Co-reporter:Lan Zhang;Leilei Fu;Shouyue Zhang;Jin Zhang;Yuqian Zhao;Yaxin Zheng;Gu He;Shengyong Yang;Bo Liu
Chemical Science (2010-Present) 2017 vol. 8(Issue 4) pp:2687-2701
Publication Date(Web):2017/03/28
DOI:10.1039/C6SC05368H
UNC-51-like kinase 1 (ULK1) is well-known to initiate autophagy, and the downregulation of ULK1 has been found in most breast cancer tissues. Thus, the activation of ULK1-modulated autophagy could be a promising strategy for breast cancer therapy. In this study, we found that ULK1 was remarkably downregulated in breast cancer tissue samples by The Cancer Genome Atlas (TCGA) analysis and tissue microarray (TMA) analysis, especially in triple negative breast cancer (TNBC). To design a ULK1 agonist, we integrated in silico screening and chemical synthesis to acquire a series of small molecule candidates. After rounds of kinase and anti-proliferative activity screening, we discovered the small molecule, LYN-1604, to be the best candidate for a ULK1 agonist. Additionally, we identified that three amino acid residues (LYS50, LEU53, and TYR89) were key to the activation site of LYN-1604 and ULK1 by site-directed mutagenesis and biochemical assays. Subsequently, we demonstrated that LYN-1604 could induce cell death, associated with autophagy by the ULK complex (ULK1-mATG13-FIP200-ATG101) in MDA-MB-231 cells. To further explore LYN-1604-induced autophagic mechanisms, we found some potential ULK1 interactors, such as ATF3, RAD21, and caspase3, by performing comparative microarray analysis. Intriguingly, we found that LYN-1604 induced cell death involved in ATF3, RAD21, and caspase3, accompanied by autophagy and apoptosis. Moreover, we demonstrated that LYN-1604 has potential for good therapeutic effects on TNBC by targeting ULK1-modulated cell death in vivo; thus making this ULK1 agonist a novel potential small-molecule drug candidate for future TNBC therapy.
Co-reporter:Jin Zhang, Xiangdong Jiang, Yingnan Jiang, Mingrui Guo, Shouyue Zhang, Jingjing Li, Jun He, Jie Liu, Jinhui Wang, Liang Ouyang
European Journal of Medicinal Chemistry 2016 Volume 108() pp:495-504
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.12.016
•The structures and biology of VEGFR and c-Met have been described.•Role of HGF/c-Met and VEGF/VEGFR in tumor signaling have been discussed.•Some small molecule inhibitors of c-Met and VEGFR have been illustrated respectively.•This review also includes data regarding dual VEGFR and c-Met small molecule inhibitors.Vascular endothelial growth factor receptor (VEGFR) is a very important receptor tyrosine kinase (RTK) that can induce angiogenesis, increase cell growth and metastasis, reduce apoptosis, alter cytoskeletal function, and affect other biologic changes. Moreover, it is identified to be deregulated in varieties of human cancers. Therefore, VEGFR turn out to be a remarkable target of significant types of anticancer drugs in clinical trials. On the other side, c-Met is the receptor of hepatocyte growth factor (HGF) and a receptor tyrosine kinase. Previous studies have shown that c-Met elicits many different signaling pathways mediating cell proliferation, migration, differentiation, and survival. Furthermore, the correlation between aberrant signaling of the HGF/c-Met pathway and aggressive tumor growth, poor prognosis in cancer patients has been established. Recent reports had shown that c-Met/HGF and VEGFR/VEGF (vascular endothelial growth factor) can act synergistically in the progression of many diseases. They were also found to be over expressed in many human cancers. Thus, in a variety of malignancies, VEGFR and c-Met receptor tyrosine kinases have acted as therapeutic targets. With the development of molecular biology techniques, further understanding of the human tumor disease pathogenesis and interrelated signaling pathways known to tumor cells, using a single target inhibitors have been difficult to achieve the desired therapeutic effect. At this point, with respect to the combination of two inhibitors, a single compound which is able to inhibit both VEGFR and c-Met may put forward the advantage of raising anticancer activity. With the strong interest in these compounds, this review represents a renewal of previous works on the development of dual VEGFR and c-Met small molecule inhibitors as novel anti-cancer agents. Newly collection derivatives have been mainly describing in their biological profiles and chemical structures.
