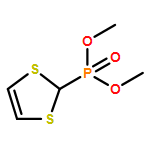
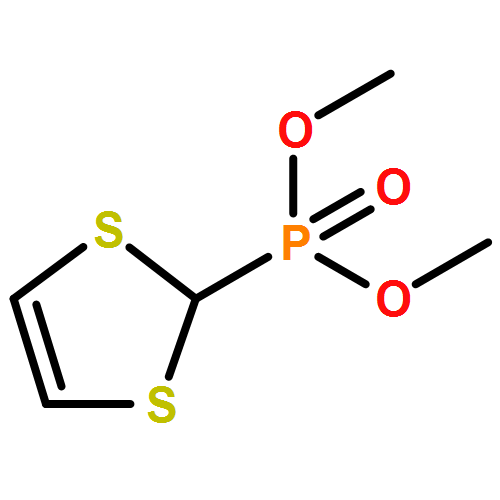
Co-reporter:Rafael Sandoval-Torrientes;Joaquín Calbo;David García-Fresnadillo;José Santos;Nazario Martín
Organic Chemistry Frontiers 2017 vol. 4(Issue 6) pp:1024-1028
Publication Date(Web):2017/05/31
DOI:10.1039/C6QO00760K
A series of new broad-absorbing dyes based on rhodanine derivatives conjugated with triarylamines using a fluorene backbone was synthesized. Spectroscopic and electrochemical studies, along with density functional theory (DFT) calculations, provided clear insight into the electronic and optical properties of the dyes, which efficiently absorb in the entire visible spectrum.
Co-reporter:Inés García-Benito;Iwan Zimmermann;Javier Urieta-Mora;Juan Aragó;Agustín Molina-Ontoria;Nazario Martín;Mohammad Khaja Nazeeruddin
Journal of Materials Chemistry A 2017 vol. 5(Issue 18) pp:8317-8324
Publication Date(Web):2017/05/10
DOI:10.1039/C7TA00997F
Engineering of inorganic–organic lead halide perovskites for photovoltaic applications has experienced significant advances in recent years. However, the use of the relatively expensive spiro-OMeTAD as a hole-transporting material (HTM) poses a challenge due to dopant-induced degradation. Herein we introduce two new three-armed and four-armed HTMs (BTT-4 and BTT-5) based on isomeric forms of benzotrithiophene (BTT). The isomerism impact on the optical, electrochemical and photophysical properties and the photovoltaic performance is systematically investigated. Perovskite solar cells (PSCs) using BTT-4 and BTT-5 as HTMs show remarkable light-to-energy conversion efficiencies of 19.0% and 18.2%, respectively, under standard measurement conditions. These results validate the readily available BTT heteroaromatic structure as a valuable core for the design of highly efficient HTMs for the preparation of PSCs.
Co-reporter:Marina Garrido;Joaquín Calbo;Laura Rodríguez-Pérez;Juan Aragó;Ma Ángeles Herranz;Nazario Martín
Chemical Communications 2017 vol. 53(Issue 92) pp:12402-12405
Publication Date(Web):2017/11/16
DOI:10.1039/C7CC07836F
Graphene nanobuds were prepared via the non-covalent anchoring of C60-based molecules endowed with one or three pyrene units, respectively. TGA, FTIR, UV-Vis and TEM investigations confirmed the formation of nanohybrids. For the two molecular derivatives, striking differences were determined in their interaction with graphene or carbon surfaces by Raman, cyclic voltammetry and molecular mechanics calculations, revealing the important role of pyrene adsorption in modulating the electronic properties of the nanohybrids.
Co-reporter:Joaquín Calbo;Rafael Viruela;Juan Aragó
Theoretical Chemistry Accounts 2017 Volume 136( Issue 6) pp:73
Publication Date(Web):23 May 2017
DOI:10.1007/s00214-017-2100-4
This work presents an analysis of the evolution of the structural, electronic and optical properties of a series of benzotrithiophene (BTT) derivatives, decorated with peripheral electron-donor bulky groups, with potential as hole-transporting materials (HTMs) in perovskite solar cells. The analysis is performed on the basis of density functional theory calculations. Theoretical calculations show that the bulky p-methoxydiphenylamine (OMeDPA) and p-methoxydiphenylamine-substituted carbazole (OMeDPAC) groups give rise to highly congested molecular structures. In contrast, the incorporation of p-methoxytriphenylamine (OMeTPA) groups leads to an almost planar structure that is suited for an optimal stacking aggregation with beneficial implications in the charge transport and in the performance of the photovoltaic device. The electronic properties calculated for neutral and cation species reveal the good electron-donor behavior of the BTT derivatives. The small reorganization energies calculated for the BTT derivatives are similar to those reported for other excellent HTMs and support the potential of the sulfur-rich BTT core to design new π-conjugated HTMs. Calculations properly account for the changes observed in the absorption spectra of the BTT derivatives as a function of the peripheral groups attached. Whereas the OMeDPA and OMeTPA groups produce an intensity increase and a red shift of the main absorption band, the OMeDPAC group shifts this band to the blue.
Co-reporter:Dr. György Szalóki;Dr. Vincent Croué;Dr. Vincent Carré; Frédéric Aubriet;Dr. Olivier Alévêque;Dr. Eric Levillain;Magali Allain;Dr. Juan Aragó; Enrique Ortí;Dr. Sébastien Goeb; Marc Sallé
Angewandte Chemie International Edition 2017 Volume 56(Issue 51) pp:16272-16276
Publication Date(Web):2017/12/18
DOI:10.1002/anie.201709483
AbstractA proof-of-concept related to the redox-control of the binding/releasing process in a host–guest system is achieved by designing a neutral and robust Pt-based redox-active metallacage involving two extended-tetrathiafulvalene (exTTF) ligands. When neutral, the cage is able to bind a planar polyaromatic guest (coronene). Remarkably, the chemical or electrochemical oxidation of the host–guest complex leads to the reversible expulsion of the guest outside the cavity, which is assigned to a drastic change of the host–guest interaction mode, illustrating the key role of counteranions along the exchange process. The reversible process is supported by various experimental data (1H NMR spectroscopy, ESI-FTICR, and spectroelectrochemistry) as well as by in-depth theoretical calculations performed at the density functional theory (DFT) level.
Co-reporter:Dr. György Szalóki;Dr. Vincent Croué;Dr. Vincent Carré; Frédéric Aubriet;Dr. Olivier Alévêque;Dr. Eric Levillain;Magali Allain;Dr. Juan Aragó; Enrique Ortí;Dr. Sébastien Goeb; Marc Sallé
Angewandte Chemie 2017 Volume 129(Issue 51) pp:16490-16494
Publication Date(Web):2017/12/18
DOI:10.1002/ange.201709483
AbstractA proof-of-concept related to the redox-control of the binding/releasing process in a host–guest system is achieved by designing a neutral and robust Pt-based redox-active metallacage involving two extended-tetrathiafulvalene (exTTF) ligands. When neutral, the cage is able to bind a planar polyaromatic guest (coronene). Remarkably, the chemical or electrochemical oxidation of the host–guest complex leads to the reversible expulsion of the guest outside the cavity, which is assigned to a drastic change of the host–guest interaction mode, illustrating the key role of counteranions along the exchange process. The reversible process is supported by various experimental data (1H NMR spectroscopy, ESI-FTICR, and spectroelectrochemistry) as well as by in-depth theoretical calculations performed at the density functional theory (DFT) level.
Co-reporter:Luis Moreira, Joaquín Calbo, Juan Aragó, Beatriz M. Illescas, Iwona Nierengarten, Béatrice Delavaux-Nicot, Enrique Ortí, Nazario Martín, and Jean-François Nierengarten
Journal of the American Chemical Society 2016 Volume 138(Issue 47) pp:15359-15367
Publication Date(Web):September 17, 2016
DOI:10.1021/jacs.6b07250
Two new conjugated porphyrin-based systems (dimers 3 and 4) endowed with suitable crown ethers have been synthesized as receptors for a fullerene-ammonium salt derivative (1). Association constants in solution have been determined by UV–vis titration experiments in CH2Cl2 at room temperature. The designed hosts are able to associate up to two fullerene-based guest molecules and present association constants as high as ∼5 × 108 M–1. Calculation of the allosteric cooperative factor α for supramolecular complexes [3·12] and [4·12] showed a negative cooperative effect in both cases. The interactions accounting for the formation of the associates are based, first, on the complementary ammonium-crown ether interaction and, second, on the π–π interactions between the porphyrin rings and the C60 moieties. Theoretical calculations have evidenced a significant decrease of the electron density in the porphyrin dimers 3 and 4 upon complexation of the first C60 molecule, in good agreement with the negative cooperativity found in these systems. This negative effect is partially compensated by the stabilizing C60–C60 interactions that take place in the more stable syn-disposition of [4·12].
Co-reporter:Sarah Keller, Antonio Pertegás, Giulia Longo, Laura Martínez, Jesús Cerdá, José M. Junquera-Hernández, Alessandro Prescimone, Edwin C. Constable, Catherine E. Housecroft, Enrique Ortí and Henk J. Bolink
Journal of Materials Chemistry A 2016 vol. 4(Issue 17) pp:3872-3872
Publication Date(Web):05 Feb 2016
DOI:10.1039/C6TC90030E
Correction for ‘Shine bright or live long: substituent effects in [Cu(N^N)(P^P)]+-based light-emitting electrochemical cells where N^N is a 6-substituted 2,2′-bipyridine’ by Sarah Keller et al., J. Mater. Chem. C, 2016, DOI: 10.1039/c5tc03725e.
Co-reporter:Sarah Keller, Antonio Pertegás, Giulia Longo, Laura Martínez, Jesús Cerdá, José M. Junquera-Hernández, Alessandro Prescimone, Edwin C. Constable, Catherine E. Housecroft, Enrique Ortí and Henk J. Bolink
Journal of Materials Chemistry A 2016 vol. 4(Issue 17) pp:3857-3871
Publication Date(Web):25 Jan 2016
DOI:10.1039/C5TC03725E
We report [Cu(P^P)(N^N)][PF6] complexes with P^P = bis(2-(diphenylphosphino)phenyl)ether (POP) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos) and N^N = 6-methyl-2,2′-bipyridine (Mebpy), 6-ethyl-2,2′-bipyridine (Etbpy), 6,6′-dimethyl-2,2′-bipyridine (Me2bpy) or 6-phenyl-2,2′-bipyridine (Phbpy). The crystal structures of [Cu(POP)(Phbpy)][PF6]·Et2O, [Cu(POP)(Etbpy)][PF6]·Et2O, [Cu(xantphos)(Me2bpy)][PF6], [Cu(xantphos)(Mebpy)][PF6]·CH2Cl2·0.4Et2O, [Cu(xantphos)(Etbpy)][PF6]·CH2Cl2·1.5H2O and [Cu(xantphos)(Phbpy)][PF6] are described; each copper(I) centre is distorted tetrahedral. In the crystallographically determined structures, the N^N domain in [Cu(xantphos)(Phbpy)]+ and [Cu(POP)(Phbpy)]+ is rotated ∼180° with respect to its orientation in [Cu(xantphos)(Mebpy)]+, [Cu(POP)(Etbpy)]+ and [Cu(xantphos)(Etbpy)]+; in each complex containing xantphos, the xanthene ‘bowl’ retains the same conformation in the solid-state structures. The two conformers resulting from the 180° rotation of the N^N ligand were optimized at the B3LYP-D3/(6-31G**+LANL2DZ) level and are close in energy for each complex. Variable temperature NMR spectroscopy evidences the presence of two conformers of [Cu(xantphos)(Phbpy)]+ in solution which are related by inversion of the xanthene unit. The complexes exhibit MLCT absorption bands in the range 378 to 388 nm, and excitation into each MLCT band leads to yellow emissions. Photoluminescence quantum yields (PLQYs) increase from solution to thin-film and powder; the highest PLQYs are observed for powdered [Cu(xantphos)(Mebpy)][PF6] (34%), [Cu(xantphos)(Etbpy)][PF6] (37%) and [Cu(xantphos)(Me2bpy)][PF6] (37%) with lifetimes of 9.6–11 μs. Density functional theory calculations predict that the emitting triplet (T1) involves an electron transfer from the Cu–P^P environment to the N^N ligand and therefore shows a 3MLCT character. T1 is calculated to be ∼0.20 eV lower in energy than the first singlet excited state (S1). The [Cu(P^P)(N^N)][PF6] ionic transition-metal (iTMC) complexes were tested in light-emitting electrochemical cells (LECs). Turn-on times are fast, and the LEC with [Cu(xantphos)(Me2bpy)][PF6] achieves a maximum efficacy of 3.0 cd A−1 (luminance = 145 cd m−2) with a lifetime of 1 h; on going to the [Cu(xantphos)(Mebpy)][PF6]-based LEC, the lifetime exceeds 15 h but at the expense of the efficacy (1.9 cd A−1). The lifetimes of LECs containing [Cu(xantphos)(Etbpy)][PF6] and [Cu(POP)(Etbpy)][PF6] exceed 40 and 80 h respectively.
Co-reporter:Mateusz Wielopolski, Magdalena Marszalek, Fulvio G. Brunetti, Damien Joly, Joaquín Calbo, Juan Aragó, Jacques-E. Moser, Robin Humphry-Baker, Shaik M. Zakeeruddin, Juan Luis Delgado, Michael Grätzel, Enrique Ortí and Nazario Martín
Journal of Materials Chemistry A 2016 vol. 4(Issue 17) pp:3798-3808
Publication Date(Web):09 Dec 2015
DOI:10.1039/C5TC03501E
The development of new light harvesting materials is a key issue for the progress of the research on organic & hybrid photovoltaics. Here, we report a new class of organic sensitizers based on the bi-fluorenylidene moiety as π-linker within the donor–π-linker–acceptor (D–π–A) scheme. The new dyes are endowed with electron donor and electron acceptor units at strategic positions in order to improve their electronic and light-harvesting properties. The comprehensive study of these compounds through the use of different experimental and theoretical techniques, provides an in-depth understanding of their electronic and photophysical properties, and reveal their interest as photovoltaic materials.
Co-reporter:Julia Buendía, Joaquín Calbo, Fátima García, Juan Aragó, Pedro M. Viruela, Enrique Ortí and Luis Sánchez
Chemical Communications 2016 vol. 52(Issue 42) pp:6907-6910
Publication Date(Web):25 Apr 2016
DOI:10.1039/C6CC02681H
The cooperative supramolecular polymerization of 1 and 2 yields P- or M-type helical aggregates depending on the absolute configuration (S or R) of the stereogenic centres attached to the side chains. The connectivity of the amide group does not affect the handedness of the helical aggregates, but determines a larger cooperativity for retroamides 1.
Co-reporter:Cathrin D. Ertl, Lidón Gil-Escrig, Jesús Cerdá, Antonio Pertegás, Henk J. Bolink, José M. Junquera-Hernández, Alessandro Prescimone, Markus Neuburger, Edwin C. Constable, Enrique Ortí and Catherine E. Housecroft
Dalton Transactions 2016 vol. 45(Issue 29) pp:11668-11681
Publication Date(Web):12 May 2016
DOI:10.1039/C6DT01325B
A series of regioisomeric cationic iridium complexes of the type [Ir(C^N)2(bpy)][PF6] (bpy = 2,2′-bipyridine) is reported. The complexes contain 2-phenylpyridine-based cyclometallating ligands with a methylsulfonyl group in either the 3-, 4- or 5-position of the phenyl ring. All the complexes have been fully characterized, including their crystal structures. In acetonitrile solution, all the compounds are green emitters with emission maxima between 493 and 517 nm. Whereas substitution meta to the Ir–C bond leads to vibrationally structured emission profiles and photoluminescence quantum yields of 74 and 77%, placing a sulfone substituent in a para position results in a broad, featureless emission band, an enhanced quantum yield of 92% and a shorter excited-state lifetime. These results suggest a larger ligand-centred (3LC) character of the emissive triplet state in the case of meta substitution and a more pronounced charge transfer (CT) character in the case of para substitution. Going from solution to the solid state (powder samples and thin films), the emission maxima are red-shifted for all the complexes, resulting in green-yellow emission. Data obtained from electrochemical measurements and density functional theory calculations parallel the photophysical trends. Light-emitting electrochemical cells (LECs) based on the complexes were fabricated and evaluated. A maximum efficiency of 4.5 lm W−1 at a maximum luminance of 940 cd m−2 was observed for the LEC with the complex incorporating the sulfone substituent in the 4-position when operated under pulsed current driving conditions.
