1054606
The acid−base equilibrium in acid (phenylphosphonic acid, methylsulfonic acid, phosphoric acid, hydrochloric acid, acetic acid, and sulfuric acid)−benzimidazole (BIm) complexes was studied by 1H NMR spectra, FTIR spectra, and density functional theory (DFT) calculations. The DFT optimized structures of the acid−BIm complexes, the vibrational frequencies of the acidic protons, and potential profiles were applied to study the equilibrium between the acids and BIm. In gas phase, it was shown that the strong sulfuric acid could completely protonate BIm and the other acids could only incompletely protonate BIm. When the polarizable continuum model of DMSO was applied, it was shown that all the acids could completely protonate BIm, except the weak acetic acid and phenylphosphonic acid. Furthermore, the potential profile for the PPoA−BIm shows double-well structure facilitating the free movement of the acidic proton between PPoA and BIm. If an explicit solvent molecule of DMSO was included into an acid−BIm complex, significant changes in equilibrium between acid and BIm are observed. The potential profiles for the MSA−BIm and HCl−BIm show very flat bottom wells, facilitating the free movement of proton between the acid and BIm. These calculations were consistent with the 1H NMR chemical shifts of the immobile protons of the benzimidazole ring in DMSO-d6 and the FTIR spectra of the acid−BIm complexes.
Co-reporter:Xiaobo Ji, Liuming Yan, Suhua Zhu, Liangmiao Zhang and Wencong Lu
The Journal of Physical Chemistry B 2008 Volume 112(Issue 49) pp:15616-15627
Publication Date(Web):November 14, 2008
DOI:10.1021/jp8066469
The methanol distribution and electroosmotic drag in hydrated poly(perfluorosulfonic) acid electrolyte membrane are studied using molecular dynamics simulations under various electric fields applied. The results indicate that the methanol molecules are preferentially distributed near the hydrophobic PFSA backbones with their methyl groups in contact with the fluorine atoms and their hydroxyl groups pointing to the hydrophilic subphase. As the hydroxyl groups of methanol forming hydrogen bonds, hydroxyl groups are more likely to accept hydrogen atoms than to donate hydrogen atoms. The calculated methanol diffusion coefficient is in good correspondence with experimental values, and the electroosmotic drag coefficient for methanol is much smaller than that of water molecules.
Co-reporter:Liuming Yan, Yi Lu and Xuejiao Li
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 7) pp:NaN5536-5536
Publication Date(Web):2016/01/07
DOI:10.1039/C5CP06638G
A density functional theory (DFT) protocol for the calculation of redox potentials of copper complexes is developed based on 13 model copper complexes. The redox potentials are calculated in terms of Gibbs free energy change of the redox reaction at the theory level of CAM-B3LYP/6-31+G(d,p)/SMD, with the overall Gibbs free energy change being partitioned into the Gibbs free energy change of the gas phase reaction and the Gibbs free energy change of solvation. In addition, the calculated Gibbs free energy change of solvation is corrected by a unified correction factor of −0.258 eV as the second-layer Gibbs free energy change of solvation and other interactions for each redox reaction. And an empirical Gibbs free energy change of solvation at −0.348 eV is applied to each water molecule if the number of inner-sphere water molecule changes during the redox reaction. Satisfactory agreements between the DFT calculated and experimental results are obtained, with a maximum absolute error at 0.197 V, a mean absolute error at 0.114 V and a standard deviation at 0.133 V. Finally, it is concluded that the accurate prediction of redox potentials is dependent on the accurate prediction of geometrical structures as well as on geometrical conservation during the redox reaction.
Co-reporter:Chao Sun, Liuming Yan, Baohua Yue, Huiting Liu and Yanfeng Gao
Journal of Materials Chemistry A 2014 - vol. 2(Issue 43) pp:NaN9293-9293
Publication Date(Web):2014/09/02
DOI:10.1039/C4TC00778F
The modulation of metal–insulator transition (MIT) temperature and phase stability of thermochromic materials based on all the transition metal doped VO2 were systematically studied using density functional theory (DFT) calculations. The free energies, formation enthalpies, and Fermi energies of transition metal doped VO2 were evaluated from DFT calculations; the cell volumes and bulk moduli were obtained by fitting the free energies to the Birch–Murnaghan equation of states; and the decomposition enthalpies and entropies of the transition metal doped VO2 were calculated using both experimental data and DFT calculations. Based on these results, the MIT temperature was associated with lattice distortion of VO2 (M1) upon doping, the expansion of cell volume and the decrease in β-angle were associated with the decrease in MIT temperature, and the shrinkage of cell volume and the increase in β-angle were associated with the increase in MIT temperature. And it was also concluded that VO2 (M1) doped with high valence cations is more stable than those doped with low valence cations. These conclusions are consistent with experimental facts that W-, Mo-, and Re- are the most studied and the most effective dopants for the reduction of MIT temperature, and La-, Hg-, and Ag-doped VO2 undergoes phase separation. In addition, DFT calculations without spin-polarization were also carried out, and the influence of spin-polarization was evaluated. Finally, scandium was proposed as a potential dopant for VO2 in view of balanced comprehensive performance.



![Benzo[uv]naphtho[2,1,8,7-defg]pentaphene](http://img.cochemist.com/ccimg/120900/120835-86-1.png)
![Benzo[uv]naphtho[2,1,8,7-defg]pentaphene](http://img.cochemist.com/ccimg/120900/120835-86-1_b.png)
![Dibenz[bc,ef]ovalene](http://img.cochemist.com/ccimg/74400/74335-52-7.png)
![Dibenz[bc,ef]ovalene](http://img.cochemist.com/ccimg/74400/74335-52-7_b.png)


![Poly([5,5'-bi-1H-benzimidazole]-2,2'-diyl-1,3-phenylene)](/data/chemimg/472200/25734-65-0.png)
![Poly([5,5'-bi-1H-benzimidazole]-2,2'-diyl-1,3-phenylene)](/data/chemimg/472200/25734-65-0_b.png)


![dibenzo[a,ghi]perylene](http://img.cochemist.com/ccimg/6600/6596-37-8.png)
![dibenzo[a,ghi]perylene](http://img.cochemist.com/ccimg/6600/6596-37-8_b.png)