Co-reporter:Jie Li, Peiqi Wang, Bihui Zhou, Jianyou Shi, Jie Liu, Xiangrong Li, Limei Fan, Yaxin Zheng, Liang Ouyang
European Journal of Medicinal Chemistry 2016 Volume 121() pp:294-299
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2016.05.057
•A new chemical series of BRD4 inhibitors were synthesized.•Some of them showed anti-BRD4 activity at micromolar level.•Compound 10d exhibited anticancer activity and induced apoptosis in leukemia cells.•A molecular docking procedure was used for in silico target identification.Bromodomains (BRDs) are protein interaction modules that selectively recognize ε -N-lysine residues, serving as key epigenetic readers and play a key role in epigenetic regulation of gene transcription. Bromodomain-containing protein 4 (BRD4), a protein containing two BRDs termed BD1 and BD2, has emerged as an attractive candidate for the development of inhibitors targeting gene transcription in several types of cancers. In this study, we made structural modifications of previously reported BRD4 inhibitors, to develop new chemical scaffold 3,4-dihydroquinoxalin-2(1H)-one. Four series of compounds (compounds 7–10) were synthesized, and the BRD4-inhibitory activity and anti-proliferative effect of these compounds were evaluated. We found compound 10d has remarkable anti-proliferative activities toward leukemia cells and could induce apoptosis by mitochondrial pathways. Notably, the analysis of molecular docking suggested that hydrophobic interaction was essential for compound 10d to bind to BD1. In conclusion, these results demonstrate the potential of compound 10d to be utilized as a BRD4 inhibitor with apoptosis inducing effect in future leukemia therapy.
Co-reporter:Hongjia Zhang, Rongsheng Tong, Lan Bai, Jianyou Shi, Liang Ouyang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 7) pp:1419-1430
Publication Date(Web):1 April 2016
DOI:10.1016/j.bmc.2016.02.030
Parkinson’s disease (PD) is a common chronic degenerative disease of the central nervous system. Due to a rapidly aging society worldwide, PD morbidity is on the rise; however, the treatment of PD with conventional drugs carries serious adverse reactions and cannot fix the root cause of PD, the degeneration of dopaminergic neurons, which limits conventional drug usage in clinical practice. In recent years, research on the pathogenesis of PD and its clinical manifestations has led to the discovery of an increasing number of novel targets in PD, including several small molecule targeted compounds. In this paper, we analyze and summarize the most recently published PD literature and review several recently discovered novel targets in PD and their small molecule targeted pharmacologically active agents based on their mechanisms of action and pharmacodynamic profiles.
Co-reporter:Dahong Yao;Peiqi Wang;Jin Zhang;Leilei Fu;Jinhui Wang
Apoptosis 2016 Volume 21( Issue 6) pp:683-698
Publication Date(Web):2016 June
DOI:10.1007/s10495-016-1237-2
Autophagy is a highly conserved lysosome-dependent degradation process that may digest some long-lived proteins and damaged organelles. As an essential homeostasis maintaining system in normal cells, autophagy plays a key role in several pathological settings, especially cancer. Metastasis, known as a crucial hallmark of cancer progression, is the primary cause of cancer lethality. The role of autophagy in metastasis is quite complex as supportive evidence has indicated both pro-metastatic and anti-metastatic functions of autophagy. Autophagy can inhibit metastasis by restricting necrosis and mediating autophagic cell death, whereas it may also promote metastasis by enhancing cancer cell fitness in response to stress. Moreover, the function of autophagy is context- and stage-dependent. Specifically, during the early steps of metastasis, autophagy mainly serves as a suppressor, while it plays a pro-metastatic role in the later steps. Here, we focus on highlighting the dual roles of autophagy in metastasis and address the molecular mechanisms involved in this process, which may provide a new insight into cancer biology. While, we also summarize several anti-metastatic agents manipulating autophagy, in the hope of shedding light on exploration of potential novel drugs for future cancer therapy.