Co-reporter:Luis Moreira, Joaquín Calbo, Rafael M. Krick Calderon, José Santos, Beatriz M. Illescas, Juan Aragó, Jean-François Nierengarten, Dirk M. Guldi, Enrique Ortí and Nazario Martín
Chemical Science 2015 vol. 6(Issue 8) pp:4426-4432
Publication Date(Web):18 May 2015
DOI:10.1039/C5SC00850F
A series of exTTF-(crown ether)2 receptors, designed to host C60, has been prepared. The size of the crown ether and the nature of the heteroatoms have been systematically changed to fine tune the association constants. Electrochemical measurements and transient absorption spectroscopy assisted in corroborating charge transfer in the ground state and in the excited state, leading to the formation of radical ion pairs featuring lifetimes in the range from 12 to 21 ps. To rationalize the nature of the exTTF-(crown ether)2·C60 stabilizing interactions, theoretical calculations have been carried out, suggesting a synergetic interplay of donor–acceptor, π–π, n–π and CH⋯π interactions, which is the basis for the affinity of our novel receptors towards C60.
Co-reporter:Alberto de Juan, Alejandro López-Moreno, Joaquín Calbo, Enrique Ortí and Emilio M. Pérez
Chemical Science 2015 vol. 6(Issue 12) pp:7008-7014
Publication Date(Web):07 Sep 2015
DOI:10.1039/C5SC02916C
Single-walled carbon nanotubes (SWNTs) are one of the most promising nanomaterials and their supramolecular chemistry has attracted a lot of attention. However, despite well over a decade of research, there is no standard method for the quantification of their noncovalent chemistry in solution/suspension. Here, we describe a simple procedure for the determination of association constants (Ka) between soluble molecules and insoluble and heterogeneous carbon nanotube samples. To test the scope of the method, we report binding constants between five different hosts and two types of SWNTs in four solvents. We have determined numeric values of Ka in the range of 1–104 M−1. Solvent effects as well as structural changes in both the host and guest result in noticeable changes of Ka. The results obtained experimentally were validated through state-of-the-art DFT calculations. The generalization of quantitative and comparable association constants data should significantly help advance the supramolecular chemistry of carbon nanotubes.
Co-reporter:Jorge S. Valera, Joaquín Calbo, Rafael Gómez, Enrique Ortí and Luis Sánchez
Chemical Communications 2015 vol. 51(Issue 50) pp:10142-10145
Publication Date(Web):13 May 2015
DOI:10.1039/C5CC03616J
The supramolecular polymerization of pyrene imidazoles 1 and 2, governed by H-bonding and C–H⋯π interactions, yields aggregates showing the characteristic bluish emission pattern of pyrene-based monomers.
Co-reporter:Belén Nieto-Ortega, Fátima García, Giovanna Longhi, Ettore Castiglioni, Joaquín Calbo, Sergio Abbate, Juan T. López Navarrete, Francisco J. Ramírez, Enrique Ortí, Luis Sánchez and Juan Casado
Chemical Communications 2015 vol. 51(Issue 48) pp:9781-9784
Publication Date(Web):30 Apr 2015
DOI:10.1039/C5CC03054D
A complete chiroptical characterization of the supramolecular polymers formed by tricarboxamides (S)-1 and (R)-1 is performed using ECD, VCD and CPL dichroic techniques. The helical aggregates show an intense CPL signal and their absolute P- or M-configuration is assigned with the help of theoretical calculations.
Co-reporter:Paula Pla, José M. Junquera-Hernández, Henk J. Bolink and Enrique Ortí
Dalton Transactions 2015 vol. 44(Issue 18) pp:8497-8505
Publication Date(Web):05 Nov 2014
DOI:10.1039/C4DT03046J
A theoretical density functional theory study has been performed on different families of cationic cyclometallated Ir(III) complexes with the general formula [Ir(C^N)2(N^N)]+ and azole-based ligands. The goal was to investigate the effect that the number and position of the nitrogen atoms of the azole ring have on the electronic structure and emission wavelength of the complex. The increase in the number of nitrogen atoms changes the relative energy of the HOMO and LUMO levels and leads to a gradual shift in the emission wavelength that can be larger than 100 nm. The direction of the shift however depends on the ligand in which the azole ring is introduced. The emission shifts to bluer wavelengths when the azole forms part of the cyclometallating C^N ligands, whereas it shifts to the red when the 5-membered ring is incorporated into the ancillary N^N ligand. The position of the nitrogen atoms in the azole ring also plays an important role in determining the emission energy. Complexes with phenyl-azole C^N ligands bearing a nitrogen in the azole position to which the phenyl is linked show a markedly blue-shifted emission compared to complexes with the same number of nitrogen atoms in the azole ring and bearing a carbon atom in that position. Therefore, when comparing the emission properties of azole-based [Ir(C^N)2(N^N)]+ complexes, not only the number of nitrogen atoms of the azole but also their position in the ring and the ligand where the azole ring is incorporated should be taken into account.
Co-reporter:Beatriz Pelado;Dr. Fawzi Abou-Chahine;Joaquín Calbo;Dr. Rubén Caballero;Dr. Pilar delaCruz;Dr. José M. Junquera-Hernández; Enrique Ortí; Nikolai V. Tkachenko; Ferno Langa
Chemistry - A European Journal 2015 Volume 21( Issue 15) pp:5814-5825
Publication Date(Web):
DOI:10.1002/chem.201406514
Abstract
The role of π-conjugated molecular bridges in through-space and through-bond electron transfer is studied by comparing two porphyrin–fullerene donor–acceptor (D–A) dyads. One dyad, ZnP–Ph–C60 (ZnP=zinc porphyrin), incorporates a phenyl bridge between D and A and behaves very similarly to analogous dyads studied previously. The second dyad, ZnP–EDOTV–C60, introduces an additional 3,4-ethylenedioxythienylvinylene (EDOTV) unit into the conjugated bridge, which increases the distance between D and A, but, at the same time, provides increased electronic communication between them. Two essential outcomes that result from the introduction of the EDOTV unit in the bridge are as follows: 1) faster charge recombination, which indicates enhanced electronic coupling between the charge-separated and ground electronic states; and 2) the disappearance of the intramolecular exciplex, which mediates photoinduced charge separation in the ZnP–Ph–C60 dyad. The latter can be interpreted as a gradual decrease in electronic coupling between locally excited singlet states of D and A when introducing the EDOTV unit into the D–A bridge.
Co-reporter:Dr. Luis Moreira;Joaquín Calbo;Dr. Beatriz M. Illescas;Dr. Juan Aragó;Dr. Iwona Nierengarten;Dr. Béatrice Delavaux-Nicot;Dr. Enrique Ortí;Dr. Nazario Martín;Dr. Jean-François Nierengarten
Angewandte Chemie International Edition 2015 Volume 54( Issue 4) pp:1255-1260
Publication Date(Web):
DOI:10.1002/anie.201409487
Abstract
A fullerene ammonium derivative has been combined with different metalloporphyrin–crown ether receptors to generate very stable supramolecules. The combination of fullerene–porphyrin and ammonium–crown ether interactions leads to a strong chelate effect as evidenced by a high effective molarity (3.16 M). The different parameters influencing the stability of the supramolecular ensembles, in particular the nature of the metal in the porphyrin moiety, have been rationalized with the help of theoretical calculations thus providing new insights into fullerene–porphyrin interactions.
Co-reporter:Dr. Luis Moreira;Joaquín Calbo;Dr. Beatriz M. Illescas;Dr. Juan Aragó;Dr. Iwona Nierengarten;Dr. Béatrice Delavaux-Nicot;Dr. Enrique Ortí;Dr. Nazario Martín;Dr. Jean-François Nierengarten
Angewandte Chemie 2015 Volume 127( Issue 4) pp:1271-1276
Publication Date(Web):
DOI:10.1002/ange.201409487
Abstract
A fullerene ammonium derivative has been combined with different metalloporphyrin–crown ether receptors to generate very stable supramolecules. The combination of fullerene–porphyrin and ammonium–crown ether interactions leads to a strong chelate effect as evidenced by a high effective molarity (3.16 M). The different parameters influencing the stability of the supramolecular ensembles, in particular the nature of the metal in the porphyrin moiety, have been rationalized with the help of theoretical calculations thus providing new insights into fullerene–porphyrin interactions.
Co-reporter:Julia Guilleme;Dr. María J. Mayoral;Joaquín Calbo;Dr. Juan Aragó;Dr. Pedro M. Viruela; Enrique Ortí; Tomás Torres;Dr. David González-Rodríguez
Angewandte Chemie International Edition 2015 Volume 54( Issue 8) pp:2543-2547
Publication Date(Web):
DOI:10.1002/anie.201411272
Abstract
A combination of spectroscopy (UV/Vis absorption, emission, and circular dichroism), microscopy (AFM and TEM), and computational studies reveal the formation of non-centrosymmetric homochiral columnar subphthalocyanine assemblies. These assemblies form through a cooperative supramolecular polymerization process driven by hydrogen-bonding between amide groups, π–π stacking, and dipolar interactions between axial BF bonds.
Co-reporter:Julia Guilleme;Dr. María J. Mayoral;Joaquín Calbo;Dr. Juan Aragó;Dr. Pedro M. Viruela; Enrique Ortí; Tomás Torres;Dr. David González-Rodríguez
Angewandte Chemie 2015 Volume 127( Issue 8) pp:2573-2577
Publication Date(Web):
DOI:10.1002/ange.201411272
Abstract
A combination of spectroscopy (UV/Vis absorption, emission, and circular dichroism), microscopy (AFM and TEM), and computational studies reveal the formation of non-centrosymmetric homochiral columnar subphthalocyanine assemblies. These assemblies form through a cooperative supramolecular polymerization process driven by hydrogen-bonding between amide groups, π–π stacking, and dipolar interactions between axial BF bonds.
Co-reporter:Gabriel E. Schneider, Antonio Pertegás, Edwin C. Constable, Catherine E. Housecroft, Nik Hostettler, Collin D. Morris, Jennifer A. Zampese, Henk J. Bolink, José M. Junquera-Hernández, Enrique Ortí and Michele Sessolo
Journal of Materials Chemistry A 2014 vol. 2(Issue 34) pp:7047-7055
Publication Date(Web):10 Jul 2014
DOI:10.1039/C4TC01171F
The synthesis and characterization of a new cationic bis-cyclometallated iridium(III) complex and its use in solid-state light-emitting electrochemical cells (LECs) are described. The complex [Ir(ppy)2(Naphbpy)][PF6], where Hppy = 2-phenylpyridine and Naphbpy = 6-(2-naphthyl)-2,2′-bipyridine, incorporates a pendant 2-naphthyl unit that π-stacks face-to-face with the adjacent ppy− ligand and acts as a peripheral bulky group. The complex presents a structureless emission centred around 595–600 nm both in solution and in thin film with relatively low photoluminescence quantum yields compared with analogous systems. Density functional theory calculations support the charge transfer character of the emitting triplet state and rationalize the low quantum yields in terms of a ligand-centred triplet localized on the 2-naphthyl unit that lies close in energy to the emitting state. LECs incorporating the [Ir(ppy)2(Naphbpy)][PF6] complex as the electroluminescent material are driven using a pulsed current operation mode and show high luminance, exceeding 300 cd m−2, and exceptional stabilities.
Co-reporter:Alejandro López-Moreno, David Clemente-Tejeda, Joaquín Calbo, Atena Naeimi, Francisco A. Bermejo, Enrique Ortí and Emilio M. Pérez
Chemical Communications 2014 vol. 50(Issue 66) pp:9372-9375
Publication Date(Web):27 Jun 2014
DOI:10.1039/C4CC04026K
We present a mild catalytic method to oxidize PAHs and, in particular, pyrene. The pyrenediones are much better electron acceptors than benzoquinone in the gas phase and present similar accepting abilities in solution.
Co-reporter:Filippo Monti, Andrea Baschieri, Isacco Gualandi, Juan J. Serrano-Pérez, José M. Junquera-Hernández, Domenica Tonelli, Andrea Mazzanti, Sara Muzzioli, Stefano Stagni, Cristina Roldan-Carmona, Antonio Pertegás, Henk J. Bolink, Enrique Ortí, Letizia Sambri, and Nicola Armaroli
Inorganic Chemistry 2014 Volume 53(Issue 14) pp:7709-7721
Publication Date(Web):July 8, 2014
DOI:10.1021/ic500999k
Ir(III) cationic complexes with cyclometalating tetrazolate ligands were prepared for the first time, following a two-step strategy based on (i) a silver-assisted cyclometalation reaction of a tetrazole derivative with IrCl3 affording a bis-cyclometalated solvato-complex P ([Ir(ptrz)2(CH3CN)2]+, Hptrz = 2-methyl-5-phenyl-2H-tetrazole); (ii) a substitution reaction with five neutral ancillary ligands to get [Ir(ptrz)2L]+, with L = 2,2′-bypiridine (1), 4,4′-di-tert-butyl-2,2′-bipyridine (2), 1,10-phenanthroline (3), and 2-(1-phenyl-1H-1,2,3-triazol-4-yl)pyridine (4), and [Ir(ptrz)2L2]+, with L = tert-butyl isocyanide (5). X-ray crystal structures of P, 2, and 3 were solved. Electrochemical and photophysical studies, along with density functional theory calculations, allowed a comprehensive rationalization of the electronic properties of 1–5. In acetonitrile at 298 K, complexes equipped with bipyridine or phenanthroline ancillary ligands (1–3) exhibit intense and structureless emission bands centered at around 540 nm, with metal-to-ligand and ligand-to-ligand charge transfer (MLCT/LLCT) character; their photoluminescence quantum yields (PLQYs) are in the range of 55–70%. By contrast, the luminescence band of 5 is weak, structured, and blue-shifted and is attributed to a ligand-centered (LC) triplet state of the tetrazolate cyclometalated ligand. The PLQY of 4 is extremely low (<0.1%) since its lowest level is a nonemissive triplet metal-centered (3MC) state. In rigid matrix at 77 K, all of the complexes exhibit intense luminescence. Ligands 1–3 are also strong emitters in solid matrices at room temperature (1% poly(methyl methacrylate) matrix and neat films), with PLQYs in the range of 27–70%. Good quality films of 2 could be obtained to make light-emitting electrochemical cells that emit bright green light and exhibit a maximum luminance of 310 cd m–2. Tetrazolate cyclometalated ligands push the emission of Ir(III) complexes to the blue, when compared to pyrazolate or triazolate analogues. More generally, among the cationic Ir(III) complexes without fluorine substituents on the cyclometalated ligands, 1–3 exhibit the highest-energy MLCT/LLCT emission bands ever reported.
