Co-reporter:William B. Kiosses;Sean Studer;Kosuke Matsuki;Tadatsugu Taniguchi;Michael B. A. Oldstone;Hugh Rosen;Nhan Nguyen;Nora Leaf;Hideo Negishi
PNAS 2016 Volume 113 (Issue 5 ) pp:1351-1356
Publication Date(Web):2016-02-02
DOI:10.1073/pnas.1525356113
Blunting immunopathology without abolishing host defense is the foundation for safe and effective modulation of infectious
and autoimmune diseases. Sphingosine 1-phosphate receptor 1 (S1PR1) agonists are effective in treating infectious and multiple
autoimmune pathologies; however, mechanisms underlying their clinical efficacy are yet to be fully elucidated. Here, we uncover
an unexpected mechanism of convergence between S1PR1 and interferon alpha receptor 1 (IFNAR1) signaling pathways. Activation
of S1PR1 signaling by pharmacological tools or endogenous ligand sphingosine-1 phosphate (S1P) inhibits type 1 IFN responses
that exacerbate numerous pathogenic conditions. Mechanistically, S1PR1 selectively suppresses the type I IFN autoamplification
loop in plasmacytoid dendritic cells (pDCs), a specialized DC subset, for robust type I IFN release. S1PR1 agonist suppression
is pertussis toxin-resistant, but inhibited by an S1PR1 C-terminal–derived transactivating transcriptional activator (Tat)-fusion
peptide that blocks receptor internalization. S1PR1 agonist treatment accelerates turnover of IFNAR1, suppresses signal transducer
and activator of transcription 1 (STAT1) phosphorylation, and down-modulates total STAT1 levels, thereby inactivating the
autoamplification loop. Inhibition of S1P-S1PR1 signaling in vivo using the selective antagonist Ex26 significantly elevates
IFN-α production in response to CpG-A. Thus, multiple lines of evidence demonstrate that S1PR1 signaling sets the sensitivity
of pDC amplification of IFN responses, thereby blunting pathogenic immune responses. These data illustrate a lipid G-protein
coupled receptor (GPCR)-IFNAR1 regulatory loop that balances effective and detrimental immune responses and elevated endogenous
S1PR1 signaling. This mechanism will likely be advantageous in individuals subject to a range of inflammatory conditions.
Co-reporter:Michael B.A. Oldstone, John R. Teijaro, Kevin B. Walsh, Hugh Rosen
Virology (5 January 2013) Volume 435(Issue 1) pp:92-101
Publication Date(Web):5 January 2013
DOI:10.1016/j.virol.2012.09.039
The cytokine storm is an aggressive immune response characterized by the recruitment of inflammatory leukocytes and exaggerated levels of cytokines and chemokines at the site of infection. Here we review evidence that cytokine storm directly contributes to the morbidity and mortality resulting from influenza virus infection and that sphingosine-1-phosphate (S1P) receptor agonists can abort cytokine storms providing significant protection against pathogenic human influenza viral infections. In experiments using murine models and the human pathogenic 2009 influenza viruses, S1P1 receptor agonist alone reduced deaths from influenza virus by over 80% as compared to lesser protection (50%) offered by the antiviral neuraminidase inhibitor oseltamivir. Optimal protection of 96% was achieved by combined therapy with the S1P1 receptor agonist and oseltamivir. The functional mechanism of S1P receptor agonist(s) action and the predominant role played by pulmonary endothelial cells as amplifiers of cytokine storm during influenza infection are described.Highlights► Cytokine storm plays a direct role in causation of injury due to influenza. ► Signaling via S1P1 receptor on pulmonary endothelial cells initiates lung pathology. ► S1P1 receptor agonist blocks the influenza-induced cytokine storm. ► S1P1 receptor agonist does not block host immune control over influenza. ► Pulmonary endothelial cells are the central regulators of cytokine storm.