Co-reporter:Hui Dong, Sicheng Song, Jingjing Li, Chunyun Xu, Haowei Zhang, Liang Ouyang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3585-3591
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.06.076
•A facile method was developed for a 15-membered library of spirooxindoles.•In silico drug discovery and computational methods were used to identify the activity properties and toxicological profiles.•In vitro, the anti-tumor activity of spirooxindoles was screened, and an ideal lead compound was found.•A reverse docking procedure was used for target identification.A facile method via 1,3-dipolar cycloaddition of substituted benzylidene-2-phenyloxazolone under mild conditions with azomethine ylides, which were generated in situ by a decarboxylative route from a common set of diverse isatins and amino acid derivatives was developed for a 15-membered library of regio- and stereoselective oxazolones-grafted spirooxindole-pyrrolidine, pyrrolizidines and pyrrolothiazoles. After screening their cytotoxic activities against a spectrum of cell-lines, compound 4h was identified as potent antitumor agent and inducing apoptosis. The present study has provided an effective entry to rapidly construct a chemical library of oxazolones-grafted spirooxindoles and developed a good lead compound for subsequent optimization.
Co-reporter:Jin Zhang, Lei-lei Fu, Mao Tian, Hao-qiu Liu, Jing-jing Li, Yan Li, Jun He, Jian Huang, Liang Ouyang, Hui-yuan Gao, Jin-hui Wang
Bioorganic & Medicinal Chemistry 2015 23(5) pp: 976-984
Publication Date(Web):
DOI:10.1016/j.bmc.2015.01.020
Co-reporter:Jinbao Wei, Jianyou Shi, Jing Zhang, Gu He, Junzhu Pan, Jun He, Rui Zhou, Li Guo, Liang Ouyang
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 14) pp:4192-4200
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmc.2013.05.006
•Three different dendritic scaffolds of naproxen conjugates are synthesized.•The different bond-type dendritic drugs are compared with release properties in vitro.•Self-immolative dendritic prodrugs release naproxen after a single enzymatic activation step.•Dendritic prodrugs exhibit significant anti-inflammatory activity in vivo.•Dendritic prodrugs cause less gastric ulceration and disruption.It has been reported that delivery systems based on dendritic prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) improved the properties of drug molecules and reduced the side effects and irritation on the gastric mucosa. To find a more effective way in NSAIDs dendritic prodrugs, in this paper, three different dendritic scaffolds of enzymatically cleavable naproxen conjugates have been synthesized in a convergent approach and well characterized by NMR and MS techniques. These self-immolative dendritic NISADs prodrugs programmed to release multiple molecules of the potent naproxen after a single enzymatic activation step, and in 50% human plasma, the drug released from the compound T3 reaching 47.3% after 24 h in vitro assay. Moreover, all prodrugs were also found to maintain more significant anti-inflammatory activity, no significant cytotoxicity against HEK293 cells and less degree of ulcerogenic potential in vivo than their monomeric counterpart naproxen. These results provided an effective entry to the development of new dendritic NSAIDs prodrugs.In this paper, three different dendritic scaffords of enzymatically cleavable naproxen conjugates have been synthesized in a convergent approach and well characterized by NMR and MS techniques. The efficient release of the active drug moiety occurred by the cleavage of different benzyl ester or amide triggers by enzymatic activation of hydrophobic self-immolative dendritic prodrugs, cleavage properties and cytotoxicity of the new conjugates are also presented. Moreover, all prodrugs were also found to possess more significant anti-inflammatory activity and less degree of ulcerogenic potential in vivo than their monomeric counterpart naproxen.
Co-reporter:Guansheng Wu;Jie Liu;Shi Zeng;Wei Huang;Bo Han
Molecular Diversity 2013 Volume 17( Issue 2) pp:271-283
Publication Date(Web):2013 May
DOI:10.1007/s11030-013-9432-3
A series of spirooxindolo-pyrrolidines, pyrrolizidines, and pyrrolothiazoles hybrid compounds were prepared in good yields by regioselective, three-component, 1,3-dipolar cycloaddition reactions between \(\alpha , \beta \)-unsaturated ketones with furanyl substituents and unstable azomethine ylides, which were generated in situ from isatin and various types of amino acids. The synthesized compounds were screened for their antibacterial activities against a spectrum of pathogens. Preliminary studies identified compound 5c as a potent antimicrobial agent against drug-resistant bacteria. In addition, molecular docking studies indicated that compound 5c showed strong interactions with the active sites of lanosterol demethylase, dihydrofolate reductase, and topoisomerase II. This study provides an effective entry to the rapidly construction of a chemical library of heterocycles and compound 5c is one potent antibacterial lead for subsequent optimization.