Co-reporter:Joaquín Calbo, Mariachiara Pastore, Edoardo Mosconi, Enrique Ortí and Filippo De Angelis
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 10) pp:4709-4719
Publication Date(Web):13 Jan 2014
DOI:10.1039/C3CP54970D
We present a first-principles DFT investigation of the adsorption geometry on the anatase (101) surface of a prototypical di-branched organic dye based on the extended tetrathiafulvalene moiety, incorporating two anchoring cyanoacrylic acid units. Reduced model systems with one and two anchoring groups have been initially studied to investigate the vibrational frequencies related to TiO2 dye adsorption. Our calculations confirm that the reduced systems can be used as reliable models to study the anchoring modes and that the conclusions extracted from the reduced systems can be extrapolated to the entire molecule. A series of molecular structures have been investigated to simulate the anchoring environment in monodentate- and bidentate-like adsorption modes. The comparison between the theoretical results and the available experimental data suggests a di-anchored monodentate adsorption mode as the most probable adsorption structure. Geometry optimizations of the di-branched model system adsorbed on a periodic slab of anatase (101) allowed us to compare the relative stability of different adsorption conformations and led to a di-anchored monodentate mode as the most stable adsorption structure. Furthermore, ab initio molecular dynamics simulations confirmed this structure as the preferred one, providing additional stabilization by effective hydrogen-bonding to surface oxygens and structure distortion from planarity. The analysis of the partial density of states for the prototypical models confirms that the doubly anchored adsorption provides improved electronic properties compared to the singly anchored structures for dye-sensitized solar cell purposes.
Co-reporter:Andreas M. Bünzli, Henk J. Bolink, Edwin C. Constable, Catherine E. Housecroft, José M. Junquera-Hernández, Markus Neuburger, Enrique Ortí, Antonio Pertegás, Juan J. Serrano-Pérez, Daniel Tordera and Jennifer A. Zampese
Dalton Transactions 2014 vol. 43(Issue 2) pp:738-750
Publication Date(Web):22 Oct 2013
DOI:10.1039/C3DT52622D
The synthesis and characterization of four iridium(III) complexes [Ir(thpy)2(N^N)][PF6] where Hthpy = 2-(2′-thienyl)pyridine and N^N are 6-phenyl-2,2′-bipyridine (1), 4,4′-di-tbutyl-2,2′-bipyridine (2), 4,4′-di-tbutyl-6-phenyl-2,2′-bipyridine (3) or 4,4′-dimethylthio-2,2′-bipyridine (4) are described. The single crystal structures of ligand 4 and the complexes containing the [Ir(thpy)2(1)]+ and [Ir(thpy)2(4)]+ cations have been determined. In [Ir(thpy)2(1)]+, the pendant phenyl ring engages in an intra-cation π-stacking interaction with one of the thienyl rings in the solid state, and undergoes hindered rotation on the NMR timescale in [Ir(thpy)2(1)]+ and [Ir(thpy)2(3)]+. The solution spectra of [Ir(thpy)2(1)][PF6] and [Ir(thpy)2(4)][PF6] show emission maxima around 640 nm and are significantly red-shifted compared with [Ir(thpy)2(2)][PF6] and [Ir(thpy)2(3)][PF6] which have structured emission bands with maxima around 550 and 590 nm. In thin films, the emission spectra of the four complexes are similar with emission peaks around 550 and 590 nm and a shoulder around 640 nm that are reminiscent of the features observed in solution. In solution, quantum yields are low, but in thin films, values range from 29% for [Ir(thpy)2(1)][PF6] to 51% for [Ir(thpy)2(4)][PF6]. Density functional theory calculations rationalize the structured emission observed for the four complexes in terms of the 3LC nature predicted for the lowest-energy triplet states that mainly involve the cyclometallated [thpy]− ligands. Support for this theoretical result comes from the observed features of the low temperature (in frozen MeCN) photoluminescence spectra of the complexes. Photoluminescence and electroluminescence spectra of the complexes in a light-emitting electrochemical cell (LEC) device configuration have been investigated. The electroluminescence spectra are similar for all [Ir(thpy)2(N^N)][PF6] complexes with emission maxima at ≈600 nm, but device performances are relatively poor probably due to the poor charge-transporting properties of the complexes.
Co-reporter:María Gallego;Joaquín Calbo;Dr. Juan Aragó;Rafael M. KrickCalderon;Ferno H. Liquido;Takahiro Iwamoto;Allison K. Greene;Edward A. Jackson;Dr. Emilio M. Pérez;Dr. Enrique Ortí;Dr. Dirk M. Guldi;Dr. Lawrence T. Scott;Dr. Nazario Martín
Angewandte Chemie International Edition 2014 Volume 53( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/anie.201400662
Co-reporter:María Gallego;Joaquín Calbo;Dr. Juan Aragó;Rafael M. KrickCalderon;Ferno H. Liquido;Takahiro Iwamoto;Allison K. Greene;Edward A. Jackson;Dr. Emilio M. Pérez;Dr. Enrique Ortí;Dr. Dirk M. Guldi;Dr. Lawrence T. Scott;Dr. Nazario Martín
Angewandte Chemie International Edition 2014 Volume 53( Issue 8) pp:2170-2175
Publication Date(Web):
DOI:10.1002/anie.201309672
Abstract
Herein, we investigate the association of a fullerene fragment, hemifullerene C30H12, with an electron-donating bowl-shaped tetrathiafulvalene derivative (truxTTF). UV/Vis titrations and DFT calculations support formation of the supramolecular complex, for which an association constant of log Ka=3.6±0.3 in CHCl3 at room temperature is calculated. Remarkably, electron transfer from truxTTF to C30H12 to form the fully charge-separated species takes place upon irradiation of the associate with light, constituting the first example in which a fullerene fragment mimics the electron-accepting behavior of fullerenes within a supramolecular complex.
Co-reporter:María Gallego;Joaquín Calbo;Dr. Juan Aragó;Rafael M. KrickCalderon;Ferno H. Liquido;Takahiro Iwamoto;Allison K. Greene;Edward A. Jackson;Dr. Emilio M. Pérez;Dr. Enrique Ortí;Dr. Dirk M. Guldi;Dr. Lawrence T. Scott;Dr. Nazario Martín
Angewandte Chemie 2014 Volume 126( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/ange.201400662
Co-reporter:María Gallego;Joaquín Calbo;Dr. Juan Aragó;Rafael M. KrickCalderon;Ferno H. Liquido;Takahiro Iwamoto;Allison K. Greene;Edward A. Jackson;Dr. Emilio M. Pérez;Dr. Enrique Ortí;Dr. Dirk M. Guldi;Dr. Lawrence T. Scott;Dr. Nazario Martín
Angewandte Chemie 2014 Volume 126( Issue 8) pp:2202-2207
Publication Date(Web):
DOI:10.1002/ange.201309672
Abstract
Herein, we investigate the association of a fullerene fragment, hemifullerene C30H12, with an electron-donating bowl-shaped tetrathiafulvalene derivative (truxTTF). UV/Vis titrations and DFT calculations support formation of the supramolecular complex, for which an association constant of log Ka=3.6±0.3 in CHCl3 at room temperature is calculated. Remarkably, electron transfer from truxTTF to C30H12 to form the fully charge-separated species takes place upon irradiation of the associate with light, constituting the first example in which a fullerene fragment mimics the electron-accepting behavior of fullerenes within a supramolecular complex.
Co-reporter:Fátima García, Rubén D. Costa, Juan Aragó, Henk J. Bolink, Enrique Ortí, and Luis Sánchez
Langmuir 2014 Volume 30(Issue 20) pp:5957-5964
Publication Date(Web):2017-2-22
DOI:10.1021/la5006117
The self-assembly of a series of nonionic amphiphilic cruciforms based on the 1,2,4,5-tetrakis(phenylethynyl)benzene (TPEB) skeleton, in which the peripheral substituents have been modified to modulate the morphology of the supramolecular structures, is reported. The presence of linear paraffinic and hydrophilic chains in TPEBs 1 and 2 gives rise to two-dimensional structures of high aspect ratio. In contrast, the incorporation of dendronized hydrophilic chains results in the formation of twisted ribbons in amphiphile 3 and impedes the organized self-assembly of TPEB 4. Theoretical calculations show that the self-assembly of these amphiphiles might be initiated with the formation of π-stacked dimeric units. Compound 2, which self-assembles into different morphologies depending on the solvent, interacts by π-stacking and also by the interdigitation of the peripheral decyl tails to generate bidimensional supramolecular structures. The steric demand exerted by the dendronized polar wedges in 3 and 4 strongly conditions their supramolecular organization. This steric demand together with the interdigitation of the decyl chains results in the self-assembly of cruciform 3 into helical aggregates. However, the lack of the paraffinic chains in 4 impedes this helical organization, and the formation of amorphous material is visualized. The joint experimental and theoretical study presented herein provides relevant guidelines for the modulated self-assembly of nonionic amphiphilic molecules.
Co-reporter:Sebastian B. Meier, Wiebke Sarfert, José M. Junquera-Hernández, Manuel Delgado, Daniel Tordera, Enrique Ortí, Henk J. Bolink, Florian Kessler, Rosario Scopelliti, Michael Grätzel, M. Khaja Nazeeruddin and Etienne Baranoff
Journal of Materials Chemistry A 2013 vol. 1(Issue 1) pp:58-68
Publication Date(Web):23 Oct 2012
DOI:10.1039/C2TC00251E
We report here a new cationic bis-cyclometallated iridium(III) complex, 1, with deep-blue emission at 440 nm and its use in Light-emitting Electrochemical Cells (LECs). The design is based on the 2′,6′-difluoro-2,3′-bipyridine skeleton as the cyclometallating ligand and a bis-imidazolium carbene-type ancillary ligand. Furthermore, bulky tert-butyl substituents are used to limit the intermolecular interactions. LECs have been driven both at constant voltage (6 V) and constant current (2.5 mA cm−2). The performances are significantly improved with the latter method, resulting overall in one of the best reported greenish-blue LECs having fast response (17 s), light intensity over 100 cd m−2 and a reasonable efficiency of almost 5 cd A−1.
Co-reporter:Pierre-Antoine Bouit, Lourdes Infantes, Joaquín Calbo, Rafael Viruela, Enrique Ortí, Juan Luis Delgado, and Nazario Martín
Organic Letters 2013 Volume 15(Issue 16) pp:4166-4169
Publication Date(Web):July 30, 2013
DOI:10.1021/ol401841u
Three new push–pull chromophores based on the 10-(1,3-dithiol-2-ylidene)anthracene core were synthesized and fully characterized. The new chromophores display broad absorption spectra, nearly covering the whole visible region, with high extinction coefficients. Electrochemistry and theoretical calculations allowed the understanding of these singular electronic properties. The molecular structures were unambiguously confirmed by X-ray diffraction.
Co-reporter:Edwin C. Constable, Markus Neuburger, Pirmin Rösel, Gabriel E. Schneider, Jennifer A. Zampese, Catherine E. Housecroft, Filippo Monti, Nicola Armaroli, Rubén D. Costa, and Enrique Ortí
Inorganic Chemistry 2013 Volume 52(Issue 2) pp:885-897
Publication Date(Web):December 26, 2012
DOI:10.1021/ic302026f
Two new heteroleptic iridium(III) complexes [Ir(ppy)2(pyr2bpy)][PF6] ([1a][PF6]) and [Ir(dfppy)2(pyr2bpy)][PF6] ([2a][PF6]), where Hppy = 2-phenylpyridine, Hdfppy = 2-(3,5-difluorophenyl)pyridine, and pyr2bpy = 5,5′-bis(pyren-1-yl)-2,2′-bipyridine, have been synthesized and fully characterized. The single-crystal structures of pyr2bpy and the complexes 4{[1a][PF6]}·2CH2Cl2·9H2O and [2a][PF6]·0.25CH2Cl2·H2O have been determined. The effect of the pyrene substituents on the electronic properties is investigated through a comprehensive photophysical and theoretical study on the two complexes in comparison to reference complexes without substituents on the ancillary ligand ([1][PF6] and [2][PF6]) and by making absorption and luminescence titrations of ligand pyr2bpy. Both theory and experiment show that the intense and broad band appearing in the 400–500 nm region of the absorption spectra of [1a][PF6] and [2a][PF6] is due to intramolecular charge-transfer (ICT) transitions from the pyrene substituents to the bipyridine ligand. [1a][PF6] and [2a][PF6] exhibit luminescence bands centered above 650 nm, attributed to a charge-transfer triplet state located on the pyr2bpy ligand, lying at lower energy than the strongly emitting Ir-ppy→bpy triplet states of the complexes lacking the pyrene fragments. Such luminescence, detected both at room temperature and 77 K, shows that the appendage of luminophoric moieties to luminescent Ir-based centers may further widen the emission tuneability of this exploited class of luminescent materials through purely electrostatic effects exerted on a properly designed N^N ancillary ligand.
Co-reporter:Filippo Monti, Florian Kessler, Manuel Delgado, Julien Frey, Federico Bazzanini, Gianluca Accorsi, Nicola Armaroli, Henk J. Bolink, Enrique Ortí, Rosario Scopelliti, Md. Khaja Nazeeruddin, and Etienne Baranoff
Inorganic Chemistry 2013 Volume 52(Issue 18) pp:10292-10305
Publication Date(Web):September 4, 2013
DOI:10.1021/ic400600d
Charged cyclometalated (C∧N) iridium(III) complexes with carbene-based ancillary ligands are a promising family of deep-blue phosphorescent compounds. Their emission properties are controlled primarily by the main C∧N ligands, in contrast to the classical design of charged complexes where N∧N ancillary ligands with low-energy π* orbitals, such as 2,2'-bipyridine, are generally used for this purpose. Herein we report two series of charged iridium complexes with various carbene-based ancillary ligands. In the first series the C∧N ligand is 2-phenylpyridine, whereas in the second one it is 2-(2,4-difluorophenyl)-pyridine. One bis-carbene (:C∧C:) and four different pyridine–carbene (N∧C:) chelators are used as bidentate ancillary ligands in each series. Synthesis, X-ray crystal structures, and photophysical and electrochemical properties of the two series of complexes are described. At room temperature, the :C∧C: complexes show much larger photoluminescence quantum yields (ΦPL) of ca. 30%, compared to the N∧C: analogues (around 1%). On the contrary, all of the investigated complexes are bright emitters in the solid state both at room temperature (1% poly(methyl methacrylate) matrix, ΦPL 30–60%) and at 77 K. Density functional theory calculations are used to rationalize the differences in the photophysical behavior observed upon change of the ancillary ligands. The N∧C:-type complexes possess a low-lying triplet metal-centered (3MC) state mainly deactivating the excited state through nonradiative processes; in contrast, no such state is present for the :C∧C: analogues. This finding is supported by temperature-dependent excited-state lifetime measurements made on representative N∧C: and :C∧C: complexes.
Co-reporter:Fátima García, Juan Aragó, Rafael Viruela, Enrique Ortí and Luis Sánchez
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 5) pp:765-772
Publication Date(Web):23 Nov 2012
DOI:10.1039/C2OB26797G
Bis(triazole)benzamide 1 has been readily synthesized by means of Cu-catalyzed 1,3-dipolar cycloaddition and its ability to bind halide anions and neutral gallic acid derivative 12GA has been theoretically and experimentally investigated. The cavity defined by the N–H amide group and the vicinal aromatic hydrogens is suitable to form H-bonding arrays with halide guests. The stability of complexes 1·Cl− and 1·Br− is very similar, as DFT calculations predict and 1H NMR titration experiments confirm. The zigzag “anti” conformation of the molecule generates two regions with complementary positive and negative potentials that favor the statistical complexation of two molecules of the neutral carboxylic acid 12GA. This guest-controlled topicity demonstrates the versatility of this class of receptor to bind species of different nature. The amide group determines the complexation of both anionic and neutral species by primary acid–base interactions.
