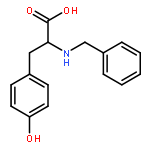
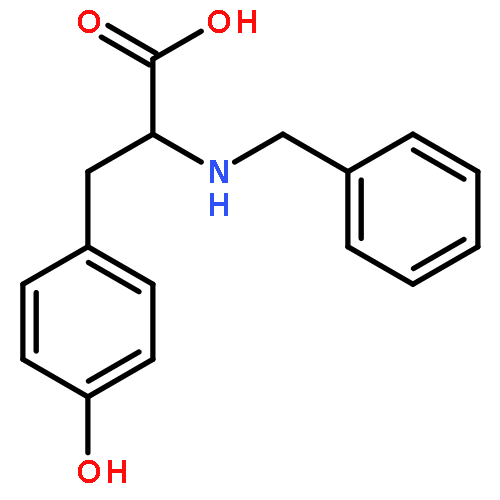
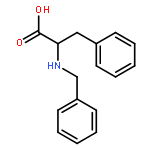
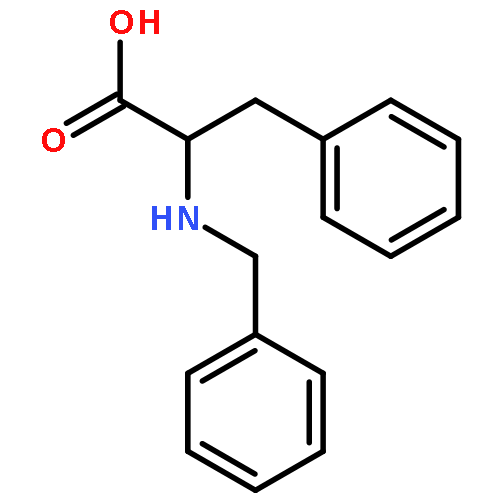
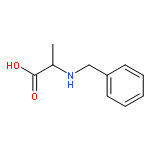
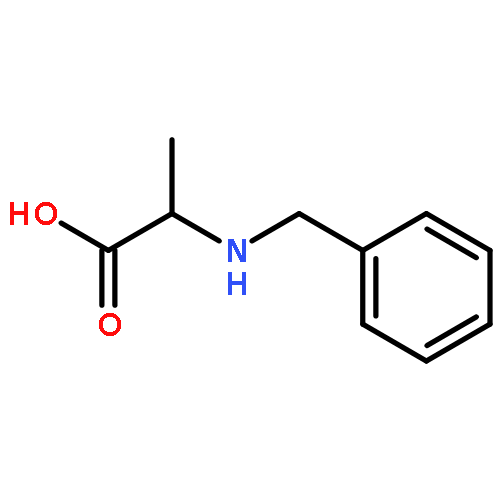
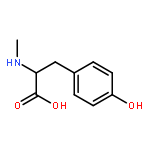
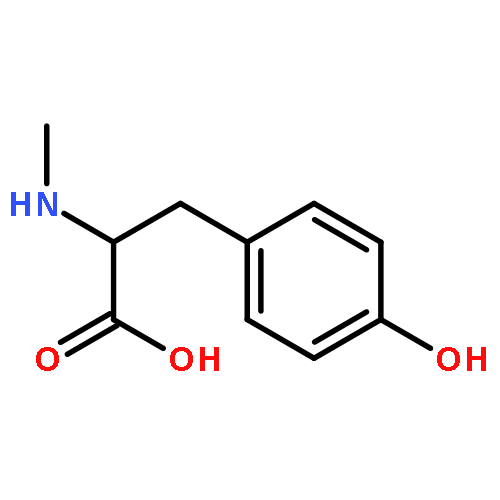
Co-reporter:J V Aldrich;S N Senadheera;N C Ross;K A Reilley;M L Ganno;S E Eans;T F Murray;J P McLaughlin
British Journal of Pharmacology 2014 Volume 171( Issue 13) pp:3212-3222
Publication Date(Web):
DOI:10.1111/bph.12664
Background and Purpose
The novel macrocyclic peptide cyclo[Phe-D-Pro-Phe-D-Trp] ([D-Trp]CJ-15,208) exhibits κ opioid (KOP) receptor antagonist activity in both in vitro and in vivo assays. The four alanine analogues of this peptide were synthesized and characterized both in vitro and in vivo to assess the contribution of different amino acid residues to the activity of [D-Trp]CJ-15,208.