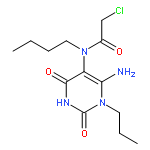
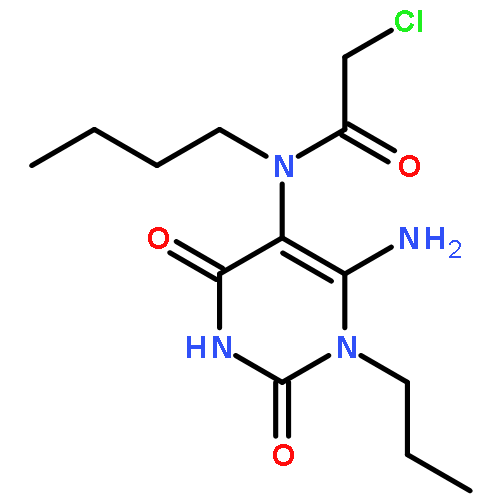
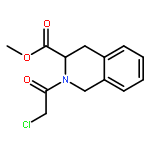
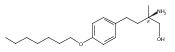
![1-(chloroacetyl)-4-[(2-fluorophenyl)sulfonyl]piperazine](http://img.cochemist.com/ccimg/923300/923204-90-4.png)
![1-(chloroacetyl)-4-[(2-fluorophenyl)sulfonyl]piperazine](http://img.cochemist.com/ccimg/923300/923204-90-4_b.png)
![N-[5-(aminosulfonyl)-2-(diethylamino)phenyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/923200/923168-91-6.png)
![N-[5-(aminosulfonyl)-2-(diethylamino)phenyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/923200/923168-91-6_b.png)
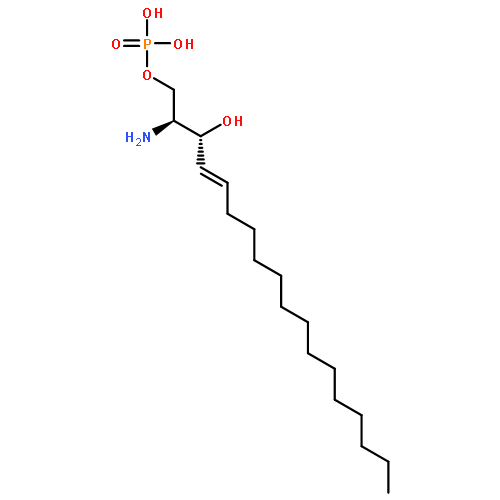
![[(3R)-3-aMino-4-[(3-hexylphenyl)aMino]-4-oxobutyl]-phosphonic acid](http://img.cochemist.com/ccimg/909800/909725-61-7.png)
![[(3R)-3-aMino-4-[(3-hexylphenyl)aMino]-4-oxobutyl]-phosphonic acid](http://img.cochemist.com/ccimg/909800/909725-61-7_b.png)
![Ethenesulfonamide,N-methyl-2-[1-(phenylmethyl)-1H-indol-5-yl]-, (1E)-](http://img.cochemist.com/ccimg/894400/894351-84-9.png)
![Ethenesulfonamide,N-methyl-2-[1-(phenylmethyl)-1H-indol-5-yl]-, (1E)-](http://img.cochemist.com/ccimg/894400/894351-84-9_b.png)
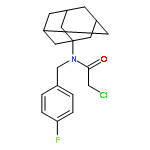
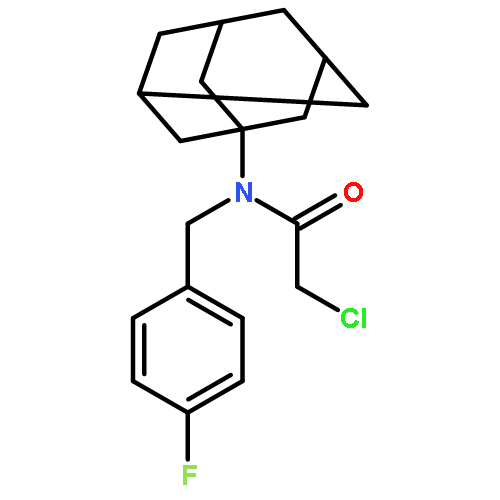
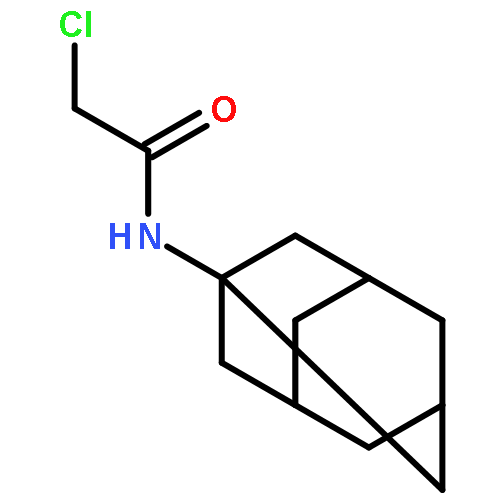
![N-[(1-adamantylamino)carbonyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/725800/725711-03-5.png)
![N-[(1-adamantylamino)carbonyl]-2-chloroacetamide](http://img.cochemist.com/ccimg/725800/725711-03-5_b.png)
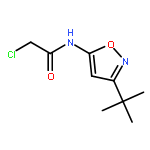
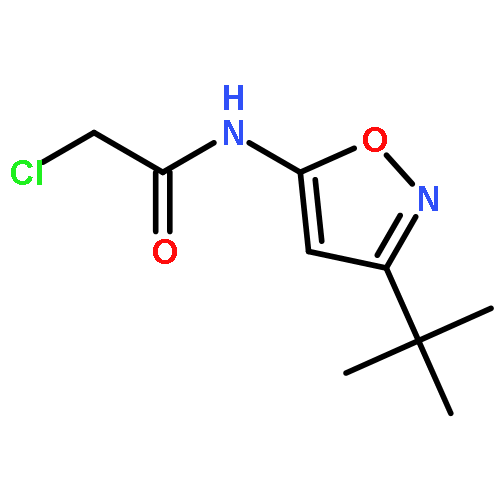
![N-[3,5-bis(trifluoromethyl)phenyl]-2-bromopropanamide](http://img.cochemist.com/ccimg/646500/646497-42-9.png)
![N-[3,5-bis(trifluoromethyl)phenyl]-2-bromopropanamide](http://img.cochemist.com/ccimg/646500/646497-42-9_b.png)