Co-reporter:Dr. Joaquín Calbo;Juan Aragó;Dr. Francisco Otón;Dr. Vega Lloveras;Dr. Marta Mas-Torrent;Dr. José Vidal-Gancedo; Jaume Veciana; Concepció Rovira; Enrique Ortí
Chemistry - A European Journal 2013 Volume 19( Issue 49) pp:16656-16664
Publication Date(Web):
DOI:10.1002/chem.201302910
Abstract
This work presents a joint theoretical and experimental characterisation of the structural and electronic properties of two tetrathiafulvalene (TTF)-based acceptor–donor–acceptor triads (BQ–TTF–BQ and BTCNQ–TTF—BTCNQ; BQ is naphthoquinone and BTCNQ is benzotetracyano-p-quinodimethane) in their neutral and reduced states. The study is performed with the use of electrochemical, electron paramagnetic resonance (EPR), and UV/Vis/NIR spectroelectrochemical techniques guided by quantum-chemical calculations. Emphasis is placed on the mixed-valence properties of both triads in their radical anion states. The electrochemical and EPR results reveal that both BQ–TTF–BQ and BTCNQ–TTF–BTCNQ triads in their radical anion states behave as class-II mixed-valence compounds with significant electronic communication between the acceptor moieties. Density functional theory calculations (BLYP35/cc-pVTZ), taking into account the solvent effects, predict charge-localised species (BQ.−–TTF–BQ and BTCNQ.−–TTF–BTCNQ) as the most stable structures for the radical anion states of both triads. A stronger localisation is found both experimentally and theoretically for the BTCNQ–TTF–BTCNQ anion, in accordance with the more electron-withdrawing character of the BTCNQ acceptor. CASSCF/CASPT2 calculations suggest that the low-energy, broad absorption bands observed experimentally for the BQ–TTF–BQ and BTCNQ–TTF–BTCNQ radical anions are associated with the intervalence charge transfer (IV-CT) electronic transition and two nearby donor-to-acceptor CT excitations. The study highlights the molecular efficiency of the electron-donor TTF unit as a molecular wire connecting two acceptor redox centres.
Co-reporter:Daniel Tordera;Andreas M. Bünzli;Antonio Pertegás;Dr. José M. Junquera-Hernández; Edwin C. Constable;Dr. Jennifer A. Zampese; Catherine E. Housecroft; Enrique Ortí;Dr. Henk J. Bolink
Chemistry - A European Journal 2013 Volume 19( Issue 26) pp:8597-8609
Publication Date(Web):
DOI:10.1002/chem.201300457
Abstract
A new approach to obtain green-emitting iridium(III) complexes is described. The synthetic approach consists of introducing a methylsulfone electron-withdrawing substituent into a 4-phenylpyrazole cyclometalating ligand in order to stabilize the highest-occupied molecular orbital (HOMO). Six new complexes have been synthesized incorporating the conjugate base of 1-(4-(methylsulfonyl)phenyl)-1 H-pyrazole as the cyclometalating ligand. The complexes show green emission and very high photoluminescence quantum yields in both diluted and concentrated films. When used as the main active component in light-emitting electrochemical cells (LECs), green electroluminance is observed. High efficiencies and luminances are obtained at low driving voltages. This approach for green emitters is an alternative to the widely used fluorine-based substituents in the cyclometalating ligands and opens new design possibilities for the synthesis of green emitters for LECs.
Co-reporter:Dr. Fulvio G. Brunetti;Helena Isla;Dr. Juan Aragó;Dr. Enrique Ortí;Dr. Emilio M. Pérez;Dr. Nazario Martín
Chemistry - A European Journal 2013 Volume 19( Issue 30) pp:9843-9848
Publication Date(Web):
DOI:10.1002/chem.201301102
Abstract
The supramolecular modification of planar graphene with the geometrically mismatched, curved 9,10-di(1,3-dithiole-2-ylidene)-9,10-dihydroanthracene (exTTF) molecule is demonstrated. The exTTF–graphene interaction is governed by π–π and CH–π interactions, with a negligible contribution from charge transfer. We amplified these weak forces through multivalent gold nanoparticles. Our results show that planarity is not a prerequisite for recognition motifs for graphene.
Co-reporter:Dr. Rubén D. Costa;Dr. Juan Aragó;Dr. Enrique Ortí;Dr. Ted M. Pappenfus;Dr. Kent R. Mann;Dr. Katarzyna Matczyszyn;Dr. Marek Samoc;Dr. José L. Zafra;Dr. Juan T. LópezNavarrete;Dr. Juan Casado
Chemistry - A European Journal 2013 Volume 19( Issue 4) pp:1476-1488
Publication Date(Web):
DOI:10.1002/chem.201202456
Abstract
The linear and non-linear optical properties of a family of dumbbell-shaped dinuclear complexes, in which an oligothiophene chain with various numbers of rings (1, 3, and 6) acts as a bridge between two homoleptic tris(2,2′-bipyridine)ruthenium(II) complexes, have been fully investigated by using a range of spectroscopic techniques (absorption and luminescence, transient absorption, Raman, and non-linear absorption), together with density functional theory calculations. Our results shed light on the impact of the synergistic collaboration between the electronic structures of the two chemical moieties on the optical properties of these materials. Experiments on the linear optical properties of these compounds indicated that the length of the oligothiophene bridge was critical for luminescent behavior. Indeed, no emission was detected for compounds with long oligothiophene bridges (compounds 3 and 4, with 3 and 6 thiophene rings, respectively), owing to the presence of the 3ππ* state of the conjugated bridge below the 3MLCT-emitting states of the end-capping RuII complexes. In contrast, the compound with the shortest bridge (2, one thiophene ring) shows excellent photophysical features. Non-linear optical experiments showed that the investigated compounds were strong non-linear absorbers in wide energy ranges. Indeed, their non-linear absorption was augmented upon increasing the length of the oligothiophene bridge. In particular, the compound with the longest oligothiophene bridge not only showed strong two-photon absorption (TPA) but also noteworthy three-photon-absorption behavior, with a cross-section value of 4×10−78 cm6 s2 at 1450 nm. This characteristic was complemented by the strong excited-state absorption (ESA) that was observed for compounds 3 and 4. As a matter of fact, the overlap between the non-linear absorption and ESA establishes compounds 3 and 4 as good candidates for optical-power-limiting applications.
Co-reporter:Helena Isla, Bruno Grimm, Emilio M. Pérez, M. Rosario Torres, M. Ángeles Herranz, Rafael Viruela, Juan Aragó, Enrique Ortí, Dirk M. Guldi and Nazario Martín
Chemical Science 2012 vol. 3(Issue 2) pp:498-508
Publication Date(Web):19 Oct 2011
DOI:10.1039/C1SC00669J
We describe the synthesis, electronic, optical and photophysical properties of a family of three electron-donor bowl-shaped organic molecules that absorb light in the whole range of the visible spectrum (up to 800 nm in one case), and associate C60 in solution with binding constants in the range of 104–102 M−1 as measured from both UV-vis and fluorescence titrations in several solvents. These molecules are π-extended derivatives of tetrathiafulvalene, based on a truxene core to which two or three units of dithiole are covalently attached. The inclusion of the bulky dithiole groups is responsible for their bowl-shape geometry, which allows them to associate with C60, and their electron-donor character. The symmetric derivative 1, with three dithiole units, absorbs light in the 370–520 nm range. Exchanging one of the dithiole groups by an electron-withdrawing group, ketone (2) and dicyanomethylene (3), results in an intramolecular push–pull effect that extends the absorption to nearly 700 nm in the case of 2, and up to 800 nm in the case of 3. Transient absorption measurements, supported by spectroelectrochemical and radiolytical experiments, reveal that upon photoexcitation of the 1∙C60 associate the fully charge-separated state 1•+∙C60•− is generated, with lifetimes of hundreds of picoseconds. Molecular-level understanding of the electronic and supramolecular properties of 1–3 is provided by density functional theory calculations.
Co-reporter:Florian Kessler, Rubén D. Costa, Davide Di Censo, Rosario Scopelliti, Enrique Ortí, Henk J. Bolink, Sebastian Meier, Wiebke Sarfert, Michael Grätzel, Md. Khaja Nazeeruddin and Etienne Baranoff
Dalton Transactions 2012 vol. 41(Issue 1) pp:180-191
Publication Date(Web):21 Oct 2011
DOI:10.1039/C1DT10698H
Herein we report a series of charged iridium complexes emitting from near-UV to red using carbene-based N⁁C: ancillary ligands. Synthesis, photophysical and electrochemical properties of this series are described in detail together with X-ray crystal structures. Density Functional Theory calculations show that the emission originates from the cyclometallated main ligand, in contrast to commonly designed charged complexes using bidentate N⁁N ancillary ligands, where the emission originates from the ancillary N⁁N ligand. The radiative process of this series of compounds is characterized by relatively low photoluminescence quantum yields in solution that is ascribed to non-radiative deactivation of the excited state by thermally accessible metal-centered states. Despite the poor photophysical properties of this series of complexes in solution, electroluminescent emission from the bluish-green to orange region of the visible spectrum is obtained when they are used as active compounds in light-emitting electrochemical cells.
Co-reporter:Dr. Pierre-Antoine Bouit;Magdalena Marszalek;Dr. Robin Humphry-Baker;Dr. Rafael Viruela;Dr. Enrique Ortí;Dr. Shaik M. Zakeeruddin;Dr. Michael Grätzel;Dr. Juan Luis Delgado;Dr. Nazario Martín
Chemistry - A European Journal 2012 Volume 18( Issue 37) pp:11621-11629
Publication Date(Web):
DOI:10.1002/chem.201201022
Abstract
Two donor–acceptor molecular tweezers incorporating the 10-(1,3-dithiol-2-ylidene)anthracene unit as donor group and two cyanoacrylic units as accepting/anchoring groups are reported as metal-free sensitizers for dye-sensitized solar cells. By changing the phenyl spacer with 3,4-ethylenedioxythiophene (EDOT) units, the absorption spectrum of the sensitizer is red-shifted with a corresponding increase in the molar absorptivity. Density functional calculations confirmed the intramolecular charge-transfer nature of the lowest-energy absorption bands. The new dyes are highly distorted from planarity and are bound to the TiO2 surface through the two anchoring groups in a unidentate binding form. A power-conversion efficiency of 3.7 % was obtained with a volatile CH3CN-based electrolyte, under air mass 1.5 global sunlight. Photovoltage decay transients and ATR-FTIR measurements allowed us to understand the photovoltaic performance, as well as the surface binding, of these new sensitizers.
Co-reporter:Raúl García, Ma Ángeles Herranz, Ma Rosario Torres, Pierre-Antoine Bouit, Juan Luis Delgado, Joaquín Calbo, Pedro M. Viruela, Enrique Ortí, and Nazario Martín
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10707-10717
Publication Date(Web):November 6, 2012
DOI:10.1021/jo302047m
A new family of π-extended tetrathiafulvalene (exTTF) donor–acceptor chromophores has been synthesized by [2 + 2] cycloaddition of TCNE with exTTF-substituted alkynes and subsequent cycloreversion. X-ray data and theoretical calculations, performed at the B3LYP/6-31G** level, show that the new chromophores exhibit highly distorted nonplanar molecular structures with largely twisted 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) units. The electronic and optical properties, investigated by UV/vis spectroscopy and electrochemical measurements, are significantly modified when the TCBD acceptor unit is substituted with a donor phenyl group, which increases the twisting of the TCBD units and reduces the conjugation between the two dicyanovinyl subunits. The introduction of phenyl substituents hampers the oxidation and reduction processes and, at the same time, largely increases the optical band gap. An effective electronic communication between the donor and acceptor units, although limited by the distorted molecular geometry, is evidenced both in the ground and in the excited electronic states. The electronic absorption spectra are characterized by low- to medium-intense charge-transfer bands that extend to the near-infrared.
Co-reporter:Juan Aragó;Dr. Rocío Ponce Ortiz ;Belén Nieto-Ortega; Víctor Hernández; Juan Casado; Antonio Facchetti; Dr. Tobin J. Marks; Dr. Pedro M. Viruela; Dr. Enrique Ortí; Dr. Juan T. López Navarrete
ChemPhysChem 2012 Volume 13( Issue 1) pp:168-176
Publication Date(Web):
DOI:10.1002/cphc.201100736
Abstract
This work investigates the evolution of the molecular, vibrational, and optical properties within a family of carbonyl-functionalized quaterthiophenes: 5,5′′′-diheptanoyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (1), 5,5′′′-diperfluorohexylcarbonyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (2), and 2,7-[bis(5-perfluorohexylcarbonylthien-2-yl)]-4H-cyclopenta[2,1-b:3,4-b′]-dithiophene-4-one (3). The analysis is performed by Raman and UV/Vis absorption/excitation/fluorescence spectroscopy in combination with density functional calculations. Theoretical calculations show that substitution with carbonyl groups and perfluorohexyl chains induces progressive quinoidization of the π-conjugated backbone in comparison to the carbonyl-free compound 5,5′′′-dimethyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (DM-4T) used as reference. Raman spectra are dominated by a strong Raman line which mainly corresponds to a combination of CC/CC stretching vibrations spreading over the whole thiophene core. This band undergoes a remarkable downshift as a consequence of the structural changes induced by the electron-withdrawing groups on the π-conjugated backbone. The band splitting on incorporation of a central carbonyl bridge evidences the formation of two structural domains in the molecule. The excitation and fluorescence spectra recorded at low temperature show well-resolved vibronic structures associated with the most intense collective CC/CC stretching mode. Optical absorption and fluorescence bands exhibit remarkable bathochromic dispersion on carbonyl functionalization, indicative of extension of π conjugation. TDDFT calculations enable a detailed description of the trends observed in the absorption spectra. Resonance Raman spectra reflect the structural changes predicted for the S0S1 electronic transition and evidence the cross-conjugated character that the central carbonyl group confers on 3.
Co-reporter:Rubén D. Costa, Filippo Monti, Gianluca Accorsi, Andrea Barbieri, Henk J. Bolink, Enrique Ortí, and Nicola Armaroli
Inorganic Chemistry 2011 Volume 50(Issue 15) pp:7229-7238
Publication Date(Web):July 5, 2011
DOI:10.1021/ic200820t
The photophysical properties of a series of charged biscyclometalated [Ir(ppy)2(N∧N)]1+ complexes, where ppyH is 2-phenylpyridine and N∧N is 2,2′-bipyridine (bpy), 6-phenyl-2,2′-bipyridine (pbpy), and 6,6′-diphenyl-2,2′-bipyridine (dpbpy) for complexes 1, 2, and 3, respectively, have been investigated in detail. The photoluminescence performance in solution decreases from 1 to 3 upon attachment of phenyl groups to the ancillary ligand. The absorption spectra recorded over time suggest that complex 3 is less stable compared to complexes 1 and 2 likely due to a nucleophilic-assisted ancillary ligand-exchange reaction. To clarify this behavior, the temperature dependence of the experimental intrinsic deactivation rate constant, kin = 1/τ, has been investigated from 77 K to room temperature. Temperature-dependent studies show that nonemitting metal-centered (MC) states are accessible at room temperature for complex 3. The experimental results are interpreted with the help of theoretical calculations performed within the density functional theory (DFT) approach. Calculations suggest that attachment of a phenyl group to the ancillary ligand (2) promotes the temperature-independent deactivation pathways, whereas attachment of a second phenyl group (3) also makes the temperature-dependent ones accessible through population of nonradiative 3MC excited states.
Co-reporter:Juan Aragó, Juan C. Sancho-García, Enrique Ortí, and David Beljonne
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 7) pp:2068-2077
Publication Date(Web):June 6, 2011
DOI:10.1021/ct200203k
This work presents a thorough quantum chemical study of the terthiophene–tetracyanoquinodimethane complex as a model for π–π donor–acceptor systems. Dispersion-corrected hybrid (B3LYP-D) and double hybrid (B2PLYP-D), hybrid meta (M06-2X and M06-HF), and recently proposed long-range corrected (LC-wPBE, CAM-B3LYP, and wB97X-D) functionals have been chosen to deal with π–π intermolecular interactions and charge-transfer excitations in a balanced way. These properties are exhaustively compared to those computed with high-level ab initio SCS-MP2 and CASPT2 methods. The wB97X-D functional exhibits the best performance. It provides reliable intermolecular distances and interaction energies and predicts a small charge transfer from the donor to the acceptor in the ground state. In addition, wB97X-D is also able to yield an accurate description of the charge-transfer excitations in comparison to benchmark CASPT2 calculations.