Experimental Approach
The peptides were synthesized by a combination of solid phase peptide synthesis and cyclization in solution. The analogues were evaluated in vitro in receptor binding and functional assays, and in vivo with mice using a tail-withdrawal assay for antinociceptive and opioid antagonist activity. Mice demonstrating extinction of cocaine conditioned-place preference (CPP) were pretreated with selected analogues to evaluate prevention of stress or cocaine-induced reinstatement of CPP.
Key Results
The alanine analogues displayed pharmacological profiles in vivo distinctly different from [D-Trp]CJ-15,208. While the analogues exhibited varying opioid receptor affinities and κ and μ opioid receptor antagonist activity in vitro, they produced potent opioid receptor-mediated antinociception (ED50 = 0.28–4.19 nmol, i.c.v.) in vivo. Three of the analogues also displayed KOP receptor antagonist activity in vivo. Pretreatment with an analogue exhibiting both KOP receptor agonist and antagonist activity in vivo prevented both cocaine- and stress-induced reinstatement of cocaine-seeking behaviour in the CPP assay in a time-dependent manner.
Conclusions and Implications
These unusual macrocyclic peptides exhibit in vivo opioid activity profiles different from the parent compound and represent novel compounds for potential development as therapeutics for drug abuse and possibly as analgesics.
Co-reporter:Jane V. Aldrich, Sanjeewa N. Senadheera, Nicolette C. Ross, Michelle L. Ganno, Shainnel O. Eans, and Jay P. McLaughlin
Journal of Natural Products 2013 Volume 76(Issue 3) pp:433-438
Publication Date(Web):January 17, 2013
DOI:10.1021/np300697k
The macrocyclic tetrapeptide natural product CJ-15,208 (cyclo[Phe-d-Pro-Phe-Trp]) exhibited both dose-dependent antinociception and kappa opioid receptor (KOR) antagonist activity after oral administration. CJ-15,208 antagonized a centrally administered KOR selective agonist, providing strong evidence it crosses the blood–brain barrier to reach KOR in the CNS. Orally administered CJ-15,208 also prevented both cocaine- and stress-induced reinstatement of extinguished cocaine-seeking behavior in the conditioned place preference assay in a time- and dose-dependent manner. Thus, CJ-15,208 is a promising lead compound with a unique activity profile for potential development, particularly as a therapeutic to prevent relapse to drug-seeking behavior in abstinent subjects.
Co-reporter: Jane V. Aldrich;Dr. Santosh S. Kulkarni;Dr. Sanjeewa N. Senadheera;Dr. Nicolette C. Ross;Kate J. Reilley;Shainnel O. Eans;Michelle L. Ganno; Thomas F. Murray;Dr. Jay P. McLaughlin
ChemMedChem 2011 Volume 6( Issue 9) pp:1739-1745
Publication Date(Web):
DOI:10.1002/cmdc.201100113
Abstract
An alanine scan was performed on the novel κ opioid receptor (KOR) peptide ligand CJ-15,208 to determine which residues contribute to the potent in vivo agonist activity observed for the parent peptide. These cyclic tetrapeptides were synthesized by a combination of solid-phase peptide synthesis of the linear precursors, followed by cyclization in solution. Like the parent peptide, each of the analogues exhibited agonist activity and KOR antagonist activity in an antinociceptive assay in vivo. Unlike the parent peptide, the agonist activity of the potent analogues was mediated predominantly, if not exclusively, by μ opioid receptors (MOR). Thus analogues 2 and 4, in which one of the phenylalanine residues was replaced by alanine, exhibited both potent MOR agonist activity and KOR antagonist activity in vivo. These peptides represent novel lead compounds for the development of peptide-based opioid analgesics.