Co-reporter:Liang Ouyang, Yuhui Huang, Yuwei Zhao, Gu He, Yongmei Xie, Jie Liu, Jun He, Bo Liu, Yuquan Wei
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 9) pp:3044-3049
Publication Date(Web):1 May 2012
DOI:10.1016/j.bmcl.2012.03.079
In this study, a novel benzothiazol- and benzooxazol-2-amine scaffold with antibacterial activity was designed and synthesized. Preliminary structure–activity relationship analysis displayed that compound 8t with a 5,6-difluorosubstituted benzothiazole was found to be a potent inhibitor of Gram-positive pathogens, and exhibited some potential against drug-resistant bacteria and without cytotoxicity in therapeutic concentrations. In addition, molecular docking studies indicated that Staphylococcus aureus methionyl-tRNA synthetase might be the possible target of these compounds. Taken together, the present study provides an effective entry to the synthesis of a good lead for subsequent optimization and a new small molecule candidate drug for antibacterial therapeutics.A series of compounds with a benzothiazol- and benzooxazol-2-amine scaffold were synthesized and screened for antibacterial activity, among which compound 8t was the most potent with MIC value of 1 μg/mL against MRSA.
Co-reporter:Liang Ouyang, Dongsheng He, Jing Zhang, Gu He, Bo Jiang, Qiang Wang, Zhengjun Chen, Junzhu Pan, Yanhua Li, Li Guo
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 12) pp:3750-3756
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmc.2011.05.004
Bone tumor is a notoriously difficult disease to manage, requiring frequent and heavy doses of systemically administered chemotherapy. Targeting anticancer drug to the bone after systemic administration may provide both greater efficacy of treatment and less frequent administration. In this paper, a series of bone targeting Asp oligopeptides 5-fluorouracil conjugates have been synthesized in a convergent approach and well characterized by NMR and MS techniques. Their hydroxyapatite (HAP) affinity, drug release and cytotoxicity characteristics were evaluated in in vitro conditions. All the prodrugs were water soluble and exhibited high affinity to HAP .The efficient release of the active drug moiety occurring by the cleavage of different linkage in physiological conditions significantly reduced the number of viable human cancer cells. From in vivo distribution, we get these compounds with high bone-selectivity and long halflife. These results provided an effective entry to the development of new bone targeting chemotherapeutic drugs.
Co-reporter:Lan Zhang, Leilei Fu, Shouyue Zhang, Jin Zhang, Yuqian Zhao, Yaxin Zheng, Gu He, Shengyong Yang, Liang Ouyang and Bo Liu
Chemical Science (2010-Present) 2017 - vol. 8(Issue 4) pp:NaN2701-2701
Publication Date(Web):2017/01/09
DOI:10.1039/C6SC05368H
UNC-51-like kinase 1 (ULK1) is well-known to initiate autophagy, and the downregulation of ULK1 has been found in most breast cancer tissues. Thus, the activation of ULK1-modulated autophagy could be a promising strategy for breast cancer therapy. In this study, we found that ULK1 was remarkably downregulated in breast cancer tissue samples by The Cancer Genome Atlas (TCGA) analysis and tissue microarray (TMA) analysis, especially in triple negative breast cancer (TNBC). To design a ULK1 agonist, we integrated in silico screening and chemical synthesis to acquire a series of small molecule candidates. After rounds of kinase and anti-proliferative activity screening, we discovered the small molecule, LYN-1604, to be the best candidate for a ULK1 agonist. Additionally, we identified that three amino acid residues (LYS50, LEU53, and TYR89) were key to the activation site of LYN-1604 and ULK1 by site-directed mutagenesis and biochemical assays. Subsequently, we demonstrated that LYN-1604 could induce cell death, associated with autophagy by the ULK complex (ULK1-mATG13-FIP200-ATG101) in MDA-MB-231 cells. To further explore LYN-1604-induced autophagic mechanisms, we found some potential ULK1 interactors, such as ATF3, RAD21, and caspase3, by performing comparative microarray analysis. Intriguingly, we found that LYN-1604 induced cell death involved in ATF3, RAD21, and caspase3, accompanied by autophagy and apoptosis. Moreover, we demonstrated that LYN-1604 has potential for good therapeutic effects on TNBC by targeting ULK1-modulated cell death in vivo; thus making this ULK1 agonist a novel potential small-molecule drug candidate for future TNBC therapy.