Co-reporter:Juan Aragó;Pedro M. Viruela
Theoretical Chemistry Accounts 2011 Volume 128( Issue 4-6) pp:521-530
Publication Date(Web):2011 March
DOI:10.1007/s00214-010-0821-8
This work presents an analysis of the evolution of the molecular, vibrational and optical properties of a family of functionalized pentacenes. The analysis is performed on the basis of DFT quantum-chemical calculations in combination with spectroscopic techniques (Raman and UV–Vis). Theoretical calculations show that the bond length C–C/C=C alternation along the peripheral oligoenic ribbons increases with electron-releasing dioxolane substituents and diminishes with electron-withdrawing chlorine atoms and cyano groups. The attachment of triisopropylsilylethynyl groups increases the complexity of the Raman spectra. The spectra present many intense features of similar intensities in the 1,200–1,600 cm−1 range which are described by a combination of C–C/C=C stretching vibrations and in-plane C–H deformations spreading over the whole pentacene backbone. The absorption spectra display absorption bands in three different energy regions of the UV–Vis electromagnetic range. The spectra are dominated by a strong absorption band measured in the 300–350 nm region, which undergoes a sizeable red-shift with the substitution pattern, and a low-intensity, three-peak band in the 500–700 nm region, which undergoes a blue- or a red-shift depending on the electronic nature of the substituents. TDDFT calculations enable a detailed description of the trends observed in the absorption spectra.
Co-reporter:Dr. José Santos;Dr. Beatriz M. Illescas;Dr. Nazario Martín;Dr. Javier Adrio;Dr. Juan C. Carretero;Dr. Rafael Viruela;Dr. Enrique Ortí;Dr. Fabian Spänig;Dr. Dirk M. Guldi
Chemistry - A European Journal 2011 Volume 17( Issue 10) pp:2957-2964
Publication Date(Web):
DOI:10.1002/chem.201002674
Abstract
The synthesis of the first fully conjugated tetrathiafulvalene–tetracyano-p-quinodimethane ((TTF)–TCNQ)-type system has been carried out by means of a Julia–Kocienski olefination reaction. In particular, a tetracyanoanthraquinodimethane (TCAQ) formyl derivative and two new sulfonylmethyl-exTTFs (exTTF=2-[9-(1,3-dithiol-2-ylidene)anthracen-10(9H)-ylidene]-1,3-dithiole)—prepared as new building blocks—were linked. A variety of experimental conditions reveal that the use of sodium hexamethyldisilazane (NaHMDS) as base in THF afforded the E olefins with excellent stereoselectivity. Theoretical calculations at the B3LYP/6-31G** level point to highly distorted exTTF and TCAQ that form an almost planar stilbene unit between them. Although calculations predicted appreciable electronic communication between the donor and the acceptor, cyclic voltammetric studies did not substantiate this effect. It was only in photophysical assays that the electronic communication emerged in the form of a charge-transfer (CT) absorption and emission. Once photoexcited (i.e., the locally excited state or excited charge-transfer state), an ultrafast, subpicosecond charge separation leads to a radical ion pair state in which the spectroscopic features of the radical cation of exTTF as well as the radical anion of TCAQ are discernable. The radical ion pair is metastable and undergoes a fast ((1.0±0.2) ps) charge recombination to reconstitute the electronic ground state. Such ultrafast charge separation and recombination processes come as a consequence of the very short vinyl linkage between the two electroactive units.
Co-reporter:Fátima García;Dr. Pedro M. Viruela;Dr. Emilio Matesanz;Dr. Enrique Ortí;Dr. Luis Sánchez
Chemistry - A European Journal 2011 Volume 17( Issue 28) pp:7755-7759
Publication Date(Web):
DOI:10.1002/chem.201100898
Co-reporter:Helena Isla ; María Gallego ; Emilio M. Pérez ; Rafael Viruela ; Enrique Ortí ;Nazario Martín
Journal of the American Chemical Society 2010 Volume 132(Issue 6) pp:1772-1773
Publication Date(Web):January 20, 2010
DOI:10.1021/ja910107m
An exTTF-based macrocyclic receptor that associates C60 with a binding constant >106 M−1 in chlorobenzene at room temperature is described. This represents an improvement of 3 orders of magnitude with respect to the previous examples of exTTF-based receptors and one of the highest binding constants toward C60 reported to date.
Co-reporter:Javier Pitarch, M. Paz Clares, Raquel Belda, Rubén D. Costa, Pilar Navarro, Enrique Ortí, Conxa Soriano and Enrique García-España
Dalton Transactions 2010 vol. 39(Issue 33) pp:7741-7746
Publication Date(Web):26 Jul 2010
DOI:10.1039/C0DT00204F
The synthesis and Zn2+ coordination properties of a new macrocycle (L1) obtained by dipodal (2 + 2) condensation of the polyamine 3-(naphthalen-2-ylmethyl)pentane-1,5-diamine with 1H-pyrazole-3,5-dicarbaldehyde are reported. pH-metric studies show that L1 bears five measurable protonation steps in the 2.0–11.0 pH range. Fluorescence emission studies indicate that the removal of the first proton from the H5L15+ species leads to a significant decrease in the emission due to a photoinduced electron transfer process. Addition of Zn2+ promotes a boat-like conformation that approaches both fluorophores and facilitates the formation of an excimer which reaches its highest emission for a 1:1 Zn2+:L1 molar ratio. Density functional theory calculations support the experimental data and suggest that the protective effect of the Zn2+ ion along with hydrogen bonding between the 1H-pyrazole moiety and one of the tertiary nitrogen atoms is responsible for this behaviour.
Co-reporter:RubénD. Costa;Gustavo Fernández Dr.;Luis Sánchez Dr.;Nazario Martín Dr.;Enrique Ortí Dr.;HenkJ. Bolink Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 32) pp:9855-9863
Publication Date(Web):
DOI:10.1002/chem.201000600
Abstract
A novel family of dumbbell-shaped dinuclear complexes in which an oligophenyleneethynylene spacer is linked to two heteroleptic iridium(III) complexes is presented. The synthesis, as well as the electrochemical and photophysical characterization of the new complexes, is reported. The experimental results are interpreted with the help of density functional theory calculations. From these studies we conclude that the lowest triplet excited state corresponds to a 3π–π* state located on the conjugated spacer. The presence of this state below the 3MLCT/3LLCT emitting states of the end-capping IrIII complexes explains the low quantum yields observed for the dinuclear complexes (one order-of-magnitude less) with respect to the mononuclear complexes. The potential application of the novel dinuclear complexes in optoelectronic devices has been tested by using them as the primary active component in double-layer light-emitting electrochemical cells (LECs). Although the luminance levels are low, the external quantum efficiency suggests that a near-quantitative internal electron-to-photon conversion occurs in the device. This indicates that the emission inside the device is highly optimized and that the self-quenching associated with the high concentration of the complex in the active layer is minimized.
Co-reporter:Juan Aragó;PedroM. Viruela ;Enrique Ortí Dr.;Reyes MalavéOsuna Dr.;Barbara Vercelli Dr.;Gianni Zotti Dr.;Víctor Hernández ;JuanT. LópezNavarrete Dr.;JohnT. Henssler Dr.;AdamJ. Matzger Dr.;Yoshitake Suzuki Dr.;Shigehiro Yamaguchi Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 18) pp:5481-5491
Publication Date(Web):
DOI:10.1002/chem.200903343
Abstract
This work presents an analysis of the structural, electrochemical, and optical properties of a family of triisopropylsilyl end-capped oligothienoacenes (TIPS-Tn-TIPS, n=4–8) by combining cyclic voltammetry, spectroscopic techniques, and quantum-chemical calculations. TIPS-Tn-TIPS compounds form stable radical cations, and dications are only obtained for the longest oligomers (n=7 and 8). Oxidation leads to the quinoidization of the conjugated backbone, from which electrons are mainly extracted. The absorption and fluorescence spectra show partially resolved vibronic structures even at room temperature, due to the rigid molecular geometry. Two well-resolved vibronic progressions are observed at low temperatures due to the vibronic coupling, with normal modes showing wavenumbers of ≈1525 and ≈480 cm−1. Optical absorption bands display remarkable bathochromic dispersion with the oligomer length, indicative of the extent of π conjugation. The optical properties of the oxidized compounds are characterized by in situ UV/Vis/NIR spectroelectrochemistry. The radical cation species show two intense absorption bands emerging at energies lower than in the neutral compounds. The formation of the dication is only detected for the heptamer and the octamer, and shows a new band at intermediate energies. Optical data are interpreted with the help of density functional theory calculations performed at the B3LYP/6-31G** level, both for the neutral and the oxidized compounds.
Co-reporter:Rocío PonceOrtiz Dr.;Juan Casado Dr.;Sra RodríguezGonzález;Víctor Hernández ;JuanT. LópezNavarrete ;PedroM. Viruela ;Enrique Ortí ;Kazuo Takimiya Dr.;Tetsuo Otsubo
Chemistry - A European Journal 2010 Volume 16( Issue 2) pp:470-484
Publication Date(Web):
DOI:10.1002/chem.200902037
Abstract
A family of quinoidal oligothiophenes, from the dimer to the hexamer, with fused bis(butoxymethyl)cyclopentane groups has been extensively investigated by means of electronic and vibrational spectroscopy, electrochemical measurements, and density functional calculations. The latter predict that the electronic ground state always corresponds to a singlet state and that, for the longest oligomers, this state has biradical character that increases with increasing oligomer length. The shortest oligomers display closed-shell quinoidal structures. Calculations also predict the existence of very low energy excited triplet states that can be populated at room temperature. Aromatization of the conjugated carbon backbone is the driving force that determines the increasing biradical character of the ground state and the appearance of low-lying triplet states. UV/Vis, Raman, IR, and electrochemical experiments support the aromatic biradical structures predicted for the ground state of the longest oligomers and reveal that population of the low-lying triplet state accounts for the magnetic activity displayed by these compounds.
Co-reporter:Rubén D. Costa;Enrique Ortí;Henk J. Bolink;Stefan Graber;Silvia Schaffner;Markus Neuburger;Catherine E. Housecroft;Edwin C. Constable
Advanced Functional Materials 2009 Volume 19( Issue 21) pp:3456-3463
Publication Date(Web):
DOI:10.1002/adfm.200900911
Abstract
The archetype ionic transition-metal complexes (iTMCs) [Ir(ppy)2(bpy)][PF6] and [Ir(ppy)2(phen)][PF6], where Hppy = 2-phenylpyridine, bpy = 2,2′-bipyridine, and phen = 1,10-phenanthroline, are used as the primary active components in light-emitting electrochemical cells (LECs). Solution and solid-state photophysical properties are reported for both complexes and are interpreted with the help of density functional theory calculations. LEC devices based on these archetype complexes exhibit long turn-on times (70 and 160 h, respectively) and low external quantum efficiencies (∼2%) when the complex is used as a pure film. The long turn-on times are attributed to the low mobility of the counterions. The performance of the devices dramatically improves when small amounts of ionic liquids (ILs) are added to the Ir-iTMC: the turn-on time improves drastically (from hours to minutes) and the device current and power efficiency increase by almost one order of magnitude. However, the improvement of the turn-on time is unfortunately accompanied by a decrease in the stability of the device from 700 h to a few hours. After a careful study of the Ir-iTMC:IL molar ratios, an optimum between turn-on time and stability is found at a ratio of 4:1. The performance of the optimized devices using these rather simple complexes is among the best reported to date. This holds great promise for devices that use specially-designed iTMCs and demonstrates the prospect for LECs as low-cost light sources.
Co-reporter:Juan Luis López, Emilio M. Pérez, Pedro M. Viruela, Rafael Viruela, Enrique Ortí and Nazario Martín
Organic Letters 2009 Volume 11(Issue 20) pp:4524-4527
Publication Date(Web):September 16, 2009
DOI:10.1021/ol901695m
We employ a combination of urea−urea hydrogen bonds and π−π stacking interactions to obtain soluble self-assembled nanotubes decorated with electron-donor TTF derivatives on the periphery. We have investigated the structure and stability of the nanotubes with a combination of experiments and high-level DFT calculations. We also demonstrate that the association process can be controlled by changes in the hydrogen-bonding ability of the solvent and electrochemically.
Co-reporter:Rubén D. Costa, Pedro M. Viruela, Henk J. Bolink, Enrique Ortí
Journal of Molecular Structure: THEOCHEM 2009 Volume 912(1–3) pp:21-26
Publication Date(Web):30 October 2009
DOI:10.1016/j.theochem.2009.02.031
The [Ir(ppy-F2)2Me4phen]+1 complex, where ppy-F2 is 2-(2′,4′-fluorophenyl)pyridine and Me4phen is 3,4,7,8-tetramethyl-1,10-phenanthroline, has been theoretically investigated by means of DFT calculations. The molecular and electronic properties calculated for [Ir(ppy-F2)2Me4phen]+1 are compared with those obtained for the simpler [Ir(ppy)(bpy)]+1 complex. The introduction of fluorine substituents in the ppy ligands and the use of phenanthroline instead of 2,2′-bipyridine as the diimine ligand increase the HOMO–LUMO energy gap and blue-shift the emission colour. The phenanthroline ligand causes the appearance of two nearly-degenerate LUMO orbitals of different symmetry in [Ir(ppy-F2)2Me4phen]+1 and determines that two almost isoenergetic doublet states are obtained for the reduced complex. Calculations predict three lowest-energy triplet excited states of different nature in an energy difference of only 0.10 eV. Emission from the 3LC π–π* state takes place at higher energies and accounts for the structured blue emission observed in solution. Emission from the lowest-energy 3MLCT state occurs in the green region and explains the structureless broad band observed in solid films.
Co-reporter:S.Shankara Gayathri Dr.;Mateusz Wielopolski;EmilioM. Pérez Dr.;Gustavo Fernández;Luis Sánchez Dr.;Rafael Viruela Dr.;Enrique Ortí Dr.;DirkM. Guldi Dr.;Nazario Martín Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 4) pp:815-819
Publication Date(Web):
DOI:10.1002/anie.200803984
Co-reporter:S.Shankara Gayathri Dr.;Mateusz Wielopolski;EmilioM. Pérez Dr.;Gustavo Fernández;Luis Sánchez Dr.;Rafael Viruela Dr.;Enrique Ortí Dr.;DirkM. Guldi Dr.;Nazario Martín Dr.
Angewandte Chemie 2009 Volume 121( Issue 4) pp:829-834
Publication Date(Web):
DOI:10.1002/ange.200803984
Co-reporter:Reyes Malavé Osuna;Víctor Hernández Dr.;Juan T. López Navarrete Dr.;Juan Aragó;Pedro M. Viruela Dr.;Enrique Ortí Dr.;Yoshitake Suzuki Dr.;Shigehiro Yamaguchi Dr.;John T. Henssler Dr.;Adam J. Matzger Dr.
ChemPhysChem 2009 Volume 10( Issue 17) pp:3069-3076
Publication Date(Web):
DOI:10.1002/cphc.200900440
Abstract
Herein, we study the π-conjugational properties of a homologous series of all-anti oligothienoacenes containing four to eight fused thiophene rings by means of FT Raman spectroscopy and DFT calculations. The theoretical analysis of the spectroscopic data provides evidence that selective enhancement of a very limited number of Raman scatterings is related to the occurrence in these oligothienoacenes of strong vibronic coupling between collective ν(CC) stretching modes in the 1600–1300 cm−1 region and the HOMO/LUMO frontier orbitals (HOMO=highest occupied molecular orbital; LUMO=lowest unoccupied molecular orbital). The correlation of the Raman spectroscopic data and theoretical results for these all-anti oligothienoacenes with those previously collected for a number of all-syn oligothienohelicenes gives further support to the expectation that cross-conjugation is dominant in heterohelicenes. Fully planar all-anti oligothienoacenes display linear π conjugation which seemingly does not reach saturation with increasing number of annulated thiophene rings in the oligomeric chain at least up to the octamer.