![N-Benzo[1,3]dioxol-5-yl-2-chloro-acetamide](http://img.cochemist.com/ccimg/227200/227199-07-7.png)
![N-Benzo[1,3]dioxol-5-yl-2-chloro-acetamide](http://img.cochemist.com/ccimg/227200/227199-07-7_b.png)
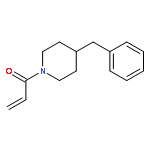
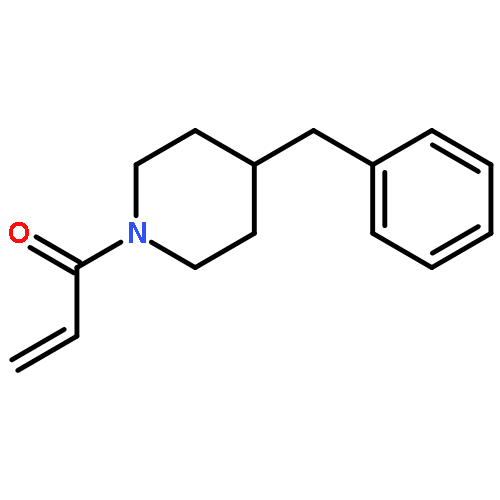












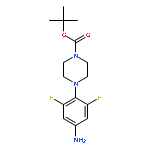
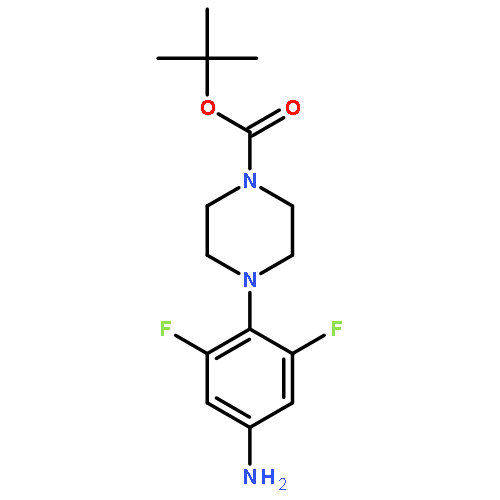




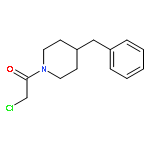
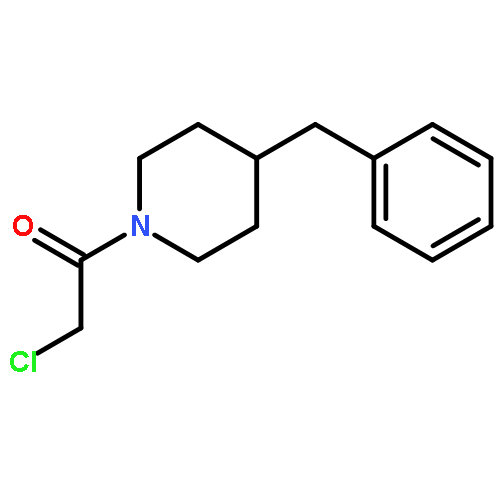


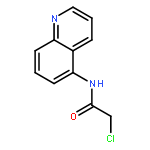
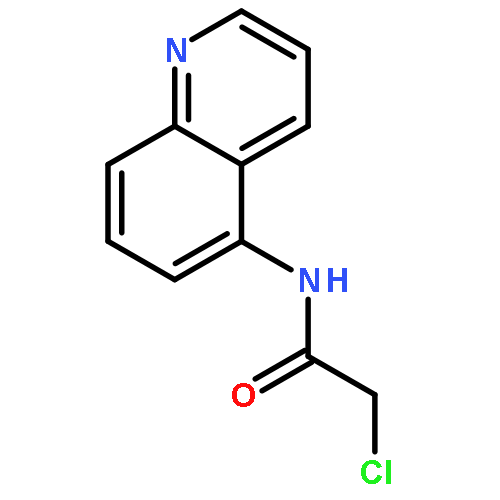
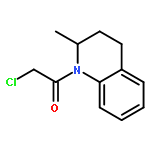
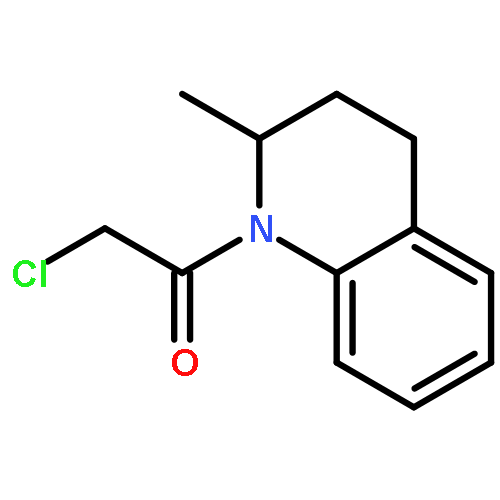