![Piperazine, 1-methyl-4-[[3-nitro-4-[(phenylmethyl)amino]phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/825700/825619-19-0.png)
![Piperazine, 1-methyl-4-[[3-nitro-4-[(phenylmethyl)amino]phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/825700/825619-19-0_b.png)


















































![5(4H)-Oxazolone, 2-phenyl-4-[(3,4,5-trimethoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/62700/62616-09-5.png)
![5(4H)-Oxazolone, 2-phenyl-4-[(3,4,5-trimethoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/62700/62616-09-5_b.png)




![5(4H)-Oxazolone, 4-[(3-chlorophenyl)methylene]-2-phenyl-](http://img.cochemist.com/ccimg/13300/13289-35-5.png)
![5(4H)-Oxazolone, 4-[(3-chlorophenyl)methylene]-2-phenyl-](http://img.cochemist.com/ccimg/13300/13289-35-5_b.png)


![5(4H)-OXAZOLONE, 2-PHENYL-4-[[4-(TRIFLUOROMETHYL)PHENYL]METHYLENE]-](http://img.cochemist.com/ccimg/7800/7757-65-5.png)
![5(4H)-OXAZOLONE, 2-PHENYL-4-[[4-(TRIFLUOROMETHYL)PHENYL]METHYLENE]-](http://img.cochemist.com/ccimg/7800/7757-65-5_b.png)
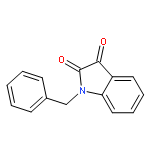
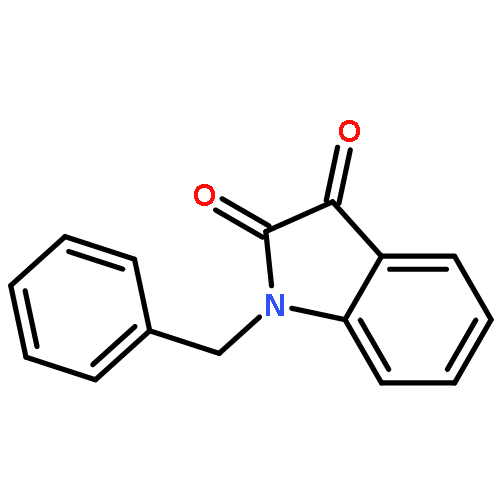
![5(4H)-Oxazolone,4-[(3,4-dichlorophenyl)methylene]-2-phenyl-](http://img.cochemist.com/ccimg/1100/1037-02-1.png)
![5(4H)-Oxazolone,4-[(3,4-dichlorophenyl)methylene]-2-phenyl-](http://img.cochemist.com/ccimg/1100/1037-02-1_b.png)


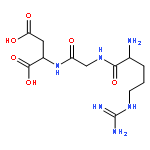

![3,11a-Epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4-dione,2,3,5a,6,10b,11-hexahydro-3-(hydroxymethyl)-10b-[(1S,4S)-3-[[4-(hydroxymethyl)-5,7-dimethyl-6,8-dioxo-2,3-dithia-5,7-diazabicyclo[2.2.2]oct-1-yl]methyl]-1H-indol-1-yl]-2-methyl-,(3S,5aR,10bS,11aS)-](http://img.cochemist.com/ccimg/1500/1403-36-7.png)
![3,11a-Epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4-dione,2,3,5a,6,10b,11-hexahydro-3-(hydroxymethyl)-10b-[(1S,4S)-3-[[4-(hydroxymethyl)-5,7-dimethyl-6,8-dioxo-2,3-dithia-5,7-diazabicyclo[2.2.2]oct-1-yl]methyl]-1H-indol-1-yl]-2-methyl-,(3S,5aR,10bS,11aS)-](http://img.cochemist.com/ccimg/1500/1403-36-7_b.png)