Co-reporter:Juan Aragó, Pedro M. Viruela, Enrique Ortí
Journal of Molecular Structure: THEOCHEM 2009 Volume 912(1–3) pp:27-31
Publication Date(Web):30 October 2009
DOI:10.1016/j.theochem.2009.03.021
In this paper, we report a theoretical study of four types of thiophene-based oligomers showing the same number of CC double bonds and very different molecular structures. The comparative study has been performed on the basis of B3LYP/6-31G∗∗ calculations. The way the thiophene rings are linked together has a remarkable influence on the molecular and electronic properties. Linear quaterthiophene and heptathienoacene show similar aromatic structures but a loss of π-conjugation is detected for the latter due to the condensation of thiophene rings. A blue shift of the most intense electronic transition is predicted for fused heptathienoacene compared with non-fused quaterthiophene. Cyclic quaterthiophene exhibits quinoid thiophene rings folded in an envelope shape and should be visualized as a sulphur-bridged, cis-transoid polyenic chain. Circularly-fused sulflower presents a loss of π-conjugation with respect to linear systems due to its highly-branched conjugated backbone. The loss of conjugation and the high symmetry of the molecule determine that sulflower shows no optical absorption in the visible or near-UV.
Co-reporter:Emilio M. Pérez, Agostina L. Capodilupo, Gustavo Fernández, Luis Sánchez, Pedro M. Viruela, Rafael Viruela, Enrique Ortí, Massimo Bietti and Nazario Martín
Chemical Communications 2008 (Issue 38) pp:4567-4569
Publication Date(Web):12 Aug 2008
DOI:10.1039/B810177A
The relative contributions of several weak intermolecular forces to the overall stability of the complexes formed between structurally related receptors and [60]fullerene are compared, revealing a discernible contribution from concave–convex complementarity.
Co-reporter:Eva M. Priego, Luis Sánchez, M. Angeles Herranz, Nazario Martín, Rafael Viruela and Enrique Ortí
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 8) pp:1201-1209
Publication Date(Web):20 Mar 2007
DOI:10.1039/B701806A
A new family of π-extended TTF analogues (3a–c) and D–π–A chromophores (5a–c), in which the electroactive units (1,3-dithiole rings and 2,2-dicyanovinyl groups) are connected through a pyridine bridge with a meta substitution pattern, is reported. The redox behavior of these compounds has been investigated by cyclic voltammetry and theoretical calculations performed at the B3P86/6-31G** level. Unlike many π-extended TTF derivatives, the 1,3-dithiole rings in compounds 3a–c do not behave independently and two oxidation processes are observed with an anodic separation ranging from 50 to 150 mV. Calculations show that electrons are equally extracted from both dithiole rings. A biradical structure is predicted for the dication state due to the near-degeneracy of the HOMO and HOMO − 1 orbitals. The presence of both donor (D) and acceptor (A) fragments in conjugates 5 results in irreversible oxidation and reduction processes associated with the 1,3-dithiole ring and with the 2,2-dicyanovinyl moiety, respectively. An electrochemical–chemical–electrochemical (ECE) process takes place for all the compounds reported. The chemical process implies the dimerization of the radical cation for compounds 5 and the oligomerization of the biradical dication for compounds 3. The ECE process therefore generates new neutral dimeric (5) or oligomeric (3) species that incorporate the TTF vinylogue core.
Co-reporter:Rocío Ponce Ortiz;Juan Casado Dr.;Víctor Hernández ;Juan T. López Navarrete ;Pedro M. Viruela ;Kazuo Takimiya Dr.;Tetsuo Otsubo
Angewandte Chemie 2007 Volume 119(Issue 47) pp:
Publication Date(Web):25 OCT 2007
DOI:10.1002/ange.200703244
Raman-Spektroskopie und quantenchemische Studien geben ein klares Bild der elektronischen Eigenschaften und Strukturen von Diradikalen. Die Analyse einer Reihe chinoider Oligothiophene ergab, dass beim längsten untersuchten System das Singulett- und Triplett-Diradikal in einem thermischen Gleichgewicht vorliegt (siehe Bild).
Co-reporter:Emilio M. Pérez Dr.;María Sierra;Luis Sánchez Dr.;M. Rosario Torres Dr.;Rafael Viruela Dr.;Pedro M. Viruela Dr. Dr.;Nazario Martín Dr.
Angewandte Chemie 2007 Volume 119(Issue 11) pp:
Publication Date(Web):24 JAN 2007
DOI:10.1002/ange.200604327
Gut behütet! Eine neue Klasse konkaver Tetrathiafulvalen(TTF)-Derivate mit ausgedehntem π-System – Truxen-TTFs – wurde synthetisiert und charakterisiert, und ihre Selbstorganisation mit Fullerenen wurde untersucht (siehe Bild). Truxen-TTFs sind die ersten von TTF abgeleiteten Elektronendonoren, die in Lösung, ohne chemisch modifiziert zu werden, als monotope Rezeptoren für Fullerene fungieren.
Co-reporter:Emilio M. Pérez Dr.;María Sierra;Luis Sánchez Dr.;M. Rosario Torres Dr.;Rafael Viruela Dr.;Pedro M. Viruela Dr. Dr.;Nazario Martín Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 11) pp:
Publication Date(Web):24 JAN 2007
DOI:10.1002/anie.200604327
The cap fits! A new class of concave π-extended tetrathiafulvalene (TTF) derivatives, truxene-TTFs, were prepared and characterized, and their self-assembly with fullerenes was investigated (see picture). Truxene-TTFs represent the first example of TTF-related electron donors that serve, without chemical modification, as monotopic receptors for fullerenes in solution.
Co-reporter:Rocío Ponce Ortiz;Juan Casado Dr.;Víctor Hernández ;Juan T. López Navarrete ;Pedro M. Viruela ;Kazuo Takimiya Dr.;Tetsuo Otsubo
Angewandte Chemie International Edition 2007 Volume 46(Issue 47) pp:
Publication Date(Web):25 OCT 2007
DOI:10.1002/anie.200703244
Raman spectroscopy in conjunction with quantum chemistry allows efficient inspection of the electronic and structural properties of biradicals. Investigations on a series of quinoidal oligothiophenes uncover a thermal equilibrium between singlet and triplet biradical states (see picture) for the longest of the investigated systems.
Co-reporter:R. Ponce Ortiz;J. Casado;V. Hernández;J. T. López Navarrete;E. Ortí;P. M. Viruela;B. Milián;S. Hotta;G. Zotti;S. Zecchin;B. Vercelli
Advanced Functional Materials 2006 Volume 16(Issue 4) pp:
Publication Date(Web):19 JAN 2006
DOI:10.1002/adfm.200500404
A series of quinoidal oligothiophenes have been investigated by means of solid-state Fourier-transform (FT)-Raman and electron spin resonance (ESR) spectroscopies complemented with density functional theory calculations. FT-Raman spectra recorded as a function of temperature show that, upon laser irradiation, the molecules undergo a reversible structural evolution from a quinoid-type pattern at low temperature to an aromatic-type pattern at high temperature. Moreover, ESR spectra show that a portion of these compounds exists in a biradical state at room temperature. These seemingly disconnected findings and others, such as conformational isomerism, are consistently explained by the consideration of biradical species associated with the presence of low-lying triplet electronic states. In addition to the well-established versatility of quinoidal oligothiophenes regarding ambipolar electrical actuation in field-effect transistors, the exhibition of dual electrical and magnetic behavior leads to the prospect of new materials that have tunable electrical, optical, and magnetic properties.
Co-reporter:Marta C. Díaz Dr.;Beatriz M. Illescas Dr.;Nazario Martín ;Igor F. Perepichka Dr.;Martin R. Bryce ;Eric Levillain Dr.;Rafael Viruela Dr. Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 10) pp:
Publication Date(Web):23 JAN 2006
DOI:10.1002/chem.200501001
The first π-extended tetrathiafulvalene (exTTF) dimer in which the two exTTF units are covalently connected by 1,3-dithiole rings has been obtained in a multistep synthetic procedure involving the Ullmann cross-coupling reaction by using copper(I) thiophene-2-carboxylate (CuTC). The electronic spectrum reveals a significant electronic interaction between the exTTF units. The electrochemical study carried out by cyclic voltammetry in solution and in thin-layer conditions, and the electrochemical simulation and spectroelectrochemical (SEC) measurements confirm the electronic communication and show that the oxidation of dimer 14 occurs as two consecutive 2 e− processes D0–D0D2+–D0D2+–D2+. Theoretical calculations, performed at the B3P86/6-31G* level, confirm the experimental findings and predict that 142+ exists as a delocalized D.+–D.+ species in the gas phase and as a localized D2+–D0 species in solution (CH3CN or CH2Cl2). Oxidation of 142+ forms the tetracation 144+ which is constituted by two aromatic anthracene units bearing four aromatic, almost orthogonal 1,3-dithiolium cations.
Se ha sintetizado el primer dímero derivado de TTF π-extendido (exTTF) en el que las dos unidades de exTTF se encuentran unidas mediante un enlace covalente a través de los anillos de 1,3-ditiol. El procedimiento sintético consta de varios pasos e implica una reacción de acoplamiento cruzado de tipo Ullmann empleando 2-tiofencarboxilato de cobre(I) (CuTC). Los espectros electrónicos revelan una interacción electrónica significativa entre las unidades de exTTF. Se ha realizado el estudio electroquímico mediante voltamperometría cíclica en disolución y en condiciones de capa fina, así como la simulación electroquímica y las medidas espectroelectroquímicas. Los datos obtenidos confirman la comunicación electrónica entre ambas unidades, y muestran que la oxidación del dímero 14 ocurre como dos procesos que involucran dos electrones D0–D0 D2+–D0D2+–D2+. Cálculos teóricos B3P86/6-31G* confirman los hechos experimentales y predicen que 142+existe como una especie deslocalizada D.+–D.+en fase gas y como una especie localizada D2+–sD0en disolución (CH3CN o CH2Cl2). La oxidación de 142+forma la especie tetracatiónica 144+, constituida por dos unidades de antraceno aromáticas con cuatro cationes 1,3-ditiolio aromáticos en disposición prácticamente ortogonal a las unidades de antraceno.
Co-reporter:Rory Berridge, Igor M. Serebryakov, Peter J. Skabara, Enrique Ortí, Rafael Viruela, Rosendo Pou-Amérigo, Simon J. Coles and Michael B. Hursthouse
Journal of Materials Chemistry A 2004 vol. 14(Issue 18) pp:2822-2830
Publication Date(Web):09 Aug 2004
DOI:10.1039/B404545A
A new family of tetrathiafulvalenes has been prepared. The materials exhibit complex redox behaviour related to the electronic influence of the 1,4-dithiin moieties embedded within the framework of the molecules. The X-ray crystal structure of compound 4 reveals an unusual non-planar conformation of the heterocyclic compound, with the TTF fragment adopting a boat conformation. Theoretical calculations, performed at the DFT level (B3P86/6-31G*), confirm the boatlike structure (C2v symmetry) as the most stable conformation for this family of tetrathiafulvalenes. Upon oxidation, electrons are extracted from the whole molecule and the radical cations and dications remain highly distorted from planarity. For the dications, the 1,4-dithiin units present foldings of ∼40° suggesting the possibility of reaching higher oxidation states in agreement with experimental results. For the tetracations, both the TTF nucleus and the 1,4-dithiin units are singly-charged and become planar. The tetracations therefore present fully-planar, π-delocalised structures and are stabilised by the gain of aromaticity of the TTF and 1,4-dithiin electron-donor units.
Co-reporter:Juan Casado Dr.;Ted M. Pappenfus Dr.;Kent R. Mann Dr. Dr.;Pedro M. Viruela Dr.;Begoña Milián;Víctor Hernández Dr.;Juan T. López Navarrete Dr.
ChemPhysChem 2004 Volume 5(Issue 4) pp:
Publication Date(Web):14 APR 2004
DOI:10.1002/cphc.200300963
The UV/Vis, infrared absorption, and Raman scattering spectra of 3′,4′-dibutyl-5,5″-bis(dicyanomethylene)-5,5″-dihydro-2,2′:5′,2″-terthiophene have been analyzed with the aid of density functional theory calculations. The compound exhibits a quinoid structure in its ground electronic state and presents an intramolecular charge transfer from the terthiophene moiety to the C(CN)2groups. The molecular system therefore consists of an electron-deficient terthiophene backbone end-capped with electron-rich C(CN)2groups. The molecule is characterized by a strong absorption in the red, due to the HOMOLUMO π–π* electronic transition of the terthiophene backbone that shifts hypsochromically on passing from the solid state to solution and with the polarity of the solvent. The analysis of the vibrational spectra confirms the structural conclusions and supports the existence of an intramolecular charge transfer. Vibrational spectra in several solvents and as a function of temperature have also been studied. Significant frequency upshifts of the vibrations involved in the π-electron-conjugated pathway have been noticed upon solution in polar solvents and with the lowering of the temperature. Finally, we propose a quinoid molecule as a reliable structural and electronic model for dication species in doped oligothiophenes or for bipolaron charged defects in doped polythiophene.
Co-reporter:Marta C. Díaz;Beatriz M. Illescas Dr.;Nazario Martín Dr.;Rafael Viruela Dr.;Pedro M. Viruela Dr. Dr.;Ortwin Brede Dr.;Israel Zilbermann Dr.;Dirk M. Guldi Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 8) pp:
Publication Date(Web):7 APR 2004
DOI:10.1002/chem.200305555
A new class of π-extended TTF-type electron donors (11 a–c) has been synthesized by Wittig–Horner olefination of bianthrone (9) with 1,3-dithiole phosphonate esters (10 a–c). In cyclic voltammetry experiments, donors 11 a–c reveal a single, electrochemically irreversible oxidation—yielding the corresponding dicationic products—at relatively low oxidation potentials (∼0.7–0.8 V). Theoretical calculations, performed at the DFT level (B3 P86/6-31 G*), predict a highly-folded C2h structure for 11 a. In the ground state, the molecule adopts a double saddle-like conformation to compensate the steric hindrance. The calculations suggest that the intramolecular charge transfer associated with the HOMOLUMO transition is responsible for an absorption band observed above 400 nm. While the radical cation 11 a.+ retains the folded C2h structure predicted for the neutral molecule as the most stable conformation, the dication 11 a2+ has a fully aromatic D2 structure, formed by an orthogonal 9,9′-bianthryl central unit to which two singly-charged dithiole rings are attached. The drastic conformational changes that compounds 11 undergo upon oxidation account for their electrochemical properties. By means of pulse radiolysis measurements, radical-induced one-electron oxidation of 11 a–c was shown to lead to the radical cation species (11 a–c.+), which were found to disproportionate with generation of the respective dication species (11 a–c2+) and the neutral molecules (11 a–c).
Una nueva familia de moléculas dadoras de electrones de tipo TTF π-extendido, altamente conjugadas, (11 a–c) se han sintetizado mediante la reacción de olefinación de Wittig–Horner de la biantrona (9) con fosfonatos de 1,3-ditiol (10 a–c). En los experimentos de voltamperometría cíclica, los dadores 11 a–c muestran una única onda de oxidación electroquímicamente irreversible—dando lugar a los productos dicatiónicos—a potenciales relativamente bajos (∼0.7–0.8 V). Cálculos teóricos, llevados a cabo a nivel DFT (B3 P86/6-31 G*), predicen una estructuraC2haltamente distorsionada para 11 a. La molécula adopta una conformación en forma de doble mariposa para aliviar el impedimento estérico. Los cálculos sugieren que la transferencia de carga intramolecular asociada a la transición HOMOLUMO es responsable de la banda de absorción observada por encima de 400 nm en el espectro electrónico. El catión radical 11 a.+retiene la estructura C2hplegada predicha para la molécula neutra como la conformación más estable. Por el contrario, el dicatión 11 a2+muestra una estructuraD2totalmente aromática,formada por una unidad central de 9,9′-biantrilo ortogonal, unida a los anillos cargados de ditiol. Los profundos cambios conformacionales que experimentan los compuestos 11 tras la oxidación explican sus propiedades electroquímicas. Medidas de radiólisis de pulso, esto es, la oxidación monoelectrónica de 11 a–c inducida por radicales, conduce a las especies catión radical (11 a–c.+), las cuales dismutan para generar las respectivas especies dicatiónicas (11 a–c2+) y la molécula neutra (11 a–c).
