X-ray crystal structure analysis of the lithiated allylic α-sulfonyl carbanions [CH2CHC(Me)SO2Ph]Li⋅diglyme, [cC6H8SO2tBu]Li⋅PMDETA and [cC7H10SO2tBu]Li⋅PMDETA showed dimeric and monomeric CIPs, having nearly planar anionic C atoms, only OLi bonds, almost planar allylic units with strong CC bond length alternation and the s-trans conformation around C1C2. They adopt a C1S conformation, which is similar to the one generally found for alkyl and aryl substituted α-sulfonyl carbanions. Cryoscopy of [EtCHCHC(Et)SO2tBu]Li in THF at 164 K revealed an equilibrium between monomers and dimers in a ratio of 83:17, which is similar to the one found by low temperature NMR spectroscopy. According to NMR spectroscopy the lone-pair orbital at C1 strongly interacts with the CC double bond. Low temperature 6Li,1H NOE experiments of [EtCHCHC(Et)SO2tBu]Li in THF point to an equilibrium between monomeric CIPs having only OLi bonds and CIPs having both OLi and C1Li bonds. Ab initio calculation of [MeCHCHC(Me)SO2Me]Li⋅(Me2O)2 gave three isomeric CIPs having the s-trans conformation and three isomeric CIPs having the s-cis conformation around the C1C2 bond. All s-trans isomers are more stable than the s-cis isomers. At all levels of theory the s-trans isomer having OLi and C1Li bonds is the most stable one followed by the isomer which has two OLi bonds. The allylic unit of the C,O,Li isomer shows strong bond length alternation and the C1 atom is in contrast to the O,Li isomer significantly pyramidalized. According to NBO analysis of the s-trans and s-cis isomers, the interaction of the lone pair at C1 with the π* orbital of the CC double bond is energetically much more favorable than that with the “empty” orbitals at the Li atom. The C1S and C1C2 conformations are determined by the stereoelectronic effects nC–σSR* interaction and allylic conjugation. 1H DNMR spectroscopy of racemic [EtCHCHC(Et)SO2tBu]Li, [iPrCHCHC(iPr)SO2tBu]Li and [EtCHC(Me)C(Et)SO2tBu]Li in [D8]THF gave estimated barriers of enantiomerization of ΔG≠=13.2 kcal mol−1 (270 K), 14.2 kcal mol−1 (291 K) and 14.2 kcal mol−1 (295 K), respectively. Deprotonation of sulfone (R)-EtCHCHCH(Et)SO2tBu (94 % ee) with nBuLi in THF at −105 °C occurred with a calculated enantioselectivity of 93 % ee and gave carbanion (M)-[EtCHCHC(Et)SO2tBu]Li, the deuteration and alkylation of which with CF3CO2D and MeOCH2I, respectively, proceeded with high enantioselectivities. Time-dependent deuteration of the enantioenriched carbanion (M)-[EtCHCHC(Et)SO2tBu]Li in THF gave a racemization barrier of ΔG≠=12.5 kcal mol−1 (168 K), which translates to a calculated half-time of racemization of t1/2=12 min at −105 °C.
Lithium–titanium exchange of tertiary α-sulfonyl carbanions with ClTi(OiPr)3 and Cl2Ti(OiPr)2 in diethyl ether gave bis(1-sulfonylalkyl)titaniums and not the corresponding (1-sulfonylalkyl)titaniums. X-ray crystal structure analysis of di(isopropoxy)bis[1-(phenylsulfonyl)cyclobutyl]titanium and di(isopropoxy)bis[1-(phenylsulfonyl)isopropyl]titanium showed asymmetric distorted octahedral complexes, having hexacoordinate Ti atoms, two C–Ti bonds, four Ti–O bonds, and two four-membered Ti–O–S–Cα rings. According to 1H NMR spectroscopy bis(1-sulfonylcycloalkyl)titaniums are non-fluxional at room temperature. This suggests that chiral bis(1-sulfonylalkyl)titaniums should be configurationally stable. The bis(1-sulfonylalkyl)titaniums are stable at room temperature towards β-H elimination. They selectively add to benzaldehyde in the presence of acetophenone but do not react with methyl iodide. The reaction of tertiary acyclic α-sulfonyl carbanions with ClTi(OiPr)3 in tetrahydrofuran (THF) gives different titanium derivatives with unspecified structures, which not only selectively react with benzaldehyde in the presence of acetophenone but are also alkylated by methyl iodide.
A modular enantioselective synthesis of spiroketals, spiroethers, and oxabicycles, each containing a dihydropyran subunit, is described. It is based on the 2,2-spiro- and 2,6-bicycloannulation of sulfoximine-substituted 2-hydroxy-dihydropyrans. Key steps of the spiroannulations are the ring-closing metathesis of the corresponding 2,2-oxadienyl and 2,6-dienyl dihydropyrans and Prins cyclization of 2-alkenyl 2-hydroxy-dihydropyrans. Ring-closing metathesis of the corresponding 2,6-dienyl dihydropyrans gave oxabicycles with oxabicyclo[4.3.1]decane skeletons. These routes were extended to the synthesis of spiroketals and spiroethers incorporating additional annulated six-membered rings. Diastereoselective Prins cyclization of mono- and bicyclic 2-alkenyl-2-hydroxy-dihydropyrans was highly selective and afforded chloro-substituted spirocycles. Substituted 2-hydroxy-dihydropyrans were obtained through cyclization of δ-hydroxy ketones, which were synthesized from enantiomerically pure sulfoximine-substituted homoallylic alcohols through lithiation and trapping of the α-lithioalkenylsulfoximines with unsaturated aldehydes, followed by allylic oxidation. Inter- and intramolecular glycosidations of the 2-hydroxy-dihydropyrans with O- and C-nucleophiles proceeded with high stereoselectivities and furnished 2,6-trans-configured glycosides. Dihydropyran oxocarbenium ions are most likely intermediates in the glycosidations. According to ab initio calculations, sulfoximine- and trimethyl-substituted dihydropyran oxocarbenium ions adopt a half-chair-like conformation. The energy difference between the oxocarbenium ion with pseudoaxial and the one with pseudoequatorial methyl groups is very small. A transition state model for their reactions with nucleophiles is proposed. It features a half-chair-like conformation, a pseudoequatorial C6 substituent, and an anti-addition of the nucleophile along an axial trajectory to C2 that produces an anti-periplanar lone pair at the O atom. A similar transition state model allows a general explanation for the trans stereoselectivity of the reactions between C6-substituted dihydropyran oxocarbenium ions and nucleophiles.
We describe a modular, enantioselective synthesis of functionalised azaspirocycles with a range of ring sizes. The synthesis exploits the special features of sulfoximines, including chirality, carbanion-stabilisation, nucleophilicity, and nucleofugacity. Diastereoselective intramolecular amination of hydroxyalkyl-substituted cycloalkenylsulfoximines by the carbamate method gave bicyclic oxazinanones containing an amino-substituted tertiary C atom. Cycloalkylation of the corresponding C,N-dianions with biselectrophiles afforded sulfoximine-substituted spirocycles. Monoalkylation of the C,N-dianions with functionalised electrophiles, having a double bond and acetal group, furnished the corresponding C-alkylated bicyclic sulfoximines. Displacement of the sulfoximine group of bicyclic and spirocyclic sulfoximines by haloformate reactions gave the corresponding halides (Cl, I). Alkylation of the bicyclic halides with functionalised cuprates and reduction of the sulfoximine-substituted bicycles, carrying an alkyl group at the Cα atom, gave starting materials for a step-wise construction of the heterocyclic ring. Ring-closing metathesis of a bicyclic C,N-dienyl derivative furnished the corresponding spirocycle with an unsaturated piperidine ring. Cyclisation of an acetal group containing bicyclic oxazinanone gave spirocycles containing O,N-acetal and enamide groups. The diastereoselective reaction of a spirocyclic O,N-acetal with an allylsilane furnished the corresponding spirocycle, carrying an allyl group at the C atom adjacent to the N atom. Attempts to lithiate a bicyclic carbamate at the CH2 group adjacent to the N atom were not successful.