Co-reporter:Nicolette C. Ross, Santosh S. Kulkarni, Jay P. McLaughlin, Jane V. Aldrich
Tetrahedron Letters 2010 Volume 51(Issue 38) pp:5020-5023
Publication Date(Web):22 September 2010
DOI:10.1016/j.tetlet.2010.07.086
The tryptophan isomers of the cyclic tetrapeptide CJ-15,208, reported to be a kappa opioid receptor (KOR) antagonist [Saito, T.; Hirai, H.; Kim, Y. J.; Kojima, Y.; Matsunaga, Y.; Nishida, H.; Sakakibara, T.; Suga, O.; Sujaku, T.; Kojima, N. J. Antibiot. (Tokyo)2002, 55, 847–854.], were synthesized to determine the tryptophan stereochemistry in the natural product. A strategy was developed to select linear precursor peptides that favor cyclization using molecular modeling, and optimized cyclization conditions are reported. The optical rotation of the l-Trp isomer is consistent with that of the natural product. Unexpectedly both isomers exhibit similar nanomolar affinity for KOR.A strategy to select linear precursor peptides favoring cyclization was developed and cyclization conditions were optimized.
Co-reporter:Kshitij A. Patkar ; Thomas F. Murray ;Jane V. Aldrich
Journal of Medicinal Chemistry 2009 Volume 52(Issue 21) pp:6814-6821
Publication Date(Web):October 6, 2009
DOI:10.1021/jm900715m
Structural modifications affecting the efficacy of analogues of the endogenous opioid peptide dynorphin (Dyn) A have focused on the N-terminal “message” sequence based on the “message-address” concept. To test the hypothesis that changes in the C-terminal “address” domain could affect efficacy, modified amino acids and cyclic constraints were incorporated into this region of the partial agonist [N-benzylTyr1]Dyn A-(1−11). Modifications in the C-terminal domain of [N-benzylTyr1]Dyn A-(1−11)NH2 resulted in increased κ opioid receptor (KOR) affinity for all of the linear analogues but did not affect the efficacy of these peptides at KOR. Cyclization between positions 5 and 8 yielded [N-benzylTyr1,cyclo(d-Asp5,Dap8)]Dyn A-(1−11)NH2 (zyklophin, 13) ( J. Med. Chem. 2005, 48, 4500−4503) with high selectivity for KOR. In contrast to the linear peptides, this peptide exhibits negligible efficacy in the adenylyl cyclase (AC) assay and is a KOR antagonist. These data are consistent with our hypothesis that appropriate modifications in the “address” domain of Dyn A analogues may affect efficacy.
Co-reporter:Wei-Jie Fang ; Yanjun Cui ; Thomas F. Murray ;Jane V. Aldrich
Journal of Medicinal Chemistry 2009 Volume 52(Issue 18) pp:5619-5625
Publication Date(Web):August 28, 2009
DOI:10.1021/jm900577k
Dynorphin A (Dyn A) is an endogenous ligand for κ opioid receptors. To restrict the conformational mobility, we synthesized several cyclic Dyn A-(1−11)NH2 analogues on solid phase utilizing ring-closing metathesis (RCM) between the side chains of allylglycine (AllGly) residues incorporated in positions 2, 5, and/or 8. Cyclizations between the side chains of AllGly gave reasonable yields (56−74%) of all of the desired cyclic peptides. Both the cis and trans isomers were obtained for all of the cyclic peptides, with the ratio of cis to trans isomers depending on the position and stereochemistry of the AllGly. Most of the cyclic Dyn A-(1−11)NH2 analogues examined exhibit low nanomolar binding affinity for κ opioid receptors (Ki = 0.84−11 nM). In two of the three cases, the configuration of the double bond has a significant influence on the opioid receptor affinities and agonist potency. All of the peptides inhibited adenylyl cyclase activity in a concentration-dependent manner with full or close to full agonist activity. These potent Dyn A analogues are the first ones cyclized by RCM.
Co-reporter:Bhaswati Sinha ; Zhengyu Cao ; Thomas F. Murray ;Jane V. Aldrich
Journal of Medicinal Chemistry 2009 Volume 52(Issue 23) pp:7372-7375
Publication Date(Web):July 21, 2009
DOI:10.1021/jm9007592
A series of potent electrophilic affinity labels (IC50 = 0.1−5 nM) containing either a bromoacetamide or isothiocyanate based on the μ opioid receptor (MOR) selective peptide dermorphin were prepared. All four analogues exhibited wash resistant inhibition of [3H]DAMGO binding at subnanomolar to nanomolar concentrations, suggesting that these analogues bind covalently to MOR. To our knowledge, these peptides are the highest affinity peptide-based affinity labels for MOR reported to date.