Co-reporter:Julia Buendía, Joaquín Calbo, Fátima García, Juan Aragó, Pedro M. Viruela, Enrique Ortí and Luis Sánchez
Chemical Communications 2016 - vol. 52(Issue 42) pp:NaN6910-6910
Publication Date(Web):2016/04/25
DOI:10.1039/C6CC02681H
The cooperative supramolecular polymerization of 1 and 2 yields P- or M-type helical aggregates depending on the absolute configuration (S or R) of the stereogenic centres attached to the side chains. The connectivity of the amide group does not affect the handedness of the helical aggregates, but determines a larger cooperativity for retroamides 1.
Co-reporter:Florian Kessler, Rubén D. Costa, Davide Di Censo, Rosario Scopelliti, Enrique Ortí, Henk J. Bolink, Sebastian Meier, Wiebke Sarfert, Michael Grätzel, Md. Khaja Nazeeruddin and Etienne Baranoff
Dalton Transactions 2012 - vol. 41(Issue 1) pp:NaN191-191
Publication Date(Web):2011/10/21
DOI:10.1039/C1DT10698H
Herein we report a series of charged iridium complexes emitting from near-UV to red using carbene-based N⁁C: ancillary ligands. Synthesis, photophysical and electrochemical properties of this series are described in detail together with X-ray crystal structures. Density Functional Theory calculations show that the emission originates from the cyclometallated main ligand, in contrast to commonly designed charged complexes using bidentate N⁁N ancillary ligands, where the emission originates from the ancillary N⁁N ligand. The radiative process of this series of compounds is characterized by relatively low photoluminescence quantum yields in solution that is ascribed to non-radiative deactivation of the excited state by thermally accessible metal-centered states. Despite the poor photophysical properties of this series of complexes in solution, electroluminescent emission from the bluish-green to orange region of the visible spectrum is obtained when they are used as active compounds in light-emitting electrochemical cells.
Co-reporter:Andreas M. Bünzli, Henk J. Bolink, Edwin C. Constable, Catherine E. Housecroft, José M. Junquera-Hernández, Markus Neuburger, Enrique Ortí, Antonio Pertegás, Juan J. Serrano-Pérez, Daniel Tordera and Jennifer A. Zampese
Dalton Transactions 2014 - vol. 43(Issue 2) pp:NaN750-750
Publication Date(Web):2013/10/22
DOI:10.1039/C3DT52622D
The synthesis and characterization of four iridium(III) complexes [Ir(thpy)2(N^N)][PF6] where Hthpy = 2-(2′-thienyl)pyridine and N^N are 6-phenyl-2,2′-bipyridine (1), 4,4′-di-tbutyl-2,2′-bipyridine (2), 4,4′-di-tbutyl-6-phenyl-2,2′-bipyridine (3) or 4,4′-dimethylthio-2,2′-bipyridine (4) are described. The single crystal structures of ligand 4 and the complexes containing the [Ir(thpy)2(1)]+ and [Ir(thpy)2(4)]+ cations have been determined. In [Ir(thpy)2(1)]+, the pendant phenyl ring engages in an intra-cation π-stacking interaction with one of the thienyl rings in the solid state, and undergoes hindered rotation on the NMR timescale in [Ir(thpy)2(1)]+ and [Ir(thpy)2(3)]+. The solution spectra of [Ir(thpy)2(1)][PF6] and [Ir(thpy)2(4)][PF6] show emission maxima around 640 nm and are significantly red-shifted compared with [Ir(thpy)2(2)][PF6] and [Ir(thpy)2(3)][PF6] which have structured emission bands with maxima around 550 and 590 nm. In thin films, the emission spectra of the four complexes are similar with emission peaks around 550 and 590 nm and a shoulder around 640 nm that are reminiscent of the features observed in solution. In solution, quantum yields are low, but in thin films, values range from 29% for [Ir(thpy)2(1)][PF6] to 51% for [Ir(thpy)2(4)][PF6]. Density functional theory calculations rationalize the structured emission observed for the four complexes in terms of the 3LC nature predicted for the lowest-energy triplet states that mainly involve the cyclometallated [thpy]− ligands. Support for this theoretical result comes from the observed features of the low temperature (in frozen MeCN) photoluminescence spectra of the complexes. Photoluminescence and electroluminescence spectra of the complexes in a light-emitting electrochemical cell (LEC) device configuration have been investigated. The electroluminescence spectra are similar for all [Ir(thpy)2(N^N)][PF6] complexes with emission maxima at ≈600 nm, but device performances are relatively poor probably due to the poor charge-transporting properties of the complexes.
Co-reporter:Cathrin D. Ertl, Lidón Gil-Escrig, Jesús Cerdá, Antonio Pertegás, Henk J. Bolink, José M. Junquera-Hernández, Alessandro Prescimone, Markus Neuburger, Edwin C. Constable, Enrique Ortí and Catherine E. Housecroft
Dalton Transactions 2016 - vol. 45(Issue 29) pp:NaN11681-11681
Publication Date(Web):2016/05/12
DOI:10.1039/C6DT01325B
A series of regioisomeric cationic iridium complexes of the type [Ir(C^N)2(bpy)][PF6] (bpy = 2,2′-bipyridine) is reported. The complexes contain 2-phenylpyridine-based cyclometallating ligands with a methylsulfonyl group in either the 3-, 4- or 5-position of the phenyl ring. All the complexes have been fully characterized, including their crystal structures. In acetonitrile solution, all the compounds are green emitters with emission maxima between 493 and 517 nm. Whereas substitution meta to the Ir–C bond leads to vibrationally structured emission profiles and photoluminescence quantum yields of 74 and 77%, placing a sulfone substituent in a para position results in a broad, featureless emission band, an enhanced quantum yield of 92% and a shorter excited-state lifetime. These results suggest a larger ligand-centred (3LC) character of the emissive triplet state in the case of meta substitution and a more pronounced charge transfer (CT) character in the case of para substitution. Going from solution to the solid state (powder samples and thin films), the emission maxima are red-shifted for all the complexes, resulting in green-yellow emission. Data obtained from electrochemical measurements and density functional theory calculations parallel the photophysical trends. Light-emitting electrochemical cells (LECs) based on the complexes were fabricated and evaluated. A maximum efficiency of 4.5 lm W−1 at a maximum luminance of 940 cd m−2 was observed for the LEC with the complex incorporating the sulfone substituent in the 4-position when operated under pulsed current driving conditions.
Co-reporter:Andreas M. Bünzli, Antonio Pertegás, Cristina Momblona, José M. Junquera-Hernández, Edwin C. Constable, Henk J. Bolink, Enrique Ortí and Catherine E. Housecroft
Dalton Transactions 2016 - vol. 45(Issue 41) pp:NaN16392-16392
Publication Date(Web):2016/09/23
DOI:10.1039/C6DT03082C
The synthesis of four cyclometallated [Ir(C^N)2(N^N)][PF6] compounds in which N^N is a substituted 2,2′-bipyridine (bpy) ligand and the naphthyl-centred ligand 2,7-bis(2-(2-(4-(pyridin-2-yl)phenoxy)ethoxy)ethoxy)naphthalene provides the two cyclometallating C^N units is reported. The iridium(III) complexes have been characterized by 1H and 13C NMR spectroscopies, mass spectrometry and elemental analysis, and their electrochemical and photophysical properties are described. Comparisons are made with a model [Ir(ppy)2(N^N)][PF6] compound (Hppy = 2-phenylpyridine). The complexes containing the naphthyl-unit exhibit similar absorption spectra and excitation at 280 nm leads to an orange emission. The incorporation of the naphthalene unit does not lead to a desirable blue contribution to the emission. Density functional theory calculations were performed to investigate the geometries of the complexes in their ground and first triplet excited states, as well as the energies and compositions of the highest-occupied and lowest unoccupied molecular orbital (HOMO and LUMO) manifolds. Trends in the HOMO–LUMO gaps agree with those observed electrochemically. The energy difference between the LUMO and the lowest unoccupied MO located on the naphthyl unit (LUMO+7) is large enough to explain why there is no contribution from the naphthyl-centred triplet excited state to the phosphorescence emission. Singlet excited states were also investigated. Light-emitting electrochemical cells (LECs) using the [Ir(C^N)2(N^N)][PF6] and [Ir(ppy)2(N^N)][PF6] complexes in the emissive layer were made and evaluated. The presence of the naphthyl-bridge between the cyclometallating units does not significantly alter the device response.
Co-reporter:Fátima García, Juan Aragó, Rafael Viruela, Enrique Ortí and Luis Sánchez
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 5) pp:NaN772-772
Publication Date(Web):2012/11/23
DOI:10.1039/C2OB26797G
Bis(triazole)benzamide 1 has been readily synthesized by means of Cu-catalyzed 1,3-dipolar cycloaddition and its ability to bind halide anions and neutral gallic acid derivative 12GA has been theoretically and experimentally investigated. The cavity defined by the N–H amide group and the vicinal aromatic hydrogens is suitable to form H-bonding arrays with halide guests. The stability of complexes 1·Cl− and 1·Br− is very similar, as DFT calculations predict and 1H NMR titration experiments confirm. The zigzag “anti” conformation of the molecule generates two regions with complementary positive and negative potentials that favor the statistical complexation of two molecules of the neutral carboxylic acid 12GA. This guest-controlled topicity demonstrates the versatility of this class of receptor to bind species of different nature. The amide group determines the complexation of both anionic and neutral species by primary acid–base interactions.
Co-reporter:Sarah Keller, Antonio Pertegás, Giulia Longo, Laura Martínez, Jesús Cerdá, José M. Junquera-Hernández, Alessandro Prescimone, Edwin C. Constable, Catherine E. Housecroft, Enrique Ortí and Henk J. Bolink
Journal of Materials Chemistry A 2016 - vol. 4(Issue 17) pp:NaN3872-3872
Publication Date(Web):2016/02/05
DOI:10.1039/C6TC90030E
Correction for ‘Shine bright or live long: substituent effects in [Cu(N^N)(P^P)]+-based light-emitting electrochemical cells where N^N is a 6-substituted 2,2′-bipyridine’ by Sarah Keller et al., J. Mater. Chem. C, 2016, DOI: 10.1039/c5tc03725e.
Co-reporter:Paula Pla, José M. Junquera-Hernández, Henk J. Bolink and Enrique Ortí
Dalton Transactions 2015 - vol. 44(Issue 18) pp:NaN8505-8505
Publication Date(Web):2014/11/05
DOI:10.1039/C4DT03046J
A theoretical density functional theory study has been performed on different families of cationic cyclometallated Ir(III) complexes with the general formula [Ir(C^N)2(N^N)]+ and azole-based ligands. The goal was to investigate the effect that the number and position of the nitrogen atoms of the azole ring have on the electronic structure and emission wavelength of the complex. The increase in the number of nitrogen atoms changes the relative energy of the HOMO and LUMO levels and leads to a gradual shift in the emission wavelength that can be larger than 100 nm. The direction of the shift however depends on the ligand in which the azole ring is introduced. The emission shifts to bluer wavelengths when the azole forms part of the cyclometallating C^N ligands, whereas it shifts to the red when the 5-membered ring is incorporated into the ancillary N^N ligand. The position of the nitrogen atoms in the azole ring also plays an important role in determining the emission energy. Complexes with phenyl-azole C^N ligands bearing a nitrogen in the azole position to which the phenyl is linked show a markedly blue-shifted emission compared to complexes with the same number of nitrogen atoms in the azole ring and bearing a carbon atom in that position. Therefore, when comparing the emission properties of azole-based [Ir(C^N)2(N^N)]+ complexes, not only the number of nitrogen atoms of the azole but also their position in the ring and the ligand where the azole ring is incorporated should be taken into account.
Co-reporter:Sarah Keller, Antonio Pertegás, Giulia Longo, Laura Martínez, Jesús Cerdá, José M. Junquera-Hernández, Alessandro Prescimone, Edwin C. Constable, Catherine E. Housecroft, Enrique Ortí and Henk J. Bolink
Journal of Materials Chemistry A 2016 - vol. 4(Issue 17) pp:NaN3871-3871
Publication Date(Web):2016/01/25
DOI:10.1039/C5TC03725E
We report [Cu(P^P)(N^N)][PF6] complexes with P^P = bis(2-(diphenylphosphino)phenyl)ether (POP) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos) and N^N = 6-methyl-2,2′-bipyridine (Mebpy), 6-ethyl-2,2′-bipyridine (Etbpy), 6,6′-dimethyl-2,2′-bipyridine (Me2bpy) or 6-phenyl-2,2′-bipyridine (Phbpy). The crystal structures of [Cu(POP)(Phbpy)][PF6]·Et2O, [Cu(POP)(Etbpy)][PF6]·Et2O, [Cu(xantphos)(Me2bpy)][PF6], [Cu(xantphos)(Mebpy)][PF6]·CH2Cl2·0.4Et2O, [Cu(xantphos)(Etbpy)][PF6]·CH2Cl2·1.5H2O and [Cu(xantphos)(Phbpy)][PF6] are described; each copper(I) centre is distorted tetrahedral. In the crystallographically determined structures, the N^N domain in [Cu(xantphos)(Phbpy)]+ and [Cu(POP)(Phbpy)]+ is rotated ∼180° with respect to its orientation in [Cu(xantphos)(Mebpy)]+, [Cu(POP)(Etbpy)]+ and [Cu(xantphos)(Etbpy)]+; in each complex containing xantphos, the xanthene ‘bowl’ retains the same conformation in the solid-state structures. The two conformers resulting from the 180° rotation of the N^N ligand were optimized at the B3LYP-D3/(6-31G**+LANL2DZ) level and are close in energy for each complex. Variable temperature NMR spectroscopy evidences the presence of two conformers of [Cu(xantphos)(Phbpy)]+ in solution which are related by inversion of the xanthene unit. The complexes exhibit MLCT absorption bands in the range 378 to 388 nm, and excitation into each MLCT band leads to yellow emissions. Photoluminescence quantum yields (PLQYs) increase from solution to thin-film and powder; the highest PLQYs are observed for powdered [Cu(xantphos)(Mebpy)][PF6] (34%), [Cu(xantphos)(Etbpy)][PF6] (37%) and [Cu(xantphos)(Me2bpy)][PF6] (37%) with lifetimes of 9.6–11 μs. Density functional theory calculations predict that the emitting triplet (T1) involves an electron transfer from the Cu–P^P environment to the N^N ligand and therefore shows a 3MLCT character. T1 is calculated to be ∼0.20 eV lower in energy than the first singlet excited state (S1). The [Cu(P^P)(N^N)][PF6] ionic transition-metal (iTMC) complexes were tested in light-emitting electrochemical cells (LECs). Turn-on times are fast, and the LEC with [Cu(xantphos)(Me2bpy)][PF6] achieves a maximum efficacy of 3.0 cd A−1 (luminance = 145 cd m−2) with a lifetime of 1 h; on going to the [Cu(xantphos)(Mebpy)][PF6]-based LEC, the lifetime exceeds 15 h but at the expense of the efficacy (1.9 cd A−1). The lifetimes of LECs containing [Cu(xantphos)(Etbpy)][PF6] and [Cu(POP)(Etbpy)][PF6] exceed 40 and 80 h respectively.