Enantiomerically pure triflones R1CH(R2)SO2CF3 have been synthesized starting from the corresponding chiral alcohols via thiols and trifluoromethylsulfanes. Key steps of the syntheses of the sulfanes are the photochemical trifluoromethylation of the thiols with CF3Hal (Hal=halide) or substitution of alkoxyphosphinediamines with CF3SSCF3. The deprotonation of RCH(Me)SO2CF3 (R=CH2Ph, iHex) with nBuLi with the formation of salts [RC(Me)SO2CF3]Li and their electrophilic capture both occurred with high enantioselectivities. Displacement of the SO2CF3 group of (S)-MeOCH2C(Me)(CH2Ph)SO2CF3 (95 % ee) by an ethyl group through the reaction with AlEt3 gave alkane MeOCH2C(Me)(CH2Ph)Et of 96 % ee. Racemization of salts [R1C(R2)SO2CF3]Li follows first-order kinetics and is mainly an enthalpic process with small negative activation entropy as revealed by polarimetry and dynamic NMR (DNMR) spectroscopy. This is in accordance with a CαS bond rotation as the rate-determining step. Lithium α-(S)-trifluoromethyl- and α-(S)-nonafluorobutylsulfonyl carbanion salts have a much higher racemization barrier than the corresponding α-(S)-tert-butylsulfonyl carbanion salts. Whereas [PhCH2C(Me)SO2tBu]Li/DMPU (DMPU = dimethylpropylurea) has a half-life of racemization at −105 °C of 2.4 h, that of [PhCH2C(Me)SO2CF3]Li at −78 °C is 30 d. DNMR spectroscopy of amides (PhCH2)2NSO2CF3 and (PhCH2)N(Ph)SO2CF3 gave NS rotational barriers that seem to be distinctly higher than those of nonfluorinated sulfonamides. NMR spectroscopy of [PhCH2C(Ph)SO2R]M (M=Li, K, NBu4; R=CF3, tBu) shows for both salts a confinement of the negative charge mainly to the Cα atom and a significant benzylic stabilization that is weaker in the trifluoromethylsulfonyl carbanion. According to crystal structure analyses, the carbanions of salts {[PhCH2C(Ph)SO2CF3]Li⋅L}2 (L=2 THF, tetramethylethylenediamine (TMEDA)) and [PhCH2C(Ph)SO2CF3]NBu4 have the typical chiral CαS conformation of α-sulfonyl carbanions, planar Cα atoms, and short CαS bonds. Ab initio calculations of [MeC(Ph)SO2tBu]− and [MeC(Ph)SO2CF3]− showed for the fluorinated carbanion stronger nCσ* and nOσ* interactions and a weaker benzylic stabilization. According to natural bond orbital (NBO) calculations of [R1C(R2)SO2R]− (R=tBu, CF3) the nCσ*SR interaction is much stronger for R=CF3. Ab initio calculations gave for [MeC(Ph)SO2tBu]Li⋅2 Me2O an O,Li,Cα contact ion pair (CIP) and for [MeC(Ph)SO2CF3]Li⋅2 Me2O an O,Li,O CIP. According to cryoscopy, [PhCH2C(Ph)SO2CF3]Li, [iHexC(Me)SO2CF3]Li, and [PhCH2C(Ph)SO2CF3]NBu4 predominantly form monomers in tetrahydrofuran (THF) at −108 °C. The NMR spectroscopic data of salts [R1(R2)SO2R3]Li (R3=tBu, CF3) indicate that the dominating monomeric CIPs are devoid of CαLi bonds.
An asymmetric synthesis of densely functionalized 7–11-membered carbocycles and 9–11-membered lactones has been developed. Its key steps are a modular assembly of sulfoximine-substituted C- and O-tethered trienes and C-tethered dienynes and their Ru-catalyzed ring-closing diene and enyne metathesis (RCDEM and RCEYM). The synthesis of the C-tethered trienes and dienynes includes the following steps: 1) hydroxyalkylation of enantiomerically pure titanated allylic sulfoximines with unsaturated aldehydes, 2) α-lithiation of alkenylsulfoximines, 3) alkylation, hydroxy-alkylation, formylation, and acylation of α-lithioalkenylsulfoximines, and 4) addition of Grignard reagents to α-formyl(acyl)alkenylsulfoximines. The sulfoximine group provided for high asymmetric induction in steps 1) and 4). RCDEM of the sulfoximine-substituted trienes with the second-generation Ru catalyst stereoselectively afforded the corresponding functionalized 7–11-membered carbocyles. RCDEM of diastereomeric silyloxy-substituted 1,6,12-trienes revealed an interesting difference in reactivity. While the (R)-diastereomer gave the 11-membered carbocyle, the (S)-diastereomer delivered in a cascade of cross metathesis and RCDEM 22-membered macrocycles. RCDEM of cyclic trienes furnished bicyclic carbocycles with a bicyclo[7.4.0]tridecane and bicyclo[9.4.0]pentadecane skeleton. Selective transformations of the sulfoximine- and bissilyloxy-substituted carbocycles were performed including deprotection, cross-coupling reaction and reduction of the sulfoximine moiety. Esterification of a sulfoximine-substituted homoallylic alcohol with unsaturated carboxylic acids gave the O-tethered trienes, RCDEM of which yielded the sulfoximine-substituted 9–11-membered lactones. RCEYM of a sulfoximine-substituted 1,7-dien-10-yne showed an unprecedented dichotomy in ring formation depending on the Ru catalyst. While the second-generation Ru catalyst gave the 9-membered exo 1,3-dienyl carbocycle, the first-generation Ru catalyst furnished a truncated 9-membered 1,3-dieny carbocycle having one CH2 unit less than the dienyne.
The treatment of exocyclic alkenylsulfoximines, which carry an α-glycinyl group at the allylic position, with HAliBu2 caused cascade hydroalumination–cyclization–reduction and delivered the corresponding enantio- and diastereopure sulfoximine-substituted bicyclic β-amino alcohols with a bicyclo[3.3.0]octane and bicyclo[4.3.0]nonane skeleton in high yields. Three consecutive stereogenic C atoms of the bicyclic β-amino alcohols were generated in the cascade reactions with high diastereoselectivities. Application of the hydroalumination–cyclization–reduction to a ketal-substituted six-membered exocyclic alkenylsulfoximine afforded the corresponding sulfoximine-substituted β-amino alcohol with aketal-functionalized bicyclo[4.3.0]nonane skeleton. Reduction of a sulfoximine-substituted β-amino alcohol gave the parent β-amino alcohol, whereas its oxidative deamination afforded the corresponding sulfonyl-substituted β-amino alcohol. The treatment of a sulfoximine-substituted β-amino alcohol with chloro- and iodoformates stereoselectively furnished the corresponding chloro- and iodo-substituted β-amino alcohols. Finally, the feasibility of a dehydration and elimination of sulfoximine-substituted β-amino alcohols with formation of the corresponding amino-substituted alkenylsulfoximine and allylic amine was demonstrated. An enantio- and diastereopure protected aggrecanase inhibitor mimic was synthesized in high yield starting from the sulfoximine-substituted bicyclic β-amino alcohol with a bicyclo[4.3.0]nonane skeleton and (R)-2-(3-benzyloxy)benzyl-4-tert-butoxy-4-oxobutanoic acid. Coupling of both building blocks gave the corresponding succinamide, the tert-butoxycarbonyl group of which was converted into the corresponding O-benzyl-hydroxycarbamoyl group.