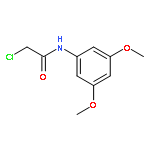
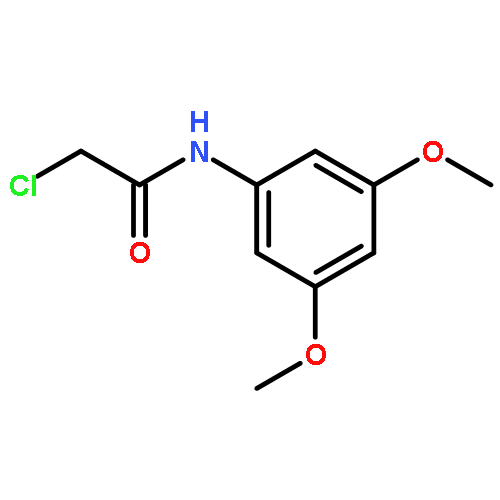
![2-[(chloroacetyl)amino]-2-deoxy-D-glucose](http://img.cochemist.com/ccimg/59900/59817-28-6.png)
![2-[(chloroacetyl)amino]-2-deoxy-D-glucose](http://img.cochemist.com/ccimg/59900/59817-28-6_b.png)
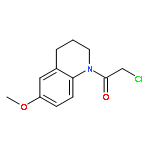
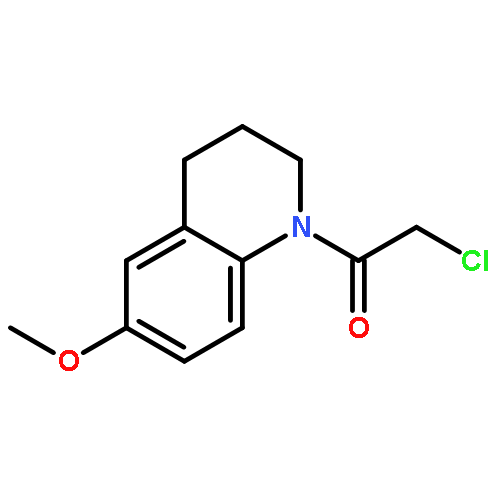
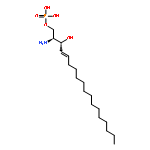




![N-(3-CHLORO-4-FLUOROPHENYL)-N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-1<WBR />,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/3300/3289-77-8.png)
![N-(3-CHLORO-4-FLUOROPHENYL)-N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-1<WBR />,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/3300/3289-77-8_b.png)
![Ethanone,2-chloro-1-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-](http://img.cochemist.com/ccimg/200/143-85-1.png)
![Ethanone,2-chloro-1-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-](http://img.cochemist.com/ccimg/200/143-85-1_b.png)










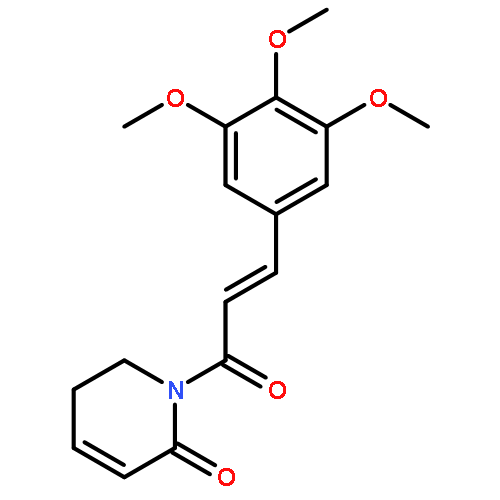
![Ethanol, 2-[[4-[5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-inden-1-yl]amino]-](http://img.cochemist.com/ccimg/1094100/1094042-01-9.png)
![Ethanol, 2-[[4-[5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-inden-1-yl]amino]-](http://img.cochemist.com/ccimg/1094100/1094042-01-9_b.png)