Co-reporter:Gabriel E. Schneider, Antonio Pertegás, Edwin C. Constable, Catherine E. Housecroft, Nik Hostettler, Collin D. Morris, Jennifer A. Zampese, Henk J. Bolink, José M. Junquera-Hernández, Enrique Ortí and Michele Sessolo
Journal of Materials Chemistry A 2014 - vol. 2(Issue 34) pp:NaN7055-7055
Publication Date(Web):2014/07/10
DOI:10.1039/C4TC01171F
The synthesis and characterization of a new cationic bis-cyclometallated iridium(III) complex and its use in solid-state light-emitting electrochemical cells (LECs) are described. The complex [Ir(ppy)2(Naphbpy)][PF6], where Hppy = 2-phenylpyridine and Naphbpy = 6-(2-naphthyl)-2,2′-bipyridine, incorporates a pendant 2-naphthyl unit that π-stacks face-to-face with the adjacent ppy− ligand and acts as a peripheral bulky group. The complex presents a structureless emission centred around 595–600 nm both in solution and in thin film with relatively low photoluminescence quantum yields compared with analogous systems. Density functional theory calculations support the charge transfer character of the emitting triplet state and rationalize the low quantum yields in terms of a ligand-centred triplet localized on the 2-naphthyl unit that lies close in energy to the emitting state. LECs incorporating the [Ir(ppy)2(Naphbpy)][PF6] complex as the electroluminescent material are driven using a pulsed current operation mode and show high luminance, exceeding 300 cd m−2, and exceptional stabilities.
Co-reporter:Belén Nieto-Ortega, Fátima García, Giovanna Longhi, Ettore Castiglioni, Joaquín Calbo, Sergio Abbate, Juan T. López Navarrete, Francisco J. Ramírez, Enrique Ortí, Luis Sánchez and Juan Casado
Chemical Communications 2015 - vol. 51(Issue 48) pp:NaN9784-9784
Publication Date(Web):2015/04/30
DOI:10.1039/C5CC03054D
A complete chiroptical characterization of the supramolecular polymers formed by tricarboxamides (S)-1 and (R)-1 is performed using ECD, VCD and CPL dichroic techniques. The helical aggregates show an intense CPL signal and their absolute P- or M-configuration is assigned with the help of theoretical calculations.
Co-reporter:Eva M. Priego, Luis Sánchez, M. Angeles Herranz, Nazario Martín, Rafael Viruela and Enrique Ortí
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 8) pp:NaN1209-1209
Publication Date(Web):2007/03/20
DOI:10.1039/B701806A
A new family of π-extended TTF analogues (3a–c) and D–π–A chromophores (5a–c), in which the electroactive units (1,3-dithiole rings and 2,2-dicyanovinyl groups) are connected through a pyridine bridge with a meta substitution pattern, is reported. The redox behavior of these compounds has been investigated by cyclic voltammetry and theoretical calculations performed at the B3P86/6-31G** level. Unlike many π-extended TTF derivatives, the 1,3-dithiole rings in compounds 3a–c do not behave independently and two oxidation processes are observed with an anodic separation ranging from 50 to 150 mV. Calculations show that electrons are equally extracted from both dithiole rings. A biradical structure is predicted for the dication state due to the near-degeneracy of the HOMO and HOMO − 1 orbitals. The presence of both donor (D) and acceptor (A) fragments in conjugates 5 results in irreversible oxidation and reduction processes associated with the 1,3-dithiole ring and with the 2,2-dicyanovinyl moiety, respectively. An electrochemical–chemical–electrochemical (ECE) process takes place for all the compounds reported. The chemical process implies the dimerization of the radical cation for compounds 5 and the oligomerization of the biradical dication for compounds 3. The ECE process therefore generates new neutral dimeric (5) or oligomeric (3) species that incorporate the TTF vinylogue core.
Co-reporter:Javier Pitarch, M. Paz Clares, Raquel Belda, Rubén D. Costa, Pilar Navarro, Enrique Ortí, Conxa Soriano and Enrique García-España
Dalton Transactions 2010 - vol. 39(Issue 33) pp:NaN7746-7746
Publication Date(Web):2010/07/26
DOI:10.1039/C0DT00204F
The synthesis and Zn2+ coordination properties of a new macrocycle (L1) obtained by dipodal (2 + 2) condensation of the polyamine 3-(naphthalen-2-ylmethyl)pentane-1,5-diamine with 1H-pyrazole-3,5-dicarbaldehyde are reported. pH-metric studies show that L1 bears five measurable protonation steps in the 2.0–11.0 pH range. Fluorescence emission studies indicate that the removal of the first proton from the H5L15+ species leads to a significant decrease in the emission due to a photoinduced electron transfer process. Addition of Zn2+ promotes a boat-like conformation that approaches both fluorophores and facilitates the formation of an excimer which reaches its highest emission for a 1:1 Zn2+:L1 molar ratio. Density functional theory calculations support the experimental data and suggest that the protective effect of the Zn2+ ion along with hydrogen bonding between the 1H-pyrazole moiety and one of the tertiary nitrogen atoms is responsible for this behaviour.
Co-reporter:Emilio M. Pérez, Agostina L. Capodilupo, Gustavo Fernández, Luis Sánchez, Pedro M. Viruela, Rafael Viruela, Enrique Ortí, Massimo Bietti and Nazario Martín
Chemical Communications 2008(Issue 38) pp:NaN4569-4569
Publication Date(Web):2008/08/12
DOI:10.1039/B810177A
The relative contributions of several weak intermolecular forces to the overall stability of the complexes formed between structurally related receptors and [60]fullerene are compared, revealing a discernible contribution from concave–convex complementarity.
Co-reporter:Luis Moreira, Joaquín Calbo, Rafael M. Krick Calderon, José Santos, Beatriz M. Illescas, Juan Aragó, Jean-François Nierengarten, Dirk M. Guldi, Enrique Ortí and Nazario Martín
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4432-4432
Publication Date(Web):2015/05/18
DOI:10.1039/C5SC00850F
A series of exTTF-(crown ether)2 receptors, designed to host C60, has been prepared. The size of the crown ether and the nature of the heteroatoms have been systematically changed to fine tune the association constants. Electrochemical measurements and transient absorption spectroscopy assisted in corroborating charge transfer in the ground state and in the excited state, leading to the formation of radical ion pairs featuring lifetimes in the range from 12 to 21 ps. To rationalize the nature of the exTTF-(crown ether)2·C60 stabilizing interactions, theoretical calculations have been carried out, suggesting a synergetic interplay of donor–acceptor, π–π, n–π and CH⋯π interactions, which is the basis for the affinity of our novel receptors towards C60.
Co-reporter:Helena Isla, Bruno Grimm, Emilio M. Pérez, M. Rosario Torres, M. Ángeles Herranz, Rafael Viruela, Juan Aragó, Enrique Ortí, Dirk M. Guldi and Nazario Martín
Chemical Science (2010-Present) 2012 - vol. 3(Issue 2) pp:NaN508-508
Publication Date(Web):2011/10/19
DOI:10.1039/C1SC00669J
We describe the synthesis, electronic, optical and photophysical properties of a family of three electron-donor bowl-shaped organic molecules that absorb light in the whole range of the visible spectrum (up to 800 nm in one case), and associate C60 in solution with binding constants in the range of 104–102 M−1 as measured from both UV-vis and fluorescence titrations in several solvents. These molecules are π-extended derivatives of tetrathiafulvalene, based on a truxene core to which two or three units of dithiole are covalently attached. The inclusion of the bulky dithiole groups is responsible for their bowl-shape geometry, which allows them to associate with C60, and their electron-donor character. The symmetric derivative 1, with three dithiole units, absorbs light in the 370–520 nm range. Exchanging one of the dithiole groups by an electron-withdrawing group, ketone (2) and dicyanomethylene (3), results in an intramolecular push–pull effect that extends the absorption to nearly 700 nm in the case of 2, and up to 800 nm in the case of 3. Transient absorption measurements, supported by spectroelectrochemical and radiolytical experiments, reveal that upon photoexcitation of the 1∙C60 associate the fully charge-separated state 1•+∙C60•− is generated, with lifetimes of hundreds of picoseconds. Molecular-level understanding of the electronic and supramolecular properties of 1–3 is provided by density functional theory calculations.
Co-reporter:Inés García-Benito, Iwan Zimmermann, Javier Urieta-Mora, Juan Aragó, Agustín Molina-Ontoria, Enrique Ortí, Nazario Martín and Mohammad Khaja Nazeeruddin
Journal of Materials Chemistry A 2017 - vol. 5(Issue 18) pp:NaN8324-8324
Publication Date(Web):2017/03/07
DOI:10.1039/C7TA00997F
Engineering of inorganic–organic lead halide perovskites for photovoltaic applications has experienced significant advances in recent years. However, the use of the relatively expensive spiro-OMeTAD as a hole-transporting material (HTM) poses a challenge due to dopant-induced degradation. Herein we introduce two new three-armed and four-armed HTMs (BTT-4 and BTT-5) based on isomeric forms of benzotrithiophene (BTT). The isomerism impact on the optical, electrochemical and photophysical properties and the photovoltaic performance is systematically investigated. Perovskite solar cells (PSCs) using BTT-4 and BTT-5 as HTMs show remarkable light-to-energy conversion efficiencies of 19.0% and 18.2%, respectively, under standard measurement conditions. These results validate the readily available BTT heteroaromatic structure as a valuable core for the design of highly efficient HTMs for the preparation of PSCs.
Co-reporter:Sebastian B. Meier, Wiebke Sarfert, José M. Junquera-Hernández, Manuel Delgado, Daniel Tordera, Enrique Ortí, Henk J. Bolink, Florian Kessler, Rosario Scopelliti, Michael Grätzel, M. Khaja Nazeeruddin and Etienne Baranoff
Journal of Materials Chemistry A 2013 - vol. 1(Issue 1) pp:NaN68-68
Publication Date(Web):2012/10/23
DOI:10.1039/C2TC00251E
We report here a new cationic bis-cyclometallated iridium(III) complex, 1, with deep-blue emission at 440 nm and its use in Light-emitting Electrochemical Cells (LECs). The design is based on the 2′,6′-difluoro-2,3′-bipyridine skeleton as the cyclometallating ligand and a bis-imidazolium carbene-type ancillary ligand. Furthermore, bulky tert-butyl substituents are used to limit the intermolecular interactions. LECs have been driven both at constant voltage (6 V) and constant current (2.5 mA cm−2). The performances are significantly improved with the latter method, resulting overall in one of the best reported greenish-blue LECs having fast response (17 s), light intensity over 100 cd m−2 and a reasonable efficiency of almost 5 cd A−1.
Co-reporter:Rafael Sandoval-Torrientes, Joaquín Calbo, David García-Fresnadillo, José Santos, Enrique Ortí and Nazario Martín
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 6) pp:NaN1028-1028
Publication Date(Web):2017/02/06
DOI:10.1039/C6QO00760K
A series of new broad-absorbing dyes based on rhodanine derivatives conjugated with triarylamines using a fluorene backbone was synthesized. Spectroscopic and electrochemical studies, along with density functional theory (DFT) calculations, provided clear insight into the electronic and optical properties of the dyes, which efficiently absorb in the entire visible spectrum.
Co-reporter:Mateusz Wielopolski, Magdalena Marszalek, Fulvio G. Brunetti, Damien Joly, Joaquín Calbo, Juan Aragó, Jacques-E. Moser, Robin Humphry-Baker, Shaik M. Zakeeruddin, Juan Luis Delgado, Michael Grätzel, Enrique Ortí and Nazario Martín
Journal of Materials Chemistry A 2016 - vol. 4(Issue 17) pp:NaN3808-3808
Publication Date(Web):2015/12/09
DOI:10.1039/C5TC03501E
The development of new light harvesting materials is a key issue for the progress of the research on organic & hybrid photovoltaics. Here, we report a new class of organic sensitizers based on the bi-fluorenylidene moiety as π-linker within the donor–π-linker–acceptor (D–π–A) scheme. The new dyes are endowed with electron donor and electron acceptor units at strategic positions in order to improve their electronic and light-harvesting properties. The comprehensive study of these compounds through the use of different experimental and theoretical techniques, provides an in-depth understanding of their electronic and photophysical properties, and reveal their interest as photovoltaic materials.
Co-reporter:Alejandro López-Moreno, David Clemente-Tejeda, Joaquín Calbo, Atena Naeimi, Francisco A. Bermejo, Enrique Ortí and Emilio M. Pérez
Chemical Communications 2014 - vol. 50(Issue 66) pp:NaN9375-9375
Publication Date(Web):2014/06/27
DOI:10.1039/C4CC04026K
We present a mild catalytic method to oxidize PAHs and, in particular, pyrene. The pyrenediones are much better electron acceptors than benzoquinone in the gas phase and present similar accepting abilities in solution.
Co-reporter:Jorge S. Valera, Joaquín Calbo, Rafael Gómez, Enrique Ortí and Luis Sánchez
Chemical Communications 2015 - vol. 51(Issue 50) pp:NaN10145-10145
Publication Date(Web):2015/05/13
DOI:10.1039/C5CC03616J
The supramolecular polymerization of pyrene imidazoles 1 and 2, governed by H-bonding and C–H⋯π interactions, yields aggregates showing the characteristic bluish emission pattern of pyrene-based monomers.
Co-reporter:Joaquín Calbo, Mariachiara Pastore, Edoardo Mosconi, Enrique Ortí and Filippo De Angelis
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 10) pp:NaN4719-4719
Publication Date(Web):2014/01/13
DOI:10.1039/C3CP54970D
We present a first-principles DFT investigation of the adsorption geometry on the anatase (101) surface of a prototypical di-branched organic dye based on the extended tetrathiafulvalene moiety, incorporating two anchoring cyanoacrylic acid units. Reduced model systems with one and two anchoring groups have been initially studied to investigate the vibrational frequencies related to TiO2 dye adsorption. Our calculations confirm that the reduced systems can be used as reliable models to study the anchoring modes and that the conclusions extracted from the reduced systems can be extrapolated to the entire molecule. A series of molecular structures have been investigated to simulate the anchoring environment in monodentate- and bidentate-like adsorption modes. The comparison between the theoretical results and the available experimental data suggests a di-anchored monodentate adsorption mode as the most probable adsorption structure. Geometry optimizations of the di-branched model system adsorbed on a periodic slab of anatase (101) allowed us to compare the relative stability of different adsorption conformations and led to a di-anchored monodentate mode as the most stable adsorption structure. Furthermore, ab initio molecular dynamics simulations confirmed this structure as the preferred one, providing additional stabilization by effective hydrogen-bonding to surface oxygens and structure distortion from planarity. The analysis of the partial density of states for the prototypical models confirms that the doubly anchored adsorption provides improved electronic properties compared to the singly anchored structures for dye-sensitized solar cell purposes.
Co-reporter:Alberto de Juan, Alejandro López-Moreno, Joaquín Calbo, Enrique Ortí and Emilio M. Pérez
Chemical Science (2010-Present) 2015 - vol. 6(Issue 12) pp:NaN7014-7014
Publication Date(Web):2015/09/07
DOI:10.1039/C5SC02916C
Single-walled carbon nanotubes (SWNTs) are one of the most promising nanomaterials and their supramolecular chemistry has attracted a lot of attention. However, despite well over a decade of research, there is no standard method for the quantification of their noncovalent chemistry in solution/suspension. Here, we describe a simple procedure for the determination of association constants (Ka) between soluble molecules and insoluble and heterogeneous carbon nanotube samples. To test the scope of the method, we report binding constants between five different hosts and two types of SWNTs in four solvents. We have determined numeric values of Ka in the range of 1–104 M−1. Solvent effects as well as structural changes in both the host and guest result in noticeable changes of Ka. The results obtained experimentally were validated through state-of-the-art DFT calculations. The generalization of quantitative and comparable association constants data should significantly help advance the supramolecular chemistry of carbon nanotubes.