Treatment of various phenylsulfoximines with nBuLi (1 equiv.) at –78 °C in THF resulted in single ortho-lithiations and gave the corresponding o-lithiosulfoximines. According to NMR spectroscopy, the o-lithiosulfoximines are generally stable at 0 °C. The o-lithiosulfoximines were efficiently trapped through deuteration, alkylation, silylation, and phosphanylation. Treatment of cyclic phenylsulfoximines also containing H atoms at their α-positions with nBuLi (1 equiv.) at –78 °C furnished the o-lithiosulfoximines with high selectivity, whereas similar treatment at –50 °C to room temperature yielded the corresponding α-lithiosulfoximines. At elevated temperatures, o-lithiosulfoximines also possessing α-H atoms underwent quantitative o,α-transmetalation to afford the corresponding α-lithiosulfoximines. Treatment of α,α-disubstituted cyclic and α,α,α-trisubstituted acyclic phenylsulfoximines with nBuLi (2 equiv.) at low temperatures led to double ortho-lithiation and furnished the corresponding o,o′-dilithiosulfoximines. At elevated temperatures, cyclic o,o′-dilithiophenylsulfoximines underwent multi-step rearrangements with formation of o,N-dilithiated benzothiazepine and benzothiazocine S-oxide derivatives in high yields. Theacyclic o,o′-dilithiophenylsulfoximine underwent a similar rearrangement and gave the corresponding o,N-dilithio-sulfinylaniline derivative. The rearrangements involve 1) elimination of the lithium sulfinamide from the o,o′-dilithiosulfoximine, 2) a Li–N addition of the lithium sulfinamide to the o-lithiobenzyne, and 3) an anionic Fries rearrangement of the o,o′-dilithiophenylsulfinamide. The rearrangements of the o,o′-dilithiophenylsulfoximines proceeded with overall retention of configuration at sulfur.
A synthesis of sulfoximine-substituted medium-ring nitrogen heterocycles (MRNHs) having a high degree of substitution has been developed. Its key steps are the modular asymmetric synthesis of sulfoximine-substituted N-tethered trienes and their Ru-catalyzed ring-closing metathesis (RCM) reaction. The highly substituted N-tethered trienes were obtained enantio- and diastereopure through 1) the diastereoselective aminoalkylation of sulfoximine-substituted allyltitanium complexes with N-tert-butylsulfonyliminoester, 2) N-allylation of homoallylic N-sulfonyl amines, 3) allylation, hydroxylalkylation, and formylation of α-lithioalkenylsulfoximines, and 4) allylation of α-formylalkenylsulfoximines. The Ru-catalyzed RCM reaction of the sulfoximine-substituted 1,7,10- and 1,7,12-trienes stereoselectively afforded the corresponding nine-, ten-, and eleven-membered MRNHs in good yields. An interesting difference in reactivity was noted in the case of a sulfoximine-substituted 1,7,10-triene and its corresponding 1,10-diene. While the triene readily underwent a RCM reaction, the diene reacted only in the presence of Ti(OiPr)4 under formation of the corresponding MRNH. The feasibility of a removal of the sulfoximine auxiliary and the N-sulfonyl protecting group from the MRNHs were demonstrated through reduction and cleavage, respectively, of a nine-membered heterocycle, both of which proceeded readily and gave the corresponding cyclic alkene and amine, respectively.
Dynamic NMR (DNMR) spectroscopy of [R1C(R2)SO2R3]Li (R1, R2 = alkyl, phenyl; R3 = Ph, tBu, adamantyl, CEt3) in [D8]THF has shown that the S-tBu, S-adamantyl, and S-CEt3 derivatives have a significantly higher enantiomerization barrier than their S-Ph analogues. Cα–S bond rotation is most likely the rate-determining step of the enantiomerization of the salts bearing a bulky group at the S atom and two substituents at the Cα atom. Ab initio calculations on [Me(Ph)SO2tBu]– gave information about the two Cα–S rotational barriers, which are dominated by steric effects. Cryoscopy of [R1C(R2)SO2tBu]Li in THF at –108 °C revealed the existence of monomers and dimers. X-ray crystal structure analysis of the monomers and dimers of [R1C(R2)SO2tBu]Li·Ln (R1 = Me, Et, tBuCH2, PhCH2, tBu; R2 = Ph, L = THF, 12-crown-4, PMDTA) and [R1C(R2)SO2Ph]Li·2diglyme [R1 = R2 = Me, Et; R1–R2 = (CH2)5] showed them to be O–Li contact ion pairs (CIPs). The monomers and dimers have a Cα–S conformation in which the lone-pair orbital at the Cα atom bisects the O–S–O angle and a significantly shortened Cα–S bond. The Cα atom of [R1C(R2)SO2R3]Li·Ln (R1 = Ph; R3 = Ph, tBu) is planar, whereas the Cα atom of [R1C(R2)SO2R3]Li·Ln (R1 = R2 = alkyl) is strongly pyramidalized in the case of R3 = Ph and most likely planar for R3 = tBu. Ab initio calculations on [MeC(Me)SO2R]– gave a pyramidalized Cα atom for R = Me and a nearly planar one for R = CF3 and tBu. The [R1C(R2)SO2tBu]Li salts were characterized by 1H, 13C, and 6Li NMR spectroscopy. 1H{1H} and 6Li{1H} NOE experiments are in accordance with the existence of O–Li CIPs. 1H and 13C NMR spectroscopy of [R1C(R2)SO2tBu]Li in [D8]THF at low temperatures showed equilibrium mixtures of up to five different species being most likely monomeric and dimeric O–Li CIPs with different configurations. According to 7Li NMR spectroscopy, the addition of HMPA to [MeC(Ph)SO2tBu]Li in [D8]THF at low temperatures causes the formation of the separated ion pair [MeC(Ph)SO2tBu]Li(HMPA)4.