Co-reporter:Jane V. Aldrich, Vivek Kumar, Thomas F. Murray, Wei Guang and Jia Bei Wang
Bioconjugate Chemistry 2009 Volume 20(Issue 2) pp:201
Publication Date(Web):January 28, 2009
DOI:10.1021/bc800420t
A general strategy for the design of dual labeled peptides was developed, and derivatives of the δ opioid receptor (DOR) selective antagonist TIPP (Tyr-Tic-Phe-PheOH) containing both an affinity label and biotin were prepared by solid-phase synthesis. Tyr-Tic-Phe-Phe(p-X)-Asp-NH(CH2CH2O)2-CH2CH2NH-biotin (where X = N═C═S or NHCOCH2Br) exhibit nanomolar DOR affinity. The ability to detect receptors labeled with these peptides following solubilization and SDS-PAGE demonstrate the applicability of this design approach for dual labeled peptide derivatives.
Co-reporter:Jane V. Aldrich;Jay P. McLaughlin
The AAPS Journal 2009 Volume 11( Issue 2) pp:312-322
Publication Date(Web):2009 June
DOI:10.1208/s12248-009-9105-4
While narcotic analgesics such as morphine, which act preferentially through mu opioid receptors, remain the gold standard in the treatment of severe pain, their use is limited by detrimental liabilities such as respiratory depression and drug dependence. Thus, there has been considerable interest in developing ligands for kappa opioid receptors (KOR) as potential analgesics and for the treatment of a variety of other disorders. These include effects mediated both by central receptors, such as antidepressant activity and a reduction in cocaine-seeking behavior, and activity resulting from the activation of peripheral receptors, such as analgesic and anti-inflammatory effects. While the vast majority of opioid receptor ligands that have progressed in preclinical development have been small molecules, significant advances have been made in recent years in identifying opioid peptide analogs that exhibit promising in vivo activity. This review will focus on possible therapeutic applications of ligands for KOR and specifically on the potential development of peptide ligands for these receptors.
Co-reporter:Kshitij A. Patkar;W. Edward Highsmith;Jane V. Aldrich
Amino Acids 2009 Volume 36( Issue 2) pp:203-207
Publication Date(Web):2009 February
DOI:10.1007/s00726-008-0048-3
Herein, we describe a general strategy for the facile synthesis of a multifunctional amino acid derivative bearing both fluorescent and photolabile groups such as the lysine derivative NvocLys(CO(CH2)5NH–NBD)OCH2CN (1) that can be used as a biophysical tool for studying protein structure. The synthetic strategy involves functionalization of the amine groups while the amino acid is attached to a solid support, followed by esterification of the carboxylic acid in solution. The solid support protects the caboxylic acid, preventing a side reaction associated with the synthesis in solution and obviating the need for chromatographic purification of several intermediates. This synthetic strategy can be used for the preparation of a variety of amino acid derivatives with unusual α-amine and side chain functionalities.
Co-reporter:Jane V. Aldrich;Kshitij A. Patkar;Jay P. McLaughlin
PNAS 2009 Volume 106 (Issue 43 ) pp:18396-18401
Publication Date(Web):2009-10-27
DOI:10.1073/pnas.0910180106
The cyclic peptide zyklophin {[N-benzylTyr1,cyclo(D-Asp5,Dap8)-dynorphin A-(1–11)NH2, Patkar KA, et al. (2005) J Med Chem 48: 4500–4503} is a selective peptide kappa opioid receptor (KOR) antagonist that shows activity following systemic administration.