The reactions of enantiopure S-tert-butyl sulfones of the type R1CH(R2)SO2tBu (≥99 % ee) with lithiumorganyl compounds gave the corresponding chiral α-sulfonyl carbanion salts [R1C(R2)SO2tBu]Li with ≥94 % ee. The enantioselectivity of the deprotonation of the phenyl- but not dialkyl-substituted sulfones is strongly dependent on the nature of the lithiumorganyl. Because of this observation and the strong decrease in enantioselectivity in the presence of TMEDA and HMPA, we propose an intramolecular proton transfer following complexation of the sulfone by RLi. Racemization of [R1C(R2)SO2tBu]Li follows first-order kinetics and seems to be mainly an enthalpic process with a small negative activation entropy, as revealed by polarimetric measurements at low temperatures. This is in accordance with Cα–S bond rotation as the rate-determining step. The salts [R1C(R2)SO2tBu]Li have half-lives of racemization in the order of several hours at –105 °C. The deuteriation of the salts at –105 °C with CF3CO2D proceeded with enantioselectivities of ≥94 % ee, the magnitude of which was not significantly affected by the presence of TMEDA and HMPA. The salts also reacted with carbon-based electrophiles at low temperatures with high enantioselectivity. The conversion of R1CH(R2)SO2tBu via [R1C(R2)SO2tBu]Li to R1C(R2,E)SO2tBu, which involves the loss of stereogenicity at the α-stereogenic center and its re-establishment upon reaction of the chiral carbanion with electrophiles, occurred with high overall enantioselectivity. Electrophiles attack the anionic C atom of [R1C(R2)SO2tBu]Li with high selectivity on the side syn to the O atoms and anti to the tert-butyl group. The reactivity of the dialkyl-substituted salts [R1C(R2)SO2tBu]Li (R1, R2 = alkyl) is significantly higher than that of the benzylic salts [RC(Ph)SO2tBu]Li (R = alkyl) and the HMPA-coordinated SIPs of [MeC(Ph)SO2tBu]Li are significantly more reactive towards EtI than the corresponding O–Li contact ion pairs.
The intermolecular phospha-Michael reaction of cyclic and acyclic alkenyl sulfoximines proceeds readily and yields the corresponding phosphanyl sulfoximines in good yield. The asymmetric induction provided by sulfoximine group in C–P bond formation is apparently only low. The configuration of three phosphanyl sulfoximine–boranes has been determined by X-ray crystal structure analysis. The ability of the phosphanyl sulfoximines to act as ligand in Pd-catalyzed asymmetric allylic alkylation was studied with pairs of diastereomeric cyclic and acyclic derivatives carrying different substituents at the N atom. This study showed that the substituent at the N atom and both the chirality of the backbone and sulfoximine group play a crucial role in determining the enantioselectivity of the allylic alkylation. The Pd-catalyzed reaction of the racemic 1,3-diphenylallyl acetate with the malonate anion in the presence of a cyclic N-benzyl-substituted phosphanyl sulfoximine gives the corresponding malonate with 97 % ee in 98 % yield. The similar alkylation with dialkyl-substituted allylic acetates proceeds only with medium enantioselectivity. The bidentate 1,5-N,P-coordination of the Pd atom by the phosphanyl sulfoximine was confirmed by NMR experiments of a π-1,3-diphenylallyl-PdII complex containing a cyclohexyl phosphanyl sulfoximine as ligand. The cyclohexane ring of the free cyclic phosphanyl sulfoximines adopts in solution and in the crystal a conformation in which the sulfoximine and phosphanyl group are both in a pseudo axial position. The coordination of the ligand to the Pd atom causes an inversion of the cyclohexane ring, which places the two groups in equatorial position.
The Pd0 complex 1 that bears the Trost ligand 2 undergoes a facile redox reaction with 1,4-biscarbonates 5 b–d and rac-22 under formation of the diamidato–PdII complex 7 and the corresponding 1,3-cycloalkadienes 8 b–d. The redox deactivation of complex 1 was the dominating pathway in the reaction of 5 b–d with HCO3− at room temperature. However, at 0 °C the six-membered biscarbonate 5 b, catalytic amounts of complex 1, and HCO3− mainly reacted in an allylic alkylation, which led to a highly selective desymmetrization of the substrate and gave alcohol 6 b with ≥99 % ee in 66 % yield. An increase of the catalyst loading in the reaction of 5 b with 1 and HCO3− afforded the bicyclic carbonate 12 b (96 % ee, 92 %). Formation of carbonate 12 b involves two consecutive inter- and intramolecular substitution reactions of the π-allyl–PdII complexes 16 b and 18 b, respectively, with O-nucleophiles and presumably proceeds through the hydrogen carbonate 17 b as key intermediate. The intermediate formation of 17 b is also indicated by the conversion of alcohol rac-6 b to carbonate 12 b upon treatment with HCO3− and 1. The Pd0-catalyzed desymmetrization of 5 b with formation of 12 b and its hydrolysis allow an efficient enantioselective synthesis of diol 13 b. The reaction of the seven-membered biscarbonate 5 c with ent-1 and HCO3− afforded carbonate ent-12 c (99 % ee, 39 %). The Pd0 complex 1 is stable in solution and suffers no intramolecular redox reaction with formation of complex 7 and dihydrogen as recently claimed for the similar Pd0 complex 9. Instead, complex 1 is rapidly oxidized by dioxygen to give the stable PdII complex 7. Thus, formation of the PdII complex 10 from 9 was most likely due to an oxidation by dioxygen. Oxidative workup (air) of the reaction mixture stemming from the desymmetrization of 5 c catalyzed by 1 gave the PdII complex 7 in high yield besides carbonate 12 c.
(E)- and (Z)-configured α-lithioalkenyl sulfoximines, which are available through lithiation of the corresponding alkenyl sulfoximines, undergo a anionic cross-coupling reaction (ACCR) with organocuprates with formation of the corresponding alkenyl cuprates and sulfinamide. The alkenyl cuprates can be trapped by electrophiles. The ACCR presumably proceeds via the formation of a higher-order sulfoximine-substituted alkenyl cuprate, which undergoes a 1,2-metal-ate rearrangement whereby the sulfoximine group acts as the nucleofuge. The parent (E)- and (Z)-configured alkenyl sulfoximines suffer upon treatment with an organocuprate a deprotonation at the α-position with formation of the corresponding α-cuprioalkenyl sulfoximines. These derivatives also enter into a similar ACCR with organocuprates. The ACCR of sulfoximines substituted homoallylic alcohols allows a stereoselective access to enantio- and diastereopure substituted homoallylic alcohols.
Sulfoximine-substituted bis(allyl)titanium complexes, which are configurationally labile at the Cα-atoms, have emerged as valuable reagents in asymmetric synthesis. Their highly selective reactions with aldehydes and N-sulfonyl imino esters allow the attainment of enantio- and diastereomerically pure sulfoximine-substituted homoallylic alcohols and homoallylic amines, respectively, which are valuable starting materials for the asymmetric synthesis of homopropargylic alcohols, 2,3-dihydrofurans, medium-sized carbocycles and lactones, unsaturated mono- and bicyclic prolines, β-amino acids, and vinyl oxiranes, respectively. The high synthetic versatility of the sulfoximine group stems from its ability to function as a chiral carbanion-stabilizing nucleofuge. © 2007 Wiley Periodicals, Inc. 18:472–481, 2007; Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hc.20331
A fully stereocontrolled synthesis of 3-oxa-15-deoxy-16-(m-tolyl)-17,18,19,20-tetranorisocarbacyclin (3-oxa-15-deoxy-TIC, 7 b) and a formal one of 15-deoxy-16-(m-tolyl)-17,18,19,20-tetranorisocarbacyclin (15-deoxy-TIC, 7 a) are described. 15-Deoxy-TIC is specific for the neuronal prostacyclin receptor (IP2) and exhibits neuroprotective activities, and the new 3-oxa-15-deoxy-TIC is expected to be metabolically more stable than 15-deoxy-TIC. The syntheses of 7 a and 7 b are based on the convergent conjugate addition–azoalkene–asymmetric olefination strategy. Key building blocks are the readily available bicyclic azoalkene 14 and the alkenylcopper derivative 15. The stereoselective conjugate addition of 15 to 14 gave hydrazone 13, which was stereoselectively converted to the bicyclic ketone 11. The key steps for the construction of the α side chain of 7 a and 7 b and the regioselective introduction of the endocyclic Δ6,6a double bond are: 1) a highly selective asymmetric olefination of ketone 11 with the chiral Horner–Wadsworth–Emmons reagent 28 and 2) a regioselective deconjugation of the α,β-unsaturated ester (E)-10 with the chiral lithium amide 29, which gave the β,γ-unsaturated ester anti-9 with high selectivity. The homoallylic alcohol 8 served at a late stage as the joint intermediate in the syntheses of 7 a and 7 b. While an etherification of 8 furnished, after hydrolysis and deprotection, 3-oxa-15-deoxy-TIC, its alkylation afforded alcohol 37, the known precursor for the synthesis of 15-deoxy-TIC.
We describe new fully stereocontrolled syntheses of the prostacyclin analogues iloprost (2), the most active component of the drugs Ilomedin and Ventavis, and 3-oxa-iloprost (3), a derivative that is expected to have a significantly higher metabolic stability than 2 perhaps allowing an oral application. The syntheses are based on the same strategy and chiral bicyclic building block as used in the synthesis of cicaprost (4), the third most potent analogue that exhibits, besides prostacyclin-like activities, antimetastatic activities. Reaction of the enantiopure C6–C13 bicyclic aldehyde 17 with Cl3CCOOH/Cl3CCOONa afforded trichlorocarbinol 24 which was converted via mesylate 25 to the C6–C14 bicyclic alkyne 9. The palladium-catalysed hydrostannylation of alkyne 9 gave with high regio- and stereoselectivity the alkenylstannane 26, Sn/Li exchange of which afforded the E-configured alkenyllithium derivative 8. Coupling of the C6–C14 building block 8 with the enantiopure C15–C20 building block, the N-methoxyamide 7, gave the C6–C20 bicyclic ketone 6 in high yield without epimerisation at C16. The configuration at C15 of iloprost (2) and 3-oxa-iloprost (3) was established through a highly diastereoselective reduction of ketone 6 with catecholborane and the chiral oxazaborolidine 28 which furnished alcohol (15S)-29. The highly stereoselective conversions of alcohol (15S)-29 to iloprost (2) and 3-oxa-iloprost (3), which include as key stereoselective steps an olefination with a chiral phosphonoacetate and a copper-mediated allylic alkylation, have already been described.
An asymmetric synthesis of cycloalkenyl and alkenyloxiranes from allylic sulfoximines and aldehydes is described. Lithiation and titanation of cyclic and acyclic allylic sulfoximines with chlorotris(diethylamino)titanium and subsequent treatment with aldehydes gave, as described previously, enantio- and diastereomerically pure, syn-configured, sulfoximine-substituted homoallylic alcohols. Treatment of the sulfoximine-substituted homoallylic alcohols with chloroethyl chloroformate resulted in a facile substitution of the sulfoximine group by a Cl atom, with formation of the corresponding alkenyl chlorohydrins. In the case of the cycloalkenyl derivatives the substitution proceeded with high diastereoselectivities with retention of configuration, while in the case of the alkenyl derivatives, medium diastereoselectivities with inversion of configuration were observed. While elimination reactions of the cycloalkenyl chlorohydrins gave the corresponding enantio- and diastereomerically pure cis-configured cycloalkenyloxiranes in good overall yields, the alkenyl chlorohydrins afforded mixtures of enantiomerically pure trans and cis isomers in which the trans isomers dominated. The solution-phase synthesis was extended to the solid phase by the synthesis of an enantiomerically pure, polymer-bound allylic sulfoximine and its conversion into an alkenyloxirane. The initial results proved that the concept of the utilization of the sulfoximine group as a traceless chiral linker is feasible and suggest that the solid-phase asymmetric synthesis of cycloalkenyloxiranes by this route should be possible. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
We describe a new method for the asymmetric synthesis, from allylic sulfoximines and aldehydes, of N,O-protected, cyclic and acyclic, β-substituted and β,β-disubstituted δ-hydroxy-β-amino acids and of N,O-protected 1,3-amino alcohols, both possessing three contiguous stereogenic centers. Treatment of enantiomerically pure, acyclic allylic sulfoximines with aldehydes after successive lithiation and titanation afforded sulfonimidoyl-substituted homoallylic alcohols with high regio- and diastereoselectivities. Diastereomerically pure, cyclic, sulfonimidoyl-substituted homoallylic alcohols were synthesized in a similar manner from the corresponding enantiomerically pure, cyclic allylic sulfoximines and isobutyraldehyde. A highly diastereoselective amination of the sulfonimidoyl-substituted homoallylic alcohols with the generation of secondary and tertiary C atoms and formation of the sulfonimidoyl-substituted, protected 1,3-amino alcohols (oxazinones) was achieved by the carbamate method, through cyclization of the corresponding carbamates after their lithiation with nBuLi. The sulfonimidoyl-substituted, monocyclic and bicyclic oxazinones were converted into protected, acyclic and cyclic, β-substituted and β,β-disubstituted β-amino acids and protected 1,3-amino alcohols by two different routes: the carbanion route and the substitution route. The carbanion route involves: (1) a double lithiation of the protected β-amino sulfoximines, (2) treatment of the dilithiated sulfoximines with electrophiles, and (3) reductive removal of the sulfonimidoyl group. By the carbanion route, double lithiation of the sulfonimidoyl-substituted oxazinones with nBuLi gave the corresponding dilithium salts, which reacted readily with a number of electrophiles to give the corresponding α-substituted sulfoximines in good yields. Reduction of the sulfoximines with Raney nickel afforded the corresponding protected monocyclic and bicyclic 1,3-amino alcohols and the protected acyclic and cyclic β-amino acids in good yields. The substitution route involves: (1) a facile substitution of the sulfonimidoyl group by a Cl atom, and (2) a substitution of the Cl atom of the protected β-amino chlorides by a cyano group. Treatment of the sulfoximines with ClCO2Me readily afforded the corresponding β-amino chlorides in good yields, and so treatment of alkyl sulfoximines with chloroformates seems to be a general method for the replacement of an N-methylsulfonimidoyl group by a Cl atom. Introduction of a cyano group was achieved through treatment of chlorides with NaCN, which gave the corresponding β-amino nitriles in good yields. Finally, hydrolysis of the nitriles afforded the protected acyclic and cyclic, β-substituted and β,β-disubstituted β-amino acids. Treatment of the protected β-amino sulfoximines with ClCO2Me gave − besides the corresponding chlorides − methyl (S)-N-phenylsulfinylcarbamate with ⩾ 99% ee in good yield. Treatment of the sulfinamide with MeMgCl afforded (S)-methyl phenyl sulfoxide with 97% ee, and this could be converted with complete retention of configuration into (S)-N,S-dimethyl-S-phenylsulfoximine, the starting material for the synthesis of the allylic sulfoximines used in this work. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