Systemic (1–3 mg/kg s.c.) as well as central (0.3–3 nmol intracerebroventricular, i.c.v.) administration of this peptide dose-dependently
antagonizes the antinociception induced by the selective KOR agonist U50,488 in C57BL/6J mice tested in the 55 °C warm water
tail withdrawal assay. Zyklophin administration had no effect on morphine- or SNC-80-mediated antinociception, suggesting
that zyklophin selectively antagonizes KOR in vivo. Additionally, the antagonism of antinociception induced by centrally (i.c.v.)
administered U50,488 following peripheral administration of zyklophin strongly suggests that the peptide crosses the blood-brain
barrier to antagonize KOR in the CNS. Most importantly, the antagonist activity of zyklophin (3 mg/kg s.c.) lasts less than
12 h, which contrasts sharply with the exceptionally long duration of antagonism reported for the established small-molecule
selective KOR antagonists such as nor-binaltorphimine (nor-BNI) that last weeks after a single administration. Systemically
administered zyklophin (3 mg/kg s.c.) also prevented stress-induced reinstatement of cocaine-seeking behavior in a conditioned
place preference assay. In conclusion, the peptide zyklophin is a KOR-selective antagonist that exhibits the desired shorter
duration of action, and represents a significant advance in the development of KOR-selective antagonists.
Co-reporter:Laksana Charoenchai ; Hongyan Wang ; Jia Bei Wang ;Jane V. Aldrich
Journal of Medicinal Chemistry 2008 Volume 51(Issue 15) pp:4385-4387
Publication Date(Web):July 15, 2008
DOI:10.1021/jm800394v
A series of cyclic analogues with a lactam linkage were prepared by solid phase peptide synthesis to explore possible biologically active conformation(s) of nociceptin/orphanin FQ (N/OFQ). cyclo[d-Asp7,Lys10]- and cyclo[Asp6,Lys10]N/OFQ(1−13)NH2 exhibit high affinity (Ki = 0.27 and 0.34 nM, respectively) and high potency in the GTPγS assay (EC50 = 1.6 and 4.1 nM, respectively) at human nociceptin/orphanin FQ peptide (NOP) receptors. These analogues exhibit 2- to 3-fold higher affinity and 2- to 5-fold higher potency than the parent peptide.
Co-reporter:Jane V. Aldrich;Vivek Kumar
International Journal of Peptide Research and Therapeutics 2008 Volume 14( Issue 4) pp:315-321
Publication Date(Web):2008 December
DOI:10.1007/s10989-008-9144-1
Solid phase synthetic methodology has been developed in our laboratory to incorporate an affinity label (a reactive functionality such as isothiocyanate or bromoacetamide) into peptides (Leelasvatanakij and Aldrich J Peptide Res 56, 80, 2000), and we have used this synthetic strategy to prepare affinity label derivatives of a variety of opioid peptides. To date side reactions have been detected only in two cases, both involving intramolecular cyclization. We have identified several peptide-based affinity labels for δ opioid receptors that exhibit wash-resistant inhibition of binding to these receptors and are valuable pharmacological tools to study opioid receptors. Even in cases where the peptide derivatives do not bind covalently to their target receptor, studying their binding has revealed subtle differences in receptor interactions with particular opioid peptide residues, especially Phe residues in the N-terminal “message” sequences. Solid phase synthetic methodology for the incorporation of other labels (e.g. biotin) into the C-terminus of peptides has also been developed in our laboratory (Kumar and Aldrich Org Lett 5, 613, 2003). These two synthetic approaches have been combined to prepare peptides containing multiple labels that can be used as tools to study peptide ligand-receptor interactions. These solid phase synthetic methodologies are versatile strategies that are applicable to the preparation of labeled peptides for a variety of targets in addition to opioid receptors.
Co-reporter:Jane V. Aldrich, Jay P. McLaughlin
Drug Discovery Today: Technologies (Spring 2012) Volume 9(Issue 1) pp:e23-e31
Publication Date(Web):1 March 2012
DOI:10.1016/j.ddtec.2011.07.007
Opioid receptors are important targets for the treatment of pain and potentially for other disease states (e.g. mood disorders and drug abuse) as well. Significant recent advances have been made in identifying opioid peptide analogs that exhibit promising in vivo activity for treatment of these maladies. This review focuses on the development and evaluation of opioid peptide analogs demonstrating activity after systemic administration, and recent clinical evaluations of opioid peptides for possible therapeutic use.

![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6_b.png)