We describe the highly selective palladium catalyzed kinetic resolutions of the racemic cyclic allylic carbonates rac-1 a–c and racemic acyclic allylic carbonates rac-3 aa and rac-3 ba through reaction with tert-butylsulfinate, tolylsulfinate, phenylsulfinate anions and 2-pyrimidinethiol by using N,N′-(1R,2R)-1,2-cyclohexanediylbis[2-(diphenylphosphino)-benzamide] (BPA) as ligand. Selectivities are expressed in yields and ee values of recovered substrate and product and in selectivity factors S. The reaction of the cyclohexenyl carbonate 1 a (≥99 % ee) with 2-pyrimidinethiol in the presence of BPA was shown to exhibit, under the conditions used, an overall pseudo-zero order kinetics in regard to the allylic substrate. Also described are the highly selective palladium catalyzed asymmetric syntheses of the cyclic and acyclic allylic tert-butylsulfones 2 aa, 2 b, 2 c, 2 d and 4 a–c, respectively, and of the cyclic and acyclic allylic 2-pyrimidyl-, 2-pyridyl-, and 4-chlorophenylsulfides 5 aa, 5 b, 5 ab, 6 aa–ac, 6 ba and 6 bb, respectively, from the corresponding racemic carbonates and sulfinate anions and thiols, respectively, in the presence of BPA. Synthesis of the E-configured allylic sulfides 6 aa, 6 ab, 6 ac and 6 bb was accompanied by the formation of minor amounts of the corresponding Z isomers. The analogous synthesis of allylic tert-butylsulfides from allylic carbonates and tert-butylthiol by using BPA could not be achieved. Reaction of the cyclopentenyl esters rac-1 da and rac-1 db with 2-pyrimidinethiol gave the allylic sulfide 5 c having only a low ee value. Similar results were obtained in the case of the reaction of the cyclohexenyl carbonate rac-1 a and of the acyclic carbonates rac-3 aa and rac-3 ba with 2-pyridinethiol and lead to the formation of the sulfides 5 ab, 6 ab, and 6 bb, respectively. The low ee values may be ascribed to the operating of a “memory effect”, that is, both enantiomers of the substrate give the substitution product with different enantioselectivities. However, in the reaction of the racemic carbonate rac-1 a as well as of the highly enriched enantiomers 1 a (≥99 % ee) and ent-1 a (≥99 % ee) with 2-pyrimidinethiol the ee values of the substrates and the substitution product remained constant until complete conversion. Similar results were obtained in the reaction of the cyclic carbonates rac-1 a, ent-1 a (≥99 % ee) and ent-1 c (≥99 % ee) with lithium tert-butylsulfinate. Thus, in the case of rac-1 a and 2-pyrimidinthiol and tert-butylsulfinate anion as nucleophiles the enantioselectivity of the substitution step is, under the conditions used, independent of the chirality of the substrate; this shows that no “memory effect” is operating in this case. Hydrolysis of the carbonates ent-1 a–c, ent-3 aa and ent-3 ba, which were obtained through kinetic resolution, afforded the enantiomerically highly enriched cyclic allylic alcohols 9 a–c (≥99 % ee) and acyclic allylic alcohols 10 a (≥99 % ee) and 10 b (99 % ee), respectively.
The structures of the lithium salts of the chiral bicyclic allylic α-sulfonyl carbanions 3−5, each possessing a norbornane skeleton and a tert-butyl group at the S atom, have been studied by 1H, 13C, 6Li, and 6Li,1H HOESY NMR spectroscopy, cryoscopy, and X-ray crystal structure analysis. Because of their relatively high endo-exo isomerization barriers, the Cα−S endo and exo diastereomers of 3−5 could be observed by NMR spectroscopy at −30 °C to −50 °C in [D8]THF. The endo diastereomer is the preferred equilibrium species under these conditions, as shown by 1H,1H HOESY experiments. Carbanion salt 3 has endo-exo isomerization barriers of ΔG#270 = 13.1±0.1 kcal/mol and 12.6±0.1 kcal/mol, while the 7-benzhydrylidene-substituted carbanion salt 5 has barriers of ΔG#288 = 13.5±0.1 kcal/mol and 13.3±0.1 kcal/mol. Cryoscopy and 6Li NMR spectroscopy of 5 in THF at −100 °C to −108 °C revealed the formation of dimers and monomers in a ratio of approximately 2:1. NMR spectroscopy of 3−5 at −90 °C to −105 °C allowed observation of the dimers and monomers of which the anions have endo conformations and also of which the anions adopt exo conformations. The NMR spectroscopic results for 3−5 are compatible with monomeric and dimeric CIPs, featuring planar allylic moieties and allylic stabilization by delocalization of the negative charge. 6Li,1H-HOESY examination of the mixture of the monomers and dimers of endo-5 and exo-5 in [D8]THF at room temperature gave only evidence for coordination of the Li atom to the O atom(s) in the CIPs. The NMR spectroscopic results for 3 were corroborated by X-ray crystal structure analysis of the monomer exo-3·PMDETA, which features (i) an essentially planar anionic C(2) atom, (ii) the exo conformation, (iii) the typical Cα−S conformation, also allowing for a stabilizing nC−σ*StBu interaction, and (iv) a single O−Li bond, but no C−Li bond. Upon treatment of the endo and exo sulfones 9, 10, and 12 with nBuLi at very low temperatures, the corresponding endo and exo carbanion salts endo-3−5 and exo-3−5, respectively, were selectively generated as mixtures of monomers and dimers, the reactions of which with electrophiles were studied. Deprotonation of the exo and endo sulfones with nBuLi proceeds stereoselectively, the exo sulfone preferentially giving the endo anion and vice versa. The diastereomeric endo and exo carbanion salts 3−5 each react with reactive electrophiles at the anionic C(2) atom syn to the sulfonyl O atoms, giving the corresponding substituted endo and exo sulfones, respectively, with significant degrees of asymmetric induction. Reactions of the endo and exo diastereomers of 3−5 with CF3COOD and MeOCH2I were faster than their endo-exo isomerization and approached kinetic quenching, while those with MeI and allyl iodide were slower, approaching the Curtin−Hammett limit and preferentially giving the exo sulfones. Deprotonation-deuteration experiments of the 7-oxa-sulfone endo-11 showed that the corresponding 7-oxa-substituted carbanion salts endo-6 and exo-6 not only can be generated at low temperatures but may also, despite their tendency to rearrange, be converted into the corresponding 7-oxa-sulfones on treatment with reactive electrophiles.
Reaction of N-alkyl, N-aryl, and N-H sulfoximines with m-chloroperbenzoic acid cleanly gives the corresponding sulfones in high yield. In the case of the cleavage of N-alkyl and N-arylsulfoximines, formation of the corresponding nitroso compounds as the other reaction product was proven. Starting from enantio- and diastereopure sulfoximines, a number of chiral sulfones, including the axially chiral sulfone 6 and the sulfonyl-functionalized homoallylic alcohol 8, have been prepared. Reaction of the enantiopure sulfoximine 30 with Merrifield resin gave the polymer-bound sulfoximine 32. Oxidative cleavage of 32 afforded the sulfone 16 in high yield. Deprotonation of the sulfoximine resin 32 and reaction of Li-32 with benzaldehyde and propanal furnished the β-hydroxysulfoximine resins 33a and 33b, respectively. Oxidative cleavage of 33a and 33b readily afforded the β-hydroxy sulfones 14a and 14b, respectively.
Enantiopure acyclic (E)- and (Z)-configured allylic sulfoximines have been synthesized from N,S-dimethyl-S-phenylsulfoximine and aldehydes by the addition− elimination−isomerization route through the intermediate generation of the corresponding (E)-configured vinylic sulfoximines. Isomerization of the vinylic sulfoximines with DBU preferentially afforded the corresponding (Z)-configured allylic sulfoximines, which were subsequently isomerized by DBU to preferentially yield the (E)-isomers. Titanation of lithiated (E)-configured allylic sulfoximines with ClTi(OiPr)3 furnished the corresponding bis(2-alkenyl)diisopropyloxytitanium(IV) complexes, which reacted with aldehydes in the presence of ClTi(OiPr)3 with high regio- and diastereoselectivities at the γ-position to give the corresponding (Z)-anti-configured δ-N-methylsulfonimidoyl-substituted homoallylic alcohols in good yields. In the absence of ClTi(OiPr)3 at low temperatures, only one allylic moiety of the bis(alkenyl)diisopropyloxytitanium complex is transferred to the aldehyde. In this way, a cyclic lithiated allylic sulfoximine has been converted with high regio- and diastereoselectivity to the corresponding homoallylic alcohols bearing a vinylic sulfonimidoyl group. Titanation of lithiated (E)- and (Z)-configured allylic sulfoximines with ClTi(NEt2)3 afforded the corresponding mono(2-alkenyl)tris(diethylamino)titanium(IV) complexes, which reacted with aldehydes with moderate to high regioselectivities and high diastereoselectivities preferentially at the α-position to give the corresponding syn-configured β-N-methylsulfonimidoyl-substituted homoallylic alcohols along with the (Z)-anti-configured δ-N-methylsulfonimidoyl-substituted homoallylic alcohols in good yields. In this way, the cyclic lithiated allylic sulfoximine was converted with high regio- and diastereoselectivity to the corresponding isomeric homoallylic alcohols bearing an allylic sulfonimidoyl group. In the case of mono(alkenyl)tris(diethylamino)titanium(IV) complexes, the regioselectivity of their reactions with aldehydes has been found to depend on the size of the substituent at the CC double bond and the aldehyde, as well as on the configuration of the double bond. Reaction of racemic lithiated N-methyl-S-(3,3-diphenyl-2-propenyl)-S-phenylsulfoximine with ClTi(OiPr)3 afforded the corresponding bis(alkenyl)diisopropyloxytitanium(IV) complex. X-ray structure analysis revealed a distorted octahedral cis,cis,cis-configured bis(2-alkenyl)diisopropyloxytitanium(IV) complex, in which the allylic moieties are coordinated in a bidentate fashion through C-α and the N atom to the Ti atom, both having the relative configuration RSSC. In solution, the titanium complex shows fluxional behavior, which is characterized by topomerization of the isopropyloxy groups and allylic moieties. The exchange of the latter occurs with retention of the configuration at C-α.
An asymmetric synthesis of isocarbacyclin (2) was achieved from ketone 7 by the olefination-isomerization-coupling process with chiral sulfoximines. The vinylic sulfoximine 6 (≥98% de) was prepared from ketone 7 and lithiosulfoximine 8 by an asymmetric olefination via an addition-elimination process. Model experiments, aiming at a rationalization of the asymmetric induction in the elimination of β-hydroxysulfoximines, with ketone 12 and lithiosulfoximine ent-8 revealed formation of the silyl ether 15 as an intermediate which eliminated LiOSiMe3 upon reaction with nBuLi under formation of (S,Z)-alkene 17 (≥98% de). Reaction of the C, O-dilithiosulfoximine 19 with ClSiMe3 led to elimination of LiOSiMe3 and also gave 17 (≥98% de). Methylation of 19, however, furnished the corresponding α-methyl-substituted β-hydroxysulfoximines, 20 and 21, in a ratio of 75:25. Isomerization of sulfoximine 6 gave the allylic sulfoximine 5 (96% de) whose absolute configuration was determined by X-ray structure analysis. Cross-coupling reaction of 5 with cuprate 23 delivered with high regioselectivity alkene 25. A similar reaction of 5 with the organocopper reagent 26, which was prepared from (benzyloxy)methylmagnesium chloride, in the presence of BF3 · OEt2 and halide afforded alkene 27. Ketone 28 is a potential starting material for the asymmetric synthesis of 3-oxaisocarbacyclin. Besides alkenes 25 and 27 sulfinamide 24 (97% ee), whose conversion to 8 has been already described, was isolated in 90% yield. The key step in the sequence leading to the construction of the ω-side chain was the deprotonation of ketone 4b with a complex of lithium (R,R)-bis(α-phenylethyl)amide and lithium chloride, 29 · LiCl, which gave enolate 3. The use of ent-29 · LiCl in the deprotonation of 4b afforded the isomeric enolate 30. Enolates 3 and 30 were trapped as the silyl ethers 31 (90% ie) and 32 (92% ie), respectively. The aldol reaction of 3 with (E)-octenal proceeded highly selective in regard to C-12 but unselective in regard to C-13 and gave aldols 34 (42%) and epi-34 (36%). It was at the stage of the aldol reaction of 3 where the unwanted diastereomers 35 and epi-35, stemming from 30, could be separated. Reduction of ketones 34 and epi-34 afforded diols 36 (≥98% de) and 37 (93% de), respectively. The Pd-catalyzed rearrangement of the allylic diacetates 39 and 41 was highly stereoselective (≥98% de) but incomplete and led to formation of mixtures of 40 and 39 as well as of 42 and 41 in ratios of 84:16 and 86:14, respectively. A two-step oxidation of alcohol 43, contaminated by 5% of the isomeric alcohol stemming from acetate 39, via aldehyde 44 gave after purification by crystallization isocarbacyclin (2) in 38% yield. Diol 45, having the undesired (15R) configuration, was selectively oxidized with dichlorodicyanobenzoquinone to enone 46 (81%).
Asymmetric total syntheses of 3-oxacarbacyclin (4) and 3-oxaisocarbacyclin (5) have been achieved by a new and common route. The key step of these syntheses is an enantioselective deprotonation of the prochiral ketone 25 with lithium (R,R)-bis(phenylethyl)amide (12) in the presence of LiCl. Treatment of the thus formed enolate 26 with ClSiEt3 gave the enol ether 27 of 92% ee in 94% yield. Deprotonation of the analogous prochiral ketone 9 with 12 in the presence of LiCl followed by reaction of the enolate 13 with ClSiEt3 led to isolation of the silyl enol ether 8b of 92% ee in 95% yield. A study of the deprotonation of 9 with the chiral lithium amides 14−19 showed that 12 in combination with LiCl is the optimal base in terms of enantioselectivity and accessibility. The ω-side chain in 4 and 5 was established by a Mukaiyama reaction of 27 with the unsaturated aldehyde 28, leading to ketone 39 of 90% de, in combination with a stereoselective Pd-catalyzed allylic rearrangement of acetate 47 to the isomeric acetate 48 and a Mitsunobu reaction of the allylic alcohol 49. The key step in the construction of the α-side chain in 4 is a Horner-Wadsworth-Emmons reaction of ketone 7c with the 8-phenylnormenthol-containing phosphonoacetate 56 which gave ester 60 of 90% de. Ester 60 was obtained diastereomerically pure by chromatography in 72% yield from 7c. Reduction of 60 furnished the allylic alcohol 62 which was converted to 4 in a standard fashion. It is at the stage of the α,β-unsaturated ester 60 where divergence into synthesis of 5 was made. Selective isomerization of 60 to the β,γ-unsaturated ester 66 of 97% ie in 91% yield was accomplished by deprotonation of 60 with 12 to enolate 65 and its subsequent regioselective protonation. By a similar reaction sequence the isomeric α,β-unsaturated ester 61 was converted to the ß,γ-unsaturated ester 69 of 97% ie in 88% yield. Reduction of 66 afforded the homoallylic alcohol 71 which was converted to 5 in a standard fashion.
The asymmetric Horner–Wadsworth–Emmons (HWE) reaction of the prochiral ketones 8a/b with the phosphonoacetates 10a, ent-10a, 10b and rac-16, which contain 8-phenylmenthol, 8-phenylnormenthol and trans-2-(triphenylsilyl)-cyclohexanol, respectively, as chiral auxiliaries, were studied. The HWE reaction of 8a with the phosphonoenolates Li-10a/b at low temperatures gave the esters 7a/b and 11a/b in high yields with diastereoselectivities up to 97:3. The (E)-configured esters 7a/b serve as starting material for the total synthesis of (+)-3-oxacarbacyclin. Similarly, the reaction of 8b with ent-Li-10a gave the esters ent-7b and ent-11b in a ratio of 95:5 in 82% yield. The olefination of 8b with rac-16 afforded a mixture of the esters rac-17 and rac-18, of yet unassigned configuration, in a ratio of 70:30. The HWE reactions of 8a/b with Li-10a, ent-Li-10a and rac-Li-10b show linear temperature–diastereoselectivity relationships. The stereochemical course of the HWE reactions can be understood in terms of a selective addition of the chiral (E)-configured phosphonoenolate from its least hindered side to the ketone 8 at the convex side. The asymmetric Peterson reaction of 8b with the silyl enolate Li-20 gave the esters ent-7b and ent-11b in a ratio of 89:11. The diastereomerically pure β-hydroxy esters 22, rac-25 and 27, which contain 8-phenyl-menthol, trans-2-(triphenylsilyl)cyclohexanol and 3-{[(3,5-dimethylphenyl)phenylsulfonyl]amino}isoborneol, respectively, as chiral auxiliaries, were prepared by a highly selective addition of the enolates Li-21, rac-Li-24 and Li-26 to the ketone 8b. The asymmetric Martin dehydration of 22, rac-25 and 27 with the sulfurane 23 proceeds with stereoselectivities ranging from 82:18 to 99:1. Interestingly, the HWE, Peterson and Martin reactions involving 8-phenylmenthol as the chiral auxiliary all proceed with the same sense of asymmetric induction.
The asymmetric synthesis of (+)-3-oxacarbacyclin (1) from cis-bicyclo[3.3.0]octane-2,5-dione has been achieved. A notable feature in this synthesis is an asymmetric Horner-Wadsworth-Emmons (HWE) reaction. A further key step is the stereoselective deprotonation of a chiral bicyclic ketone, having a high local symmetry about the carbonyl group, with lithium (R,R)-bis(phenylethyl)amide in the presence of lithium chloride. The route described also allows the synthesis of the anti-thrombotic and anti-metastatic prostacyclin analogs cicaprost and eptaloprost. The flexibility of this route should also provide access to further side-chain modified analogs of 1.
![BENZENE, [(1-ETHYL-1-METHYLPROPYL)SULFONYL]-](http://img.cochemist.com/ccimg/813500/813436-95-2.png)
![BENZENE, [(1-ETHYL-1-METHYLPROPYL)SULFONYL]-](http://img.cochemist.com/ccimg/813500/813436-95-2_b.png)
![Benzene, [(1-ethylpropyl)sulfonyl]-](http://img.cochemist.com/ccimg/813500/813436-63-4.png)
![Benzene, [(1-ethylpropyl)sulfonyl]-](http://img.cochemist.com/ccimg/813500/813436-63-4_b.png)
![Cyclopentene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3R)-](http://img.cochemist.com/ccimg/627900/627854-37-9.png)
![Cyclopentene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3R)-](http://img.cochemist.com/ccimg/627900/627854-37-9_b.png)
![Cyclooctene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3R)-](http://img.cochemist.com/ccimg/627900/627854-06-2.png)
![Cyclooctene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3R)-](http://img.cochemist.com/ccimg/627900/627854-06-2_b.png)
![Cyclohexene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3R)-](http://img.cochemist.com/ccimg/627900/627853-95-6.png)
![Cyclohexene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3R)-](http://img.cochemist.com/ccimg/627900/627853-95-6_b.png)
![Cycloheptene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3S)-](http://img.cochemist.com/ccimg/204600/204578-96-1.png)
![Cycloheptene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3S)-](http://img.cochemist.com/ccimg/204600/204578-96-1_b.png)
![Cyclohexene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3S)-](http://img.cochemist.com/ccimg/204600/204578-95-0.png)
![Cyclohexene, 3-[(1,1-dimethylethyl)sulfonyl]-, (3S)-](http://img.cochemist.com/ccimg/204600/204578-95-0_b.png)
![3-Heptene, 5-[(1,1-dimethylethyl)sulfonyl]-, (3E,5R)-](http://img.cochemist.com/ccimg/204600/204578-91-6.png)
![3-Heptene, 5-[(1,1-dimethylethyl)sulfonyl]-, (3E,5R)-](http://img.cochemist.com/ccimg/204600/204578-91-6_b.png)

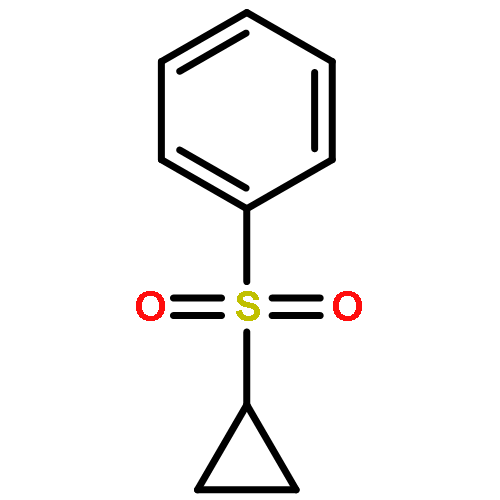
![Benzene, [(1,1-dimethyl-2-propenyl)sulfonyl]-](http://img.cochemist.com/ccimg/72900/72863-20-8.png)
![Benzene, [(1,1-dimethyl-2-propenyl)sulfonyl]-](http://img.cochemist.com/ccimg/72900/72863-20-8_b.png)
![Propane, 2-methyl-2-[(1-methylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/69500/69489-93-6.png)
![Propane, 2-methyl-2-[(1-methylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/69500/69489-93-6_b.png)




![Methanamine,N-[(S)-methyloxidophenyl-l4-sulfanylidene]-](http://img.cochemist.com/ccimg/34000/33993-53-2.png)
![Methanamine,N-[(S)-methyloxidophenyl-l4-sulfanylidene]-](http://img.cochemist.com/ccimg/34000/33993-53-2_b.png)
![Silane, [(1-ethoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/18300/18295-66-4.png)
![Silane, [(1-ethoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/18300/18295-66-4_b.png)
![Benzene, [(bromomethoxy)methyl]-](http://img.cochemist.com/ccimg/17700/17690-16-3.png)
![Benzene, [(bromomethoxy)methyl]-](http://img.cochemist.com/ccimg/17700/17690-16-3